

Abstract
Background and Purpose
This study examined the effects of imidazoline I2 receptor agonists on the development of tolerance to and physical dependence on repeated morphine treatment in rats.
Experimental Approach
Two groups of rats (n = 9 per group) were trained to lever press for sucrose (10%) presentation under a fixed‐ratio 10 schedule. The rate‐suppressing effects of the opioid receptor ligands morphine and naltrexone and the I2 receptor agonist 2‐BFI were examined weekly in rats treated with either daily morphine (20 mg·kg−1, s.c.), alone or in combination with 2‐BFI (10 mg·kg−1) for 3 weeks. Changes in body weight were measured following naltrexone tests in both groups of rats. In separate experiments, the antinociceptive effects of morphine were assessed using a warm‐water tail‐withdrawal procedure in rats before and after daily treatments (7 days) with morphine (32 mg·kg−1, i.p.) alone or in combination with various doses of the I2 receptor agonists 2‐BFI, BU224 and CR4056.
Key Results
Daily treatment for 3 weeks, with morphine in combination with 2‐BFI produced significantly less tolerance to the rate‐suppressing effects of morphine and produced a decreased sensitivity to the rate‐suppressing effects of naltrexone as well as decreased naltrexone‐induced weight loss, compared with morphine‐alone group. Repeated treatment for 7 days with morphine produced antinociceptive tolerance, which was attenuated by co‐administration with 2‐BFI, BU224 or CR4056.
Conclusions and Implications
Imidazoline I2 receptor agonists attenuated the development of tolerance to and physical dependence on morphine, further supporting the therapeutic potential of combining I2 receptor agonists and opioids for pain treatment.
Abbreviations
- 2‐BFI
2‐(2‐benzofuranyl)‐2‐imidazoline
- BU224
2‐(4,5‐dihydroimidazol‐2‐yl) quinolone
- CR4056
2‐phenyl‐6‐(1H‐imidazol‐1yl) quinazoline
- MPE
maximal possible effect
- CL
confidence limits
Tables of Links
| TARGETS |
|---|
| GPCRs |
| μ opioid receptors |
| LIGANDS |
|---|
| Morphine |
| Naltrexone |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
Pain, both as a symptom and as a disease, imparts high health cost and economic loss to society. Clinically, there are many analgesics that are used either by prescription or over‐the‐counter for pain management. However, these medications are not adequate to achieve full pain relief, leaving a large population of pain patients undertreated. For example, although opioids are very effective against many painful conditions, their use are often limited due to many unwanted effects associated with repeated use, particularly constipation, physical dependence, abuse and overdose. In addition, repeated exposure to opioids leads to the development of tolerance, which is characterized by a decrease in antinociceptive efficacy (Self and Nestler, 1995). Tolerance to opioids can be overcome by escalating doses in patients to achieve equivalent pain relief, which can exacerbate the unwanted effects and lead to drug addiction and overdose. Although extensive resources have been devoted to developing new analgesics with novel mechanisms of action over many decades, clinically significant advances are still lacking (Kissin, 2010). Alternatively, combination therapy could also be a valuable strategy, which requires the use of two or more drugs at the same time to treat clinical pain. This approach has long been used to treat many disease conditions including cancer and cardiovascular disorders, and accumulating evidence also suggests the usefulness of this strategy for pain management (Smith, 2008; Orrù et al., 2014). The goal of combination therapy is to increase the therapeutic (i.e. analgesic) effectiveness of drugs, such as the opioids, while reduce dose‐limiting side effects, such as the development of tolerance and physical dependence (Smith, 2008). This is a promising drug development approach for new analgesics with better therapeutic profiles.
In preclinical studies, we and others have shown that imidazoline I2 receptor agonists are efficacious analgesics for several chronic pain conditions (Ferrari et al., 2011; Li and Zhang, 2011, 2012; Meregalli et al., 2012; Li et al., 2014). In addition, the available data suggest the potential advantage of combining I2 receptor agonists and other analgesics for pain management. For example, in animal models of acute nociception, such as hot water tail immersion and radiant tail flick, although selective I2 receptor agonists alone have little or no antinociceptive effects, they consistently enhance the effects of morphine (Sanchez‐Blazquez et al., 2000; Gentili et al., 2006; Thorn et al., 2011; Sampson et al., 2012). In pain assays where I2 receptor agonists are effective, such as the writhing test and inflammatory pain induced by complete Freund's adjuvant, the I2 receptor agonists synergically enhance the antinociceptive effects of opioids (Li et al., 2011; Thorn et al., 2015). These results suggest that I2 receptor agonists may be useful candidates for combination therapy with opioids in the treatment of pain. However, little is known of the effects of I2 receptor agonists on the unwanted effects of opioids, particularly the development of tolerance and dependence. Boronat et al. (1998) demonstrated that I2 receptor ligands prevent tolerance to morphine‐induced antinociception in rats and may produce neuroprotective effects. In addition, the selective I2 receptor agonist 2‐(2‐benzofuranyl)‐2‐imidazoline (2‐BFI) reduced the development of tolerance to morphine‐induced firing rate suppression and withdrawal‐induced hyperactivity of locus coeruleus neurons in anaesthetized rats (Ruiz‐Durantez et al., 2003). Taken together, these results suggest that I2 receptor agonists can enhance the antinociceptive effects of opioids and may also reduce the development of tolerance. Nevertheless, many aspects remain unclear. For example, there is only one study using a single dose of I2 receptor ligands that examined their effect on morphine tolerance. Further, the magnitude of I2 receptor agonist–morphine interaction is also not known. More importantly, the effects of selective I2 receptor agonists on morphine dependence have not been fully described. The purpose of the current study is to fill this gap in our knowledge by systematically examining the effects of selective I2 receptor agonists on the development of tolerance to and physical and behavioural dependence on morphine.
Methods
Animals
All animal care and experimental procedures were conducted in accordance with guidelines from the International Association for the Study of Pain (1983) and with the 2011 Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources on Life Sciences, National Research Council, National Academy of Sciences, Washington DC). They were approved by the Institutional Animal Care and Use Committee, University at Buffalo, the State University of New York. All studies involving animals complied with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Sixty‐six adult male Sprague–Dawley rats (Harlan, Indianapolis, IN, USA) were used in these studies. They were housed individually on a 12/12‐h light/dark cycle (all experiments were conducted during the light period). Animals had free access to water and food (standard rodent chows) except during experimental sessions. For the schedule‐controlled responding study, access to food was limited to 10 g per day in their home cages for several days to facilitate lever press training. Thereafter, body weights were allowed to increase at an age‐appropriate rate and then maintained at 340–350 g by providing food in the home cage after daily sessions. These rats had free access to water in the home cage and also earned 10% sucrose solution during experimental sessions. For rats used in the warm‐water tail‐withdrawal studies, free access to food and water was made available in their home cages.
Schedule‐controlled responding
We chose to perform schedule‐controlled operant responding studies because this procedure has been extensively used to characterize the pharmacology of opioids and is highly sensitive to detect alterations in opioid effects due to both the development of tolerance and dependence (Holtzman and Villarreal, 1973; Gerak and France, 1997; Smith and Picker, 1998; Brandt et al., 2001). In this procedure, tolerance is shown by an increase of the ED50 value of opioids to suppress rate of responding, while dependence is demonstrated by an increased sensitivity to the rate‐suppressing effects of opioid antagonists, such as naltrexone (Adams and Holtzman, 1990). As previously described in the work of An et al. 2012, experiments were conducted in commercially available chambers located within sound‐attenuating, ventilated enclosures (Med Associates Inc., St. Albans, VT, USA). Chambers contained two response levers; responses on the inactive (right) lever were recorded and had no programmed consequence. Data were collected using a Med Associates interface with med‐pc (version 4) software. Rats were trained to press a lever for presentation of a reward of 10% sucrose solution (100 μL) under a multiple‐cycle procedure. Each cycle began with a 10 min pretreatment period, during which the chamber was dark and responses had no programmed consequence, followed by a 5 min response period, during which a light above the active (left) lever was illuminated and rats could receive a maximum of 5 sucrose presentations by responding on the active lever. Initially, a single response produced a sucrose presentation; as performance improved, the response requirement was progressively increased across days to a final fixed ratio of 10. The light was terminated after delivery of 5 sucrose presentations or after 5 min had elapsed, whichever occurred first. Daily sessions consisted of five cycles, and rats had to satisfy the following criteria for five consecutive sessions before testing began: the daily response rate, averaged across all five cycles within a session, did not vary by more than ±20% of the average daily response rate of the previous five training sessions; and the average response rate among the five cycles of a daily session did not vary by more than ±20%. After the first test, all tests were preceded by at least one training session that satisfied the same criteria. Experiments were conducted each day, consisting of testing days and no‐drug training sessions supplementing the non‐testing days. Drugs were tested using a cumulative dosing procedure in which the dose of drug was increased by 0.25 or 0.5 log unit per cycle (i.e. 15 min inter‐injection intervals).
Warm‐water tail‐withdrawal procedure
The warm‐water tail‐withdrawal procedure was conducted as described previously (Thorn et al., 2011). Prior to initiation of the studies, rats were habituated to the procedure room, the experimenter handling and the experimental procedure. Two Dual Poly Pro water baths were used (model RS‐PB‐200; Revolutionary Science, Lindstrom, Minnesota, USA). Each water bath has two chambers with the inside dimensions of 32 cm × 17 cm × 13.3 cm (L × W × H). Tap water was heated to the pre‐set temperature (44°, 48° or 52°C) and remained stable throughout the experimental session with a range of no more than 0.4°C. The readings of the digital display on the water bath were regularly compared with an Oakton® water‐resistant digital thermometer (Oakton Instruments, Vernon Hills, Illinois, USA) to ensure temperature accuracy. Tail‐withdrawal latencies were recorded with a hand‐operated digital stopwatch (resolution = 1/100 s). A multiple‐cycle procedure was used to determine the dose–effect curves of the study drugs with an inter‐cycle time of 15 min. Briefly, rats were gently restrained, and the distal 5 to 10 cm of the tail was immersed in the water baths with different temperatures (44°, 48° and 52°C). Testing with different temperatures varied non‐systematically among rats and across cycles. When an animal failed to remove its tail within 20 s, the experimenter removed the tail from the water, and a latency of 20 s was recorded. Test sessions began with control (no drug) determinations for each temperature. For each cycle (e.g. 15 min), tail‐withdrawal latencies were measured for each of the three temperatures with ~1 min between determinations. Tests were conducted no more than once per week to minimize the possibility of inter‐test interactions. Dose–effect relationships were determined using a cumulative dosing procedure with the first cycle administered with vehicle followed by cumulative dose increasing by 0.25 or 0.5 log unit in the following cycles. For drug combination studies, the pretreatment drug was administered with the first dose of the opioids during the first minute of the cycle, and increasing doses of the opioids were administered during the following cycles.
Experimental design
For the schedule‐controlled responding studies, the experimental design is outlined in Figure 1. The dose–effect relationship of morphine, the highly selective I2 receptor agonist 2‐BFI (Thorn et al., 2012) and the opioid receptor antagonist naltrexone were determined for their effects on the rate of responding 48 h apart in two groups of rats (n = 9 per group). Following these initial tests (week 0), rats then underwent chronic daily treatment in which one group received a once‐daily injection of morphine alone (20 mg·kg−1, s.c.) while the other group received morphine in combination with 2‐BFI (20 + 10 mg·kg−1, s.c., respectively). The dose of 2‐BFI (10 mg·kg−1) was chosen because this dose is behaviorally active and has been shown to enhance the antinociceptive effects of morphine (Li et al., 2011; Thorn et al., 2011, 2012). The effects of morphine and naltrexone were redetermined following 1, 2 and 3 weeks of daily treatment. After 3 weeks of chronic treatment, the daily injections were terminated, and the effects of morphine and naltrexone were then assessed at weeks 5 and 7. The effects of 2‐BFI were examined at weeks 3 and 7. In addition to studying schedule‐controlled operant responding, naltrexone‐precipitated changes in body weight were also assessed following each weekly naltrexone test, as weight loss has been validated as a reliable indicator of morphine withdrawal (Akera and Brody, 1968; Goode, 1971). Immediately following the naltrexone test, the rats were placed into an observation cage for 30 min. The body weights for each animal were recorded prior to the operant responding session and again following the 30 min observation period. The initial dose range (week 0) for the naltrexone operant responding test increased up to a maximum dose of 3.2 mg·kg−1 and was progressively decreased each week as sensitization to the rate‐suppressing effects occurred. When the highest dose of naltrexone during the cumulative dosing test session became less than 1 mg·kg−1, a supplementary dose was then administered to achieve a total dose of 1 mg·kg−1 naltrexone to observe changes in body weight and observable withdrawal signs (Supporting Information).
Figure 1.
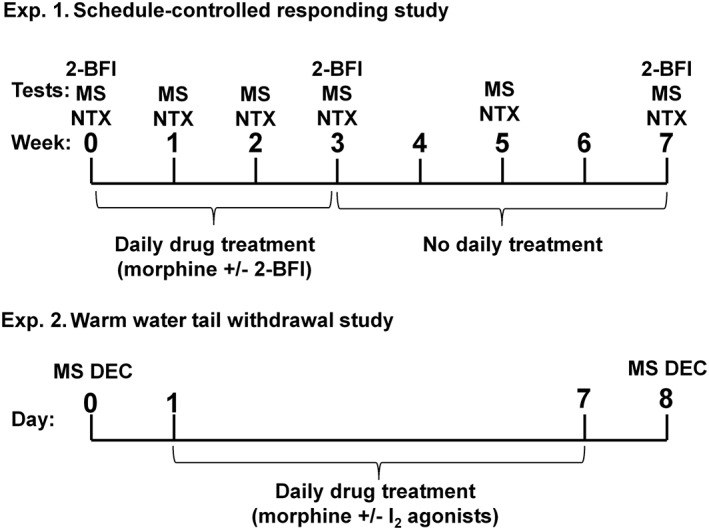
Experimental design for the schedule‐controlled responding study (top) and warm‐water tail‐withdrawal study (bottom). Top: Two groups of rats were treated daily with either morphine alone (morphine treatment) or morphine in combination with 2‐BFI (combo treatment) for 3 weeks. The rate‐suppressing effects of morphine (MS) and naltrexone (NTX) were examined on weeks 0, 1, 2, 3, 5 and 7, while the rate‐suppressing effects of 2‐BFI were examined on weeks 0, 3 and 7. (n = 9 per group.) Bottom: Different groups of rats were first tested with morphine alone, which was followed by7 days of daily morphine treatment (32 mg·kg−1). The morphine dose–effect curve was re‐determined the following day.
For the warm‐water tail‐withdrawal experiments, different groups of rats (n = 6 per group) were used to determine the dose–effect relationship of morphine. These rats were then treated once daily with morphine alone (32 mg·kg−1, i.p.) or morphine in combination with selected doses of I2 receptor agonists for 7 days, and the dose–effect relationship of morphine was then redetermined 24 h after the last daily treatment. 2‐BFI, 2‐(4,5‐dihydroimidazol‐2‐yl) quinolone (BU224) and 2‐phenyl‐6‐(1H‐imidazol‐1yl) quinazoline (CR4056) were selected because all have been shown to be highly selective I2 receptor agonists and are routinely used in studies to understand I2 receptor pharmacology (Ferrari et al., 2011; Thorn et al., 2012).
Data and statistical analyses
These studies comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). The rate of responding for 10% sucrose solution was expressed as a percentage of control responding rate, which was determined during the no‐drug training sessions prior to testing. Potencies of morphine and 2‐BFI were obtained by estimating the ED50 values using linear regression with prism (GraphPad Software, Inc., San Diego, CA, USA), along with 95% confidence limits (95% CL). Dose ratios of morphine and 2‐BFI were calculated for each individual animal and then averaged for group mean (±95% CL) to estimate potency differences each week compared with the control conditions. Dose ratios of morphine and 2‐BFI were analyzed using two‐way repeated measures ANOVA (time × treatment) followed by post hoc Bonferroni's test. The effects of naltrexone on rate of responding were analyzed using two‐way repeated measures ANOVA (dose × treatment) followed by post hoc Bonferroni's test. Changes in body weight following naltrexone administration were expressed as % body weight loss using the following formula: % body weight loss = [(post‐naltrexone body weight − pre‐naltrexone body weight)/(pre‐naltrexone body weight)] × 100. The % body weight loss was calculated for each individual subject and then averaged to obtain a group mean. Body weight loss data were analyzed using two‐way repeated measures ANOVA (time × treatment) followed by post hoc Bonferroni's test. P < 0.05 was considered statistically significant for all tests.
As previously described by Thorn et al. (2011), tail‐withdrawal latency was expressed as a percentage of the maximal possible effect (MPE) using the following formula: % MPE = [(test latency − control latency)/(20 s − control latency)] × 100, where the control latency was defined as the latency determined in the absence of drug. The MPE was calculated for each individual subject and then averaged to obtain a group mean. Within the dose range studied, morphine did not produce an effect of >50% MPE in 52°C water; thus, only data from 48°C water was used for data analysis. Potencies were obtained by estimating the dose required to produce 50% of the MPE (ED50) using linear regression, along with 95% CL. Dose ratios of morphine before and after daily treatment of morphine in the absence or presence of imidazoline I2 receptor agonists were calculated for each individual animal and then averaged for group mean (±95% CL) to estimate potency differences. The dose ratio data were analyzed using one‐way ANOVA followed by post hoc Bonferroni's test to determine the statistical significances.
Materials
2‐BFI hydrochloride, BU224 hydrochloride and CR4056 were synthesized according to described procedures (Jarry et al., 1997; Ishihara and Togo, 2007). Naltrexone hydrochloride was purchased from Sigma‐Aldrich (St. Louis, MO, USA). Morphine sulfate was provided by Research Technology Branch, National Institute on Drug Abuse, National Institutes of Health (Rockville, MD, USA). Unless otherwise noted, all drugs were dissolved in physiological saline and administered either i.p. or s.c. CR4056 was dissolved in 20% DMSO with saline and a drop of HCl. Doses are expressed as mg of the form indicated earlier per kg body weight. Injection volumes were 1 mL·kg−1.
Results
Under control conditions, morphine dose‐dependently decreased the rate of responding under a fixed‐ratio 10 schedule of sucrose presentation. Daily treatment with morphine (20 mg·kg−1, s.c.) resulted in a progressively increased rightward shift of the morphine dose–effect relationship and dose ratio of morphine (ED50 each week/ED50 control, 95% CL) = 3.11 [2.18, 4.04], 4.48 [1.68, 7.27] and 6.33 [3.08, 9.58] for weeks 1, 2 and 3 respectively) (top left panel, Figure 2). When daily treatment with morphine was terminated after week 3, the dose–effect relationship of morphine returned towards the control level, and the dose ratio of morphine decreased from 6.33 [3.08, 9.58] on week 3 to 2.38 [1.18, 3.58] and 2.07 [0.73, 3.40] on weeks 5 and 7 respectively (bottom left panel, Figure 2). In contrast, daily treatment with morphine (20 mg·kg−1, s.c.) in combination with 2‐BFI (10 mg·kg−1, s.c.) for 3 weeks resulted in less of a rightward shift of the morphine dose–effect relationship as compared with the group treated with morphine alone. For the combination group, the dose ratio of morphine was 1.94 [1.13, 2.75], 2.54 [1.66, 3.41] and 3.66 [2.60, 4.73] for weeks 1, 2 and 3 (top panels, Figure 2). When daily treatment with the combination was terminated, the dose–effect relationship of morphine returned towards the control level, and the dose ratio of morphine decreased from 3.66 [2.60, 4.73] on week 3 to 1.69 [1.22, 2.16] and 1.22 [0.79, 1.65] on weeks 5 and 7 respectively (bottom right panel, Figure 2). Furthermore, the dose ratio of morphine was plotted as a function of time (Figure 3). Two‐way ANOVA revealed significant interaction between treatment (morphine vs. combo) and time (F[2, 25] = 12.29, P < 0.0001). Post hoc analyses indicated that the treatment groups are significantly different on weeks 2 and 3.
Figure 2.
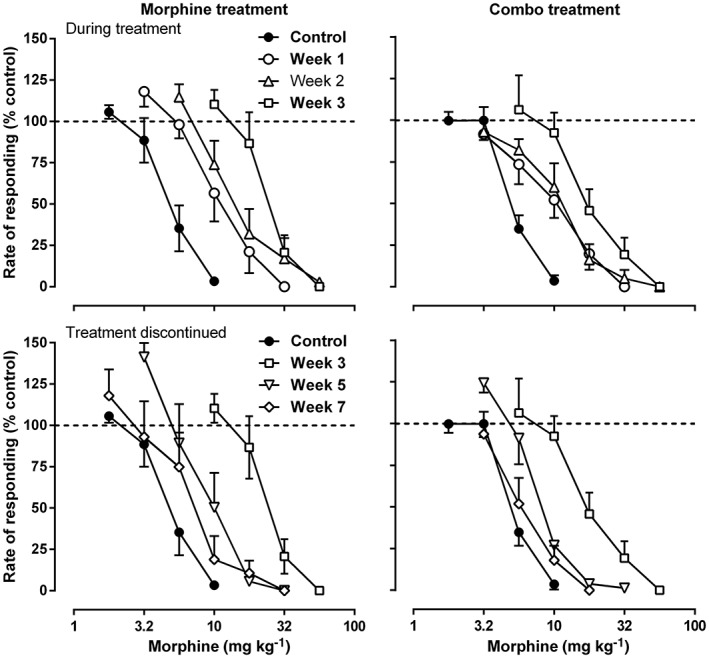
Rate‐suppressing effects of morphine before (top), during (top) and after (bottom) daily treatment of either morphine alone (left) or morphine in combination with 2‐BFI (right). Ordinate: responding rate as a percentage of control responding rate. Abscissa: dose of morphine (mg·kg−1); n = 9 per group. Linear regression was used to estimate the ED50 (±95% CL) values of morphine across the different weeks.
Figure 3.
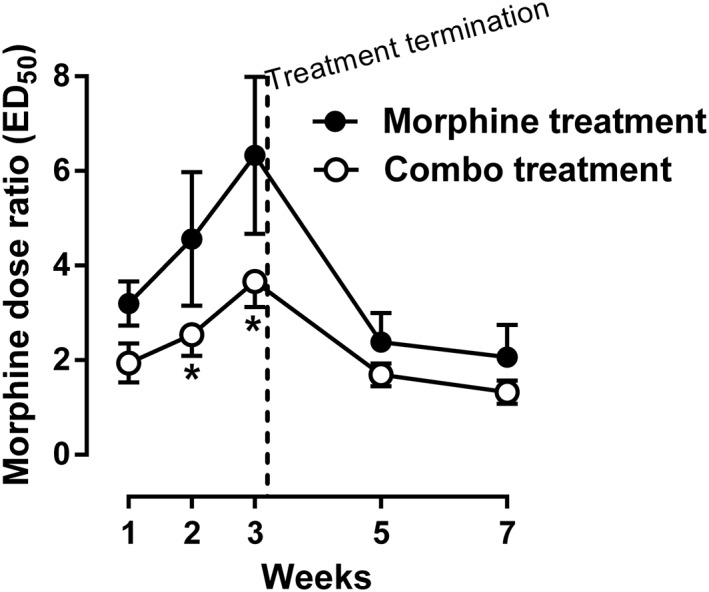
Dose ratio of morphine during and after chronic treatment of either morphine alone or morphine in combination with 2‐BFI compared with control on week 0. Data were expressed as the dose ratio of morphine and plotted as a function of time (weeks). *P < 0.05; significantly different from the morphine treatment group; two‐way repeated measures ANOVA, followed by Bonferroni's test; n = 9 per group.
2‐BFI dose‐dependently produced rate‐suppressing effects under control conditions (Figure 4). However, daily treatment with morphine did not result in a shift of the 2‐BFI dose–effect relationship or change in the 2‐BFI dose ratio on week 3 (1.47 [1.07, 1.86]) or week 7 (1.20 [1.02, 1.38]) (left panel, Figure 4). Although the group that received the combination treatment appeared to produce a slight rightward shift of the 2‐BFI dose–effect relationship on week 3, the dose ratio of 2‐BFI was not significantly different on week 3 (1.89 [1.18, 2.60]) or week 7 (1.12 [0.87, 1.37]) compared with the morphine treatment group (right panel, Figure 4).
Figure 4.
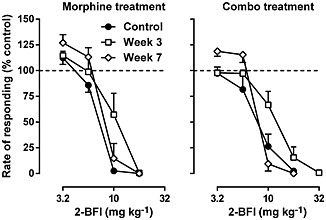
Rate‐suppressing effects of 2‐BFI before, during and after daily treatment of either morphine alone (left) or morphine in combination with 2‐BFI (right). Ordinate: responding rate as a percentage of control responding rate. Abscissa; dose of 2‐BFI (mg·kg−1). (n = 9 per group.) Linear regression was used to estimate the ED50 (±95% CL) values of morphine across the different weeks.
Initially, naltrexone did not produce any effect on the rate of responding up to a dose of 3.2 mg·kg−1, i.p. (top left panel, Figure 5). However, daily treatment with morphine produced a marked increase in the rate‐suppressing effects of naltrexone during the 3 weeks of treatment (top panels, bottom left panel, Figure 5). After 1 week of daily treatment with morphine in combination with 2‐BFI, naltrexone produced similar rate‐suppressing effects, compared with the morphine‐only treatment group (top middle panel, Figure 5). Surprisingly, the rate‐suppressing effects of naltrexone were progressively decreased during weeks 2 and 3 of daily treatment with the combination, such that on week 3, the effects of naltrexone were similar to the control level (top panels, bottom left panel, Figure 5). After daily treatment was terminated, naltrexone did not produce any rate‐suppressing effects in either group on weeks 5 and 7 (bottom middle and right panels, Figure 5). Two‐way ANOVA revealed a significant interaction between treatment and naltrexone dose for week 2 (F[4, 41] = 4.26, P < 0.01) and week 3 (F[3, 42] = 5.83, P < 0.01), but not for control or the other weeks. Post hoc analyses indicated a significant effect between treatment groups on week 3 for the 0.1 and 0.32 mg·kg−1 doses of naltrexone (bottom left panel, Figure 5). Similar to responding rate, under control conditions, naltrexone failed to produce effects on body weight. Daily morphine treatment resulted in a greater than 2% body weight loss following naltrexone administration on weeks 1, 2 and 3 (Figure 6). In contrast, the body weight loss induced by naltrexone was significantly less in the group treated with the combination than in the group treated with morphine alone. Interestingly, the effects of naltrexone in the combination group were progressively decreased across the 3 weeks of daily treatment, consistent with the rate‐suppressing effects of naltrexone shown in Figure 5. When daily treatment was terminated, naltrexone failed to produce changes in body weight in both groups of animals on weeks 5 and 7. Two‐way ANOVA revealed significant main effects of treatment (F[1, 60] = 45.02, P < 0.0001), time (F[4, 60] = 35.52, P < 0.0001) and a significant interaction between treatment and time (F[4, 60] = 3.20, P < 0.05). Post hoc analyses indicated a significant difference between treatment groups on weeks 1, 2 and 3, suggesting that the body weight loss in the combination group was significantly less than that in the morphine‐only group. Under this drug treatment regimen, no robust, observable, withdrawal signs were found even after the cumulative 1 mg·kg−1 naltrexone treatment (Supporting Information Figure S1).
Figure 5.
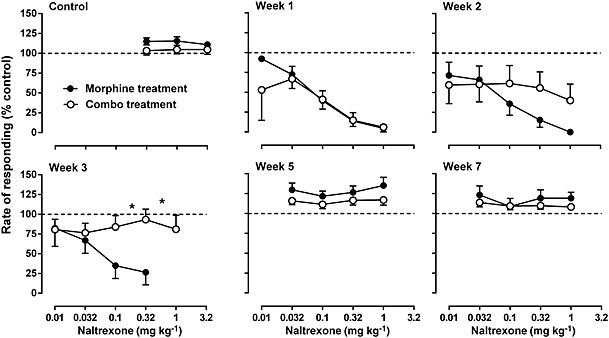
Rate‐suppressing effects of naltrexone before (top left), during (top middle and right, bottom left) and after (bottom middle and right) daily treatment of either morphine alone or morphine in combination with 2‐BFI. Daily treatment was terminated following the week 3 measurement. Ordinate: responding rate as a percentage of control responding rate. Abscissa: dose of naltrexone (mg·kg−1). *P < 0.05; significantly different from daily treatment with morphine alone; two‐way repeated measures ANOVA followed by Bonferroni's test; n = 9 per group.
Figure 6.
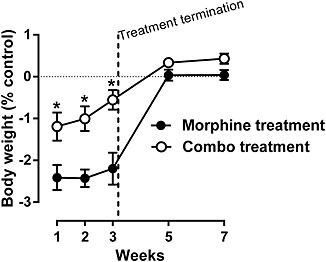
Naltrexone‐precipitated body weight loss during and after chronic treatment of either morphine alone or morphine in combination with 2‐BFI. The starting body weights before the first naltrexone test (week 1) were 331 ± 5.1 and 333 ± 5.1 g for morphine treatment and combo treatment groups respectively. Data were expressed as the % body weight loss and plotted as a function of time (weeks). *P < 0.05; significantly different from the morphine treatment group; two‐way repeated measures ANOVA followed by Bonferroni's test; n = 9 per group.
Morphine dose‐dependently produced antinociceptive effects in the warm‐water tail‐withdrawal procedure (Figure 7). Repeated treatment (7 days) of 32 mg·kg−1 morphine resulted in a marked rightward shift of the morphine dose–effect relationship, increasing the ED50 value of morphine from 4.76 [3.03, 6.50] to 51.77 [30.35, 73.19] (Figure 7). This increase in the morphine ED50 value corresponded to a greater than 10‐fold change (Figure 8). However, repeated treatment with morphine in combination with the I2 receptor agonists 2‐BFI, BU224 or CR4056 significantly decreased the morphine dose ratios, compared with the morphine‐only control (one‐way ANOVA: F[7, 44] = 3.23, P < 0.01) (Figure 8). Post hoc analyses indicated that all doses of the I2 receptor agonists produced a significant effect compared with control, except the dose of 3.2 mg·kg−1, i.p. CR4056.
Figure 7.
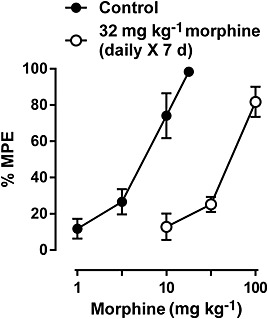
Antinociceptive effects of morphine before and after 7 days of daily morphine treatment. Ordinate: % maximal possible effect. Abscissa: dose of morphine (mg·kg−1). (n = 6 per group.) Linear regression was used to estimate the ED50 (±95% CL) values of morphine across the different weeks.
Figure 8.
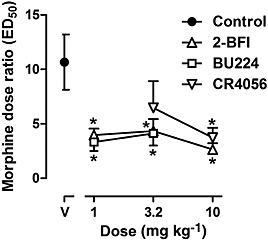
Dose ratio of morphine comparing before and after 7 days of daily treatment of morphine alone or morphine in combination with I2 receptor agonists. Each data point represents the dose ratio results from one group of animals. Data were expressed as the dose ratio of morphine and plotted as a function of dose of combined I2 receptor agonist (mg·kg−1). *P < 0.05, significantly different from the morphine treatment control group; two‐way repeated measures ANOVA followed by Bonferroni's test; n = 6 per group.
Discussion
The primary findings of the current study were that the selective imidazoline I2 receptor agonist 2‐BFI significantly attenuated the development of tolerance to the rate‐suppressing effects of morphine as well as the development of physical dependence on morphine. In addition, the selective I2 receptor agonists 2‐BFI, BU224 and CR4056 all attenuated the development of tolerance to the antinociceptive effects of morphine. These results further support the notion of combining imidazoline I2 receptor agonists with opioids for pain treatment.
Schedule‐controlled operant responding is a highly sensitive procedure to detect alterations in opioid effects due to both the development of tolerance and dependence (Holtzman and Villarreal, 1973; Gerak and France, 1997; Smith and Picker, 1998; Brandt et al., 2001). Consistent with the literature, we found that repeated treatment with morphine produced a significant rightward shift of the morphine dose–effect relationship and greater than sixfold increase of the morphine ED50 value, chanrateristics of the development of tolerance (Figures 2, 3). In contrast, repeated treatment with morphine in combination with 2‐BFI resulted in less tolerance to morphine than rats treated with morphine alone, as evidenced by a less than fourfold increase of the morphine ED50 value (Figures 2, 3). No tolerance to the rate‐suppressing effects of 2‐BFI were observed with either group (Figure 4). These results suggest that 2‐BFI attenuates the development of tolerance to the rate‐suppressing effects of morphine. However, it is well established that the development of tolerance can vary depending on the behavioural endpoint measured. For example, tolerance developed more readily to the antinociceptive effects than to the respiratory depressant effects of μ‐agonists (Paronis and Woods, 1997; Brandt and France, 2000). Tolerance has been attributed to alterations in the number and sensitivity of receptors mediating the effects of drugs. It is conceivable that discrepancies in the degree to which tolerance develops to various effects of drugs are because certain procedures or drug‐induced effects are more sensitive than others to these alterations of receptor population or sensitivity. Therefore, by investigating the effects of I2 receptor agonists during repeated treatment using multiple approaches will be particularly valuable in this regard. For this reason, we chose to study the effects of selective I2 receptor agonists on the development of tolerance to the antinociceptive effects of morphine in a warm‐water tail‐withdrawal procedure in rats.
Consistent with published results, we found that daily treatment with morphine for 7 days resulted in a significant rightward shift of the morphine dose–effect relationship and greater than 10‐fold increase in the morphine ED50 value, indicating that significant tolerance had occurred (Figures 7, 8). In contrast, daily treatment for 7 days of morphine in combination with the I2 receptor agonists 2‐BFI, BU224 or CR4056 resulted in less tolerance to morphine than rats treated with morphine alone, as shown by a less than fivefold increase in the morphine ED50 value (Figures 7, 8). These results suggest that imidazoline I2 receptor agonists attenuate the development of tolerance to the antinociceptive effects of morphine.
Repeated exposure to morphine and many other opioid agonists not only leads to the development of tolerance but can also produce physical dependence. Physical dependence can be defined as a condition in which the body has adjusted to the presence of a drug, resulting in physical symptoms of withdrawal upon discontinuation of drug treatment or administration of a pharmacological antagonist. To address the effects of I2 receptor agonists on the development of physical dependence on morphine, we studied the rate‐suppressing effects of the opioid receptor antagonist naltrexone and naltrexone‐induced body weight loss in rats treated with either morphine alone or morphine in combination with 2‐BFI. As expected, repeated treatment with morphine produced a marked increase in the sensitivity to the rate‐suppressing effects of naltrexone, a well‐characterized indication of opioid dependence (Figure 5). In striking contrast, the rate‐suppressing effects of naltrexone were progressively decreased in animals treated with morphine in combination with 2‐BFI (Figure 5). Furthermore, repeated morphine treatment produced significant body weight loss following naltrexone treatment, which was markedly decreased in the morphine and 2‐BFI combination group (Figure 6). In addition to the rate‐suppressing effects of naltrexone and naltrexone‐induced body weight loss, we also studied directly observable signs of opioid withdrawal. Although repeated morphine treatment resulted in a mildly increased occurrence of directly observed signs of withdrawal, there were no significant differences between treatment groups (Supporting Information). This is likely to be due to the morphine‐dosing regimen in these animals not being sufficient enough to induce a dependence level that can produce a significant amount of naltrexone‐induced physical withdrawal signs. In support of this hypothesis, rats did not exhibit any signs of spontaneous withdrawal following termination of daily treatment after week 3. However, concomitant treatment of morphine and 2‐BFI diminished the development of dependence on morphine as shown by a decreased sensitivity to the rate‐suppressing effects of naltrexone and naltrexone‐induced body weight loss. Both of these measurements are profoundly sensitive to changes in opioid effects due to the development of dependence (Goode, 1971; Gellert and Sparber, 1974; Adams and Holtzman, 1990). These results are consistent with an earlier study that found acute administration of the I2 receptor agonist BU224 partly attenuated the physical withdrawal signs in morphine‐dependent rats (Hudson et al., 1999).
What is particularly interesting is that there is strong evidence supporting the proposition that I2 receptor agonists enhance the antinociceptive effects of opioids. An earlier study found that selective I2 receptor agonists enhance the effects of morphine in a mouse hot water tail immersion test (Sanchez‐Blazquez et al., 2000). In addition, our group has demonstrated that I2 receptor agonists markedly potentiated the antinociceptive effects of morphine and tramadol in a rat warm‐water tail‐withdrawal test (Thorn et al., 2011) and also enhanced the effects of morphine in a hypotonic saline‐induced writhing test (Li et al., 2011). Combined with the current study, these results suggest that I2 receptor agonists significantly enhance opioid‐induced antinociception while concurrently reducing the development of tolerance to and dependence on opioids. This finding has important clinical implications. Firstly, the combination of I2 receptor agonists with opioids as an adjuvant pharmacotherapy to treat pain would maximize the beneficial effects (pain relief) while minimizing the adverse effects (tolerance and dependence), suggesting a very promising clinical treatment with a high therapeutic index. Secondly, because I2 receptor agonists markedly potentiate the antinociceptive effects of opioids, lower doses of opioids will be required to achieve adequate pain relief. This alone could possibly minimize opioid‐induced adverse effects. However, our studies suggest that I2 receptor agonists reduce the development of tolerance to and dependence on morphine at morphine doses sufficient to induce these adverse effects. If combination therapy of opioids with I2 receptor agonists results in lower doses of opioids needed to achieve adequate pain relief, I2 receptor agonists may be particularly more effective in suppressing the development of tolerance to and dependence on opioids at these lower doses.
The manner in which I2 receptor agonists inhibit the development of tolerance to and dependence on opioids remains unclear. The ability of I2 receptor agonists to potentiate opioid analgesia while also reducing tolerance and dependence on opioids argues against a simple pharmacokinetic explanation. If I2 receptor agonists were simply increasing opioid levels through an indirect effect on metabolism and/or elimination, similar effects would be expected for tolerance, dependence and analgesia. All three I2 receptor agonists used in the current study have high affinities at I2 receptors, and previous functional studies confirmed their similar pharmacological activities (Thorn et al., 2012). Because these agonists have at least 1000‐fold selectivity for I2 receptors over α2 adrenoreceptors (Thorn et al., 2012), the observed modulatory effects were more likely to be due to I2 receptor activation. Furthermore, an earlier study demonstrated that the selective I2 receptor agonist phenyzoline potentiated morphine analgesia in mice, and the modulatory effects of phenyzoline were blocked by the I2 receptor/α2 adrenoreceptor antagonist idazoxan, but not by the I1 receptor/α2 adrenoreceptor antagonist efaroxan or the α2 adrenoreceptor antagonist yohimbine (Gentili et al., 2006). These studies suggest that the ability of I2 receptor agonists to enhance opioid‐induced analgesia is an I2 receptor‐mediated effect. However, future studies aimed at investigating the mechanism of I2 receptor agonist‐induced reduction of the development of tolerance to and dependence on opioids would be beneficial.
In summary, this study has found that the selective imidazoline I2 receptor agonist 2‐BFI significantly attenuated the development of tolerance to the rate‐suppressing effects of morphine as well as the development of physical dependence on morphine. In addition, the selective I2 receptor agonists 2‐BFI, BU224 and CR4056 also attenuated the development of tolerance to the antinociceptive effects of morphine. Taken together, these results support the therapeutic potential of combining imidazoline I2 receptor agonists with opioids for pain treatment.
Author contributions
D.T. and J.L. designed the research study. D.T. and J.L. performed the study. D.T. and J.L prepared the manuscript. Y.Z. provided the compounds and participated in the manuscript preparation. D.T., Y.Z. and J.L. approved the final version of the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organizations engaged with supporting research
Supporting information
Figure S1 Average scores of withdrawal signs in rats treated with morphine alone or the combination of morphine and 2‐BFI. Withdrawal scores present data collected during the test sessions wherein 17 different observable signs were observed by two trained observers who were unaware of the treatments. In both groups, the observable signs were rarely seen and there were no differences between the two groups. Tx, treatment.
Supporting info item
Acknowledgements
This work was supported by the National Institute on Drug Abuse of the National Institutes of Health (awards no. R01DA034806 and R21DA033426). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Thorn, D. A. , Zhang, Y. , and Li, J.‐X. (2016) Effects of the imidazoline I2 receptor agonist 2‐BFI on the development of tolerance to and behavioural/physical dependence on morphine in rats. British Journal of Pharmacology, 173: 1363–1372. doi: 10.1111/bph.13435.
References
- Adams JU, Holtzman SG (1990). Tolerance and dependence after continuous morphine infusion from osmotic pumps measured by operant responding in rats. Psychopharmacology (Berl) 100: 451–458. [DOI] [PubMed] [Google Scholar]
- Akera T, Brody TM (1968). The addiction cycle to narcotics in the rat and its relation to catecholamines. Biochem Pharmacol 17: 675–688. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: G Protein‐Coupled Receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An XF, Zhang Y, Winter JC, Li JX (2012). Effects of imidazoline I2 receptor agonists and morphine on schedule controlled responding in rats. Pharmacol Biochem Behav 101: 354–359. [DOI] [PubMed] [Google Scholar]
- Boronat MA, Olmos G, Garcia‐Sevilla JA (1998). Attenuation of tolerance to opioid‐induced antinociception and protection against morphine‐induced decrease of neurofilament proteins by idazoxan and other I‐2‐imidazoline ligands. Br J Pharmacol 125: 175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt MR, France CP (2000). Chronic l‐α‐acetylmethadol (LAAM) in rhesus monkeys: tolerance and cross‐tolerance to the antinociceptive, ventilator, and rate‐decreasing effects of opioids. J Pharmacol Exp Ther 294: 168–178. [PubMed] [Google Scholar]
- Brandt MR, Furness MS, Rice KC, Fischer BD, Negus SS (2001). Studies of tolerance and dependence with the δ‐opioid agonist SNC80 in rhesus monkeys responding under a schedule of food presentation. J Pharmacol Exp Ther 299: 629–637. [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPH, Giembycz MA, et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP . Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari F, Fiorentino S, Mennuni L, Garofalo P, Letari O, Mandelli S (2011). Analgesic efficacy of CR4056, a novel imidazoline‐2 receptor ligand, in rat models of inflammatory and neuropathic pain. J Pain Res 4: 111–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellert VF, Sparber SB (1974). Utilization of operant technics to assess degree of opiate dependence after pellet implantation and naloxone administration: a comparison with body weight changes. Fed Proc 33: 501. [Google Scholar]
- Gentili F, Cardinaletti C, Carrieri A, Ghelfi F, Mattioli L, Perfumi M, et al. (2006). Involvement of I2‐imidazoline binding sites in positive and negative morphine analgesia modulatory effects. Eur J Pharmacol 553: 73–81. [DOI] [PubMed] [Google Scholar]
- Gerak LR, France CP (1997). Changes in sensitivity to the rate‐decreasing effects of opioids in pigeons treated acutely or chronically with l‐α‐acetylmethadol. J Pharmacol Exp Ther 281: 799–809. [PubMed] [Google Scholar]
- Goode PG (1971). An implanted reservoir of morphine solution for rapid induction of physical dependence in rats. Br J Pharmacol 41: 558–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman SG, Villarreal JE (1973). Operant behavior in the morphine‐dependent rhesus monkey. J Pharmacol Exp Ther 184: 528–541. [PubMed] [Google Scholar]
- Hudson AL, Gough R, Tyacke R, Lione L, Lalies M, Lewis J, et al. (1999). Novel selective compounds for the investigation of imidazoline receptors. Ann N Y Acad Sci 881: 81–91. [DOI] [PubMed] [Google Scholar]
- Ishihara M, Togo H (2007). Direct oxidative conversion of aldehydes and alcohols to 2 imidazolines and 2‐oxazolines using molecular iodine. Tetrahedron 63: 1474–1480. [Google Scholar]
- Jarry C, Forfar I, Bosc J, Renard P, Scalbert E, Guardiola B (1997). 5‐(Aryloxymethyl)oxazoline. US Patent 5,686,477. Adir e Compagnie.
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). NC3Rs Reporting Guidelines Working Group. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissin I (2010). The development of new analgesics over the past 50 years: a lack of real breakthrough drugs. Anesth Analg 110: 780–789. [DOI] [PubMed] [Google Scholar]
- Li JX, Zhang Y (2011). Imidazoline I2 receptors: target for new analgesics? Eur J Pharmacol 658: 49–56. [DOI] [PubMed] [Google Scholar]
- Li JX, Zhang Y (2012). Emerging drug targets for pain treatment. Eur J Pharmacol 681: 1–5. [DOI] [PubMed] [Google Scholar]
- Li JX, Zhang Y, Winter JC (2011). Morphine‐induced antinociception in the rat: supra‐additive interactions with imidazoline I(2) receptor ligands. Eur J Pharmacol 669: 59–65. [DOI] [PubMed] [Google Scholar]
- Li JX, Thorn DA, Qiu Y, Peng BW, Zhang Y (2014). Anti‐hyperalgesic effects of imidazoline I2 receptor ligands in rat models of inflammatory and neuropathic pain. Br J Pharmacol 171: 1580–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meregalli C, Ceresa C, Canta A, Carozzi VA, Chiorazzi A, Sala B (2012). CR4056, a new analgesic I2 ligand, is highly effective against bortezomib‐induced painful neuropathy in rats. J Pain Res 5: 151–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrù A, Marchese G, Casu G, Casu MA, Kasture S, Cottiglia F, et al. (2014). Withania somnifera root extract prolongs analgesia and suppresses hyperalgesia in mice treated with morphine. Phytomedicine 21: 745–752. [DOI] [PubMed] [Google Scholar]
- Paronis CA, Woods JH (1997). Ventilation in morphine‐maintained rhesus monkeys. II: tolerance to the antinociceptive but not the ventilatory effects of morphine. J Pharmacol Exp Ther 282: 355–362. [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SPH, Buneman OP, et al. (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucleic Acids Res 42: D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz‐Durantez E, Torrecilla M, Pineda J, Ugedo L (2003). Attenuation of acute and chronic effects of morphine by the imidazoline receptor ligand 2‐(2‐benzofuranyl)‐2‐imidazoline in rat locus coeruleus neurons. Br J Pharmacol 138: 494–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson C, Zhang Y, Del Bello F, Li JX (2012). Effects of imidazoline I2 receptor ligands on acute nociception in rats. Neuroreport 23: 73–77. [DOI] [PubMed] [Google Scholar]
- Sanchez‐Blazquez P, Boronat MA, Olmos G, Garcia‐Sevilla JA, Garzon J (2000). Activation of I(2)‐imidazoline receptors enhances supraspinal morphine analgesia in mice: a model to detect agonist and antagonist activities at these receptors. Br J Pharmacol 130: 146–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Self DW, Nestler EJ (1995). Molecular mechanisms of drug reinforcement and addiction. Annu Rev Neurosci 18: 463–495. [DOI] [PubMed] [Google Scholar]
- Smith HS (2008). Combination opioid analgesics. Pain Physician 11: 201–214. [PubMed] [Google Scholar]
- Smith MA, Picker MJ (1998). Tolerance and cross‐tolerance to the rate‐suppressing effects of opioids in butorphanol‐treated rats: influence of maintenance dose and relative efficacy at the mu receptor. Psychopharmacology (Berl) 140: 57–68. [DOI] [PubMed] [Google Scholar]
- Thorn DA, Zhang Y, Peng BW, Winter JC, Li JX (2011). Effects of imidazoline I(2) receptor ligands on morphine‐ and tramadol‐induced antinociception in rats. Eur J Pharmacol 670: 435–440. [DOI] [PubMed] [Google Scholar]
- Thorn DA, An XF, Zhang Y, Pigini M, Li JX (2012). Characterization of the hypothermic effects of imidazoline I(2) receptor agonists in rats. Br J Pharmacol 166: 1936–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorn DA, Siemian JN, Zhang Y, Li JX (2015). Anti‐hyperalgesic effects of imidazoline I2 receptor ligands in a rat model of inflammatory pain: interactions with oxycodone. Psychopharmacology (Berl) 232: 3309–3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Average scores of withdrawal signs in rats treated with morphine alone or the combination of morphine and 2‐BFI. Withdrawal scores present data collected during the test sessions wherein 17 different observable signs were observed by two trained observers who were unaware of the treatments. In both groups, the observable signs were rarely seen and there were no differences between the two groups. Tx, treatment.
Supporting info item