

Abstract
‘Induced‐fit’ binding of drugs to a target may lead to high affinity, selectivity and a long residence time, and this mechanism has been proposed to apply to many drugs with high clinical efficacy. It is a multistep process that initially involves the binding of a drug to its target to form a loose RL complex and a subsequent isomerization/conformational change to yield a tighter binding R'L state. Equations with the same mathematical form may also describe the binding of bivalent antibodies and related synthetic drugs. Based on a selected range of ‘microscopic’ rate constants and variables such as the ligand concentration and incubation time, we have simulated the experimental manifestations that may go along with induced‐fit binding. Overall, they validate different experimental procedures that have been used over the years to identify such binding mechanisms. However, they also reveal that each of these manifestations only becomes perceptible at particular combinations of rate constants. The simulations also show that the durable nature of R'L and the propensity of R'L to be formed repeatedly before the ligand dissociates will increase the residence time. This review may help pharmacologists and medicinal chemists obtain preliminary indications for identifying an induced‐fit mechanism.
Abbreviations
- [L]
concentration of ligand in bulk of solution
- RL and R'L
loose and tight binding ligand‐receptor complexes
- △Go
difference in Gibbs free energy between R and R'L
- k1, k2… k1A … k1M…
microscopic rate constants
- kobs, kon, koff
macroscopic rate constants for association and dissociation
- KD
‘thermodynamic’ equilibrium dissociation constant based on △Go
- KD*
[L] at which this observed binding is half‐maximal at equilibrium
- appKD*
[L] at which the observed binding is half‐maximal under earlier non‐equilibrium conditions
- p
‐Log
Tables of Links
| TARGETS |
| β2‐adrenoceptor |
| AT1 receptor |
| CCR5 |
| LIGANDS | |||
| Angiotensin II | EXP3174 | Maraviroc | Salmeterol |
| Aplaviroc | Gleevec | Olmesartan | Telmisartan |
| Candesartan | Losartan | Parathyroid hormone | Valsartan |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
There is currently much interest in identifying medicines with long residence times at their targets. This property has been correlated with an improved therapeutic profile for drugs without mechanism‐based toxicity (Swinney, 2004, 2006; Copeland et al., 2006; Tummino and Copeland, 2008; Zhang and Monsma, 2009). In this work, we will discuss the mechanisms and binding characteristics through which long residence times can be achieved and also discuss protocols to distinguish among these mechanisms.
Although targets also include other proteins or protein complexes such as enzymes and ion channels, they will be referred to as receptors, R, unless inappropriate. The simplest mode of ligand–receptor (L–R) interaction is a single‐step, reversible bimolecular process in which one complex, RL, is produced (Figure 1a). In compliance with Occam's razor principle, this model is most commonly used for analysing experimental data and for pharmacokinetics–pharmacodynamics simulations. However, recent real‐time spectroscopic measurements and molecular modelling approaches provide a picture in which the initially formed bimolecular complexes go through multiple states via small conformational adjustments (Garvey, 2010; Copeland, 2011; Dror et al., 2011). For simplicity, these procedures are very often reduced to a two‐step process (Figure 1b) in which the initial binding is followed by an ‘induced‐fit’‐type conformational change/isomerization of the RL complex (governed by the forward and reverse isomerization rate constants k3 and k4 respectively) to yield a more stable R'L state (Swinney, 2004; Tummino and Copeland, 2008; Lu and Tonge, 2010; Núñez et al., 2012). An identical mechanism was initially proposed by Del Castillo and Katz (1957. The complete thermodynamic cycle model shown in Figure 1c also permits R'L to be accessed by a ‘conformational selection’ mechanism in which the receptor is able to adopt the two conformations (R and R') by itself (Leff, 1995). The ligand can then bind to both of them with different affinities and this allows the distinction between agonists, inverse agonists and neutral antagonists. Yet, when the focus is on binding kinetics, this model is often simplified to invoke binding to R' only (Strickland et al., 1975; Tummino and Copeland, 2008). Here, we will only focus on the ‘induced‐fit’ mechanism, because it is now thought to account for the binding of most of the ligands with high affinity and clinical efficacy (Copeland, 2010). A list of ligands that bind via these two mechanisms was recently elaborated (Copeland, 2011). There may be even more examples that we are not aware of.
Figure 1.
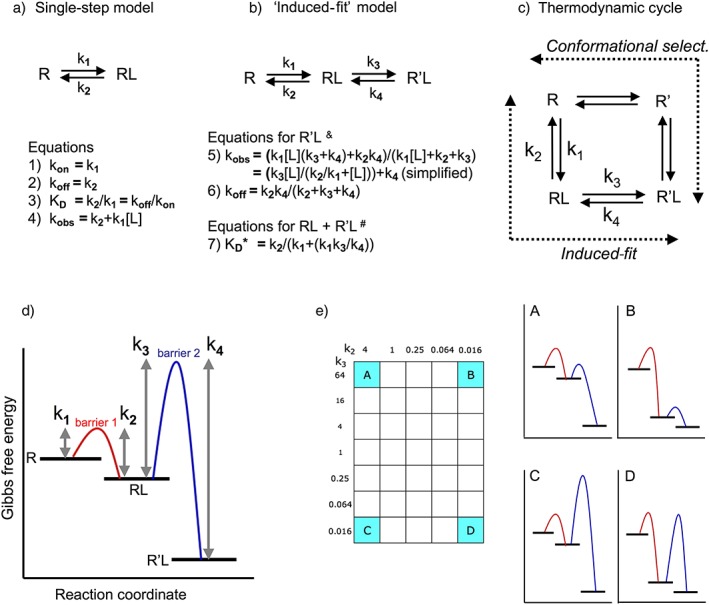
Schematic representation of (panels a to c) the binding mechanisms that are considered in this review and (panels d and e) free energy reaction coordinate diagrams of the distinct two‐step, binding situations. (a) Single‐step bimolecular binding process that obeys the law of mass action. (b) Induced‐fit model in where such bimolecular binding is followed by an isomerization of the initial RL complex into a more stable R'L complex (pertinent when k4 < k2, equations from Strickland et al. 1975; Tummino and Copeland, 2008; Lu and Tonge 2010). &Those equations apply to the timewise evolution of R'L. Because kobs and koff are macroscopic first‐order rate constants, they can be used for the analysis of association and dissociation curves when mono‐exponential; otherwise, they only apply to the slower component if such curves can be analysed in terms of a bi‐exponential model. # K D* equals [L] at which the observed binding (in where both RL and R'L participate) is half‐maximal at equilibrium. K D* acts as a ‘macroscopic’/pseudo‐binding ‘affinity’ constant. (c) More complete thermodynamic cycle model in which the receptor, R, undergoes a conformational change to yield R' either after binding of the L (according to the ‘induced‐fit’ model) or before (according to the ‘conformational selection’ model). These mechanisms correspond to the two lanes (designated by the dashed arrows) of the cycle that allow a bidirectional flow between R and R'L. Although the difference in free energy between R and R'L is fixed, all microscopic kinetic constants may differ. Please see Tummino and Copeland, 2008; Copeland, 2010, 2011 for more information. (d) General description of a two‐step reversible binding processes (Lu and Tonge, 2010; Copeland, 2011). R, RL and R'L correspond to plateaus with decreasing Gibbs free energy. To proceed from one plateau to an adjacent one, the ‘system’ must overcome an energy barrier in the appropriate direction. This barrier is known as the transition state. The difference in height between each barrier and the adjacent plateaus is inversely related to the magnitude of the corresponding ‘microscopic’ rate constants that are necessary to attain that barrier. The difference in free energy between the ground state R and the final R'L state, ∆G0 is related to those microscopic rate constants by the equation ∆G0 = −RTln K D in where K D (the ‘thermodynamic’ equilibrium KD) equals (k2.k4)/(k1.k3), R the ideal gas constant and T the temperature in degrees Kelvin. (e) Free energy reaction coordinate diagrams for the ligands that correspond to the cardinal points (A to D) of the two‐dimensional ‘kinetic space’ showing the investigated k2–k3 combinations (further referred to as the ‘grid’). The diagrams are for illustrative purposes only; the differences between the plateaus and the elevation of the energy barriers only provide a qualitative distinction between the microkinetic K D values and rate constants.
There is a growing interest by medicinal chemists in the development of drugs whose long residence time relies on two‐step/multistep binding. Yet, this information is well‐dispersed and may, therefore, be difficult and time‐consuming to come by. The present simulations mimic experimental manifestations that show up in real‐life radioligand binding conditions and also shed light on the link between them and the necessary combinations of microscopic rate constants. This review may help to obtain preliminary indications in favour of an induced‐fit‐like binding and motivate pharmacologists and medicinal chemists to adopt additional, more elaborate approaches to acquire deeper insights into this mechanism.
Constants, equations and simulations
In line with the ‘retrograde induced‐fit mechanism for drug–target dissociation’ model by Copeland (2011, we assume that the same physical processes are implicated in the bidirectional transit between R and R'L. The corresponding two‐step binding mechanism shown in Figure 1b is governed by four ‘microscopic’ rate constants that apply to the individual steps: k1 (in M−1⋅min−1), k2 (in min−1), k3 (in min−1) and k4 (in min−1). The difference in Gibbs free energy between the ground state R and the final R'L state, ∆G0 (Figure 1d), is related to these microscopic rate constants by ∆G0 = −RTln K D. K D (the ‘thermodynamic’ equilibrium K D) equals (k2.k4)/(k1.k3), R is the ideal gas constant and T is the temperature in degrees Kelvin. In line with other studies (Dahl and Akerud, 2013; Yin et al., 2013), ∆G0 was kept constant for all ligands. The (k2.k4)/(k1.k3) ratio was arbitrarily set to 4 × 10–9 M, that is, sufficiently below the k2/k1 ratio of each ligand (Supporting Information Table S2). The k1 was also kept constant at 1 × 106 M−1⋅min−1. In principle, k1 may vary between ~6 × 104 M−1⋅min−1 and the diffusion limit of ~6 × 1010 M−1⋅min−1 (Zhang and Monsma, 2009), but because the matching association rate depends as much on the ligand concentration, [L], as on k1, both can compensate for one another (Copeland et al., 2006). The binding properties are then only controlled by k2 and k3 because k4 = K D.k1.k3/k2 (values given in Supporting Information Table S2). The choice of k2–k3 combinations was inspired by the consideration that their ratio controls the balancing act between the two potential fates of RL, that is, to (re)dissociate or to (re)evolve to R'L (Vauquelin, 2013; Vauquelin et al., 2014). Explored combinations k2 and k3 are assigned by individual cases of the ‘grid’ presented in Figure 1e. The free‐energy reaction coordinate diagrams (Figure 1e) are only presented for the ligands with the most divergent k3/k2 ratios. Simulated experimental data apply to these ligands and also to others (corresponding cases are highlighted in the grid at the left side of each simulated experiment) when sufficiently relevant. Of note is that the present microscopic rate constants were selected to illustrate peculiar experimental observations that can be made within the usual time frame of classical filtration‐based radioligand binding experiments (i.e. from minutes to hours), but the same manifestations can potentially also be observed at much shorter time scales.
We will restrict our analysis to association and dissociation experiments with radiolabelled L as well as traditional ‘organ‐bath’‐type experiments with L as an antagonist, because they provide the most straightforward indications in favour of two‐step/multistep binding in the literature. In this respect, biphasic association and/or dissociation curves have often been advanced as indications of multi‐step binding in early radioligand binding studies (e.g. Kloog and Sokolovsky, 1977; Galper et al., 1977, 1982; Scheibe et al., 1984; van Giersbergen et al., 1991; Hirschberg and Schimerlik, 1994). In most of them, great care was taken to rule out concurrent labelling of distinct receptor species. For calculating the evolution of [RL] and [R'L] with time for a two‐step induced‐fit binding process, we consecutively solved the differential equations that govern the changes of each mode of receptor occupancy (shown in Supporting Information Table S2) in parallel over very small time intervals. Thereby, it becomes possible to follow how the prevalence of each bound species changes with time (Vauquelin et al., 2001a). For the experiments shown in Figure 2, L is the antagonist and A is a subsequently added one‐step (for simplicity) binding agonist. For the experiments shown in Figure 6, L is the radioligand and M is an unlabelled ligand that undergoes one‐step binding to both R and RL.
Figure 2.
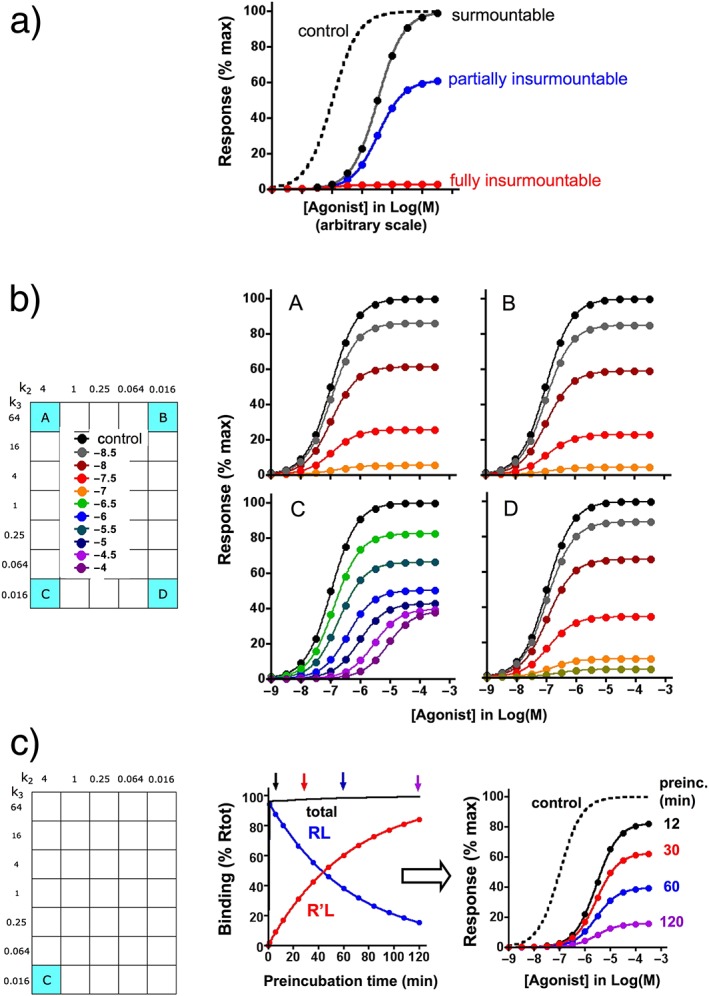
Insurmountable binding examples. Differential equations for the present simulations are given in the Supporting Information. For the sake of simplicity, it is assumed that the agonist A–receptor interaction can be represented by a fast reversible one‐step bimolecular process and also that the response takes place without delay, remains stable for the duration of the experiment and is proportional to the extent of receptor occupancy by the agonist (i.e. in the absence of receptor reserve). (a) General representation of data that can be obtained when receptors are pre‐incubated with a competitive antagonist and when the mixture is then briefly incubated with different concentrations agonist (abscissa) (Vauquelin et al., 2002a, 2002b; Kenakin et al., 2006). Very fast dissociating antagonists produce a rightward shift of the agonist concentration–response curve without affecting the maximal response; they are surmountable. Insurmountable antagonists produce a decline in the maximal response; they are fully insurmountable for irreversible antagonists and only partially insurmountable (with only a partial decline in the maximal response along with a rightward shift for the remaining response) for slow‐dissociating antagonists. In the case of induced‐fit binding, partial insurmountability may also provide a good estimate of the ratio of slow (insurmountable) and fast‐ dissociating (surmountable) antagonist–receptor complexes. Yet, the incubation with agonist has to be sufficiently long to permit quasi‐full dissociation of the surmountable complexes but short enough to prevent sizable dissociation of the insurmountable complexes. Of note is that the response is governed by the Operational model by Black and Leff (1983 in the case of ‘receptor reserve’; insurmountable binding may then still be observed, but its magnitude will be less than in the present examples (Vauquelin et al., 2002a, 2002b; Kenakin et al., 2006). (b) Distinction between the insurmountable profiles A, B, C and D when they act as antagonists. For the simulations, receptors are pre‐incubated for 1 h with different concentrations of antagonist (given as Log(M) at the left) and then further incubated with increasing concentrations (abscissa) of agonist (with k1A = 1 × 107 M−1⋅min−1 and k2A = 1 min−1) for 3 min, at which time the response is measured. Partial insurmountability is clearly demonstrated for ligand C only. (c) Ligand C: effect of the preincubation time on the degree of insurmountability. For the simulations, receptors are incubated for 12 min, 30 min, 1 h and 2 h (arrows on top of the association binding curve at the left side) with a maximally effective concentration of antagonist (0.1 mM) and then further incubated with increasing concentrations of agonist for 3 min, at which time the response is measured. The resulting agonist concentration–response curves are shown at the right side. The degree of insurmountability provides an estimation of the R'L/RL ratio, which increases with the preincubation time.
Figure 6.
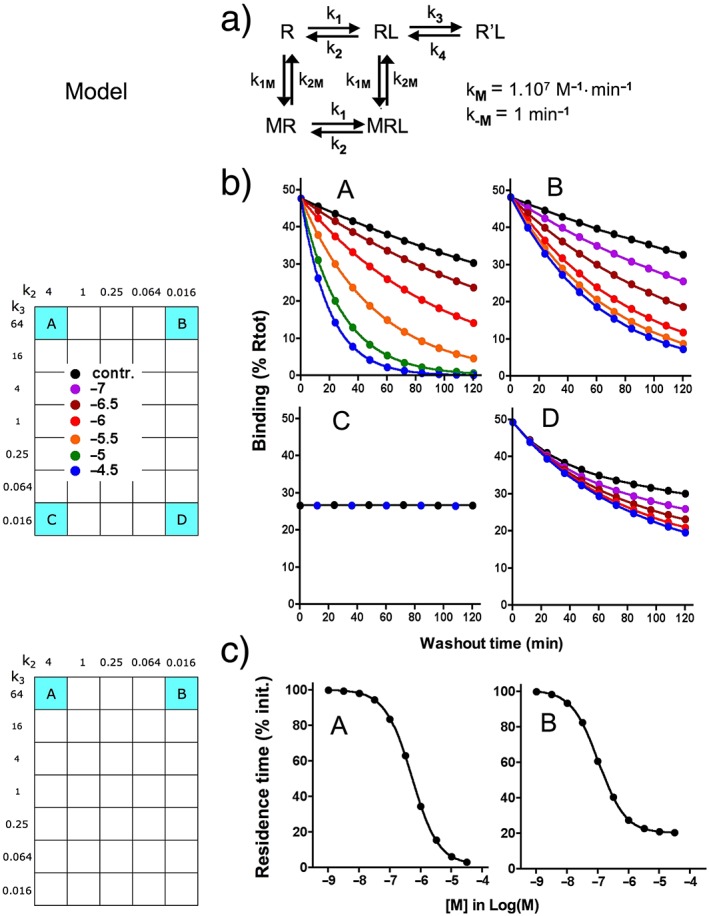
Dissociation of A to D: effect of an unlabelled monovalent ligand M that only interferes with the conversion of RL into R'L. (a) Model: binding of M to R and RL are reversible bimolecular processes with association (k1M) and dissociation (k2M) rate constants (listed). Bound M does not interfere with the binding of A to D but prevents the formation of R'L. Differential equations for the present simulations are given in the Supporting Information. (b) Simulated dissociation curves for A to D are shown for 120 min washout during which remaining binding (i.e. R'L for A and C and RL + R'L for B and D) is recorded at different time intervals. The washout starts with ‘total’ = 50% Rtot and is triggered by setting [L] = 0; it is carried out in medium only (control) or in medium containing the indicated concentrations of M. (c) Dissociation data for A and B are analysed according to a one‐phase exponential dissociation paradigm, and the residence time of those ligands (τ = 1/koff) is plotted as % of τ in naïve medium and against [M] (abscissa) in the washout. This type of analysis was not performed for C (because no effect of M) and D because, as shown in panel b, the dissociation curves evolve from a biphasic to a monophasic pattern when [M] increases.
Experimental (or simulated) association and dissociation data are usually analysed in terms of mono‐ or bi‐exponential paradigms and, according to the IUPHAR guidelines (Neubig et al., 2003), the rate constants (kobs and kon for association and koff for dissociation) derived from these analyses are categorized as ‘macroscopic’. For one‐step binding, kon equals k1, koff equals k2 and the pseudo‐first‐order rate constant, kobs, equals koff + kon.[L] (Hulme and Trevethick, 2010). The timewise evolution of R'L can only be described by kobs and koff (corresponding to equations 5 and 6 in Figure 1b). These can be obtained by analysing association and dissociation curves when mono‐exponential; otherwise, they only apply to the slower component of such curves. The hyperbolic relationship between kobs and [L] (Strickland et al., 1975) precludes the utilization of kon as a second‐order rate constant. The concentration of ligand, [L], at which this observed binding is half‐maximal at equilibrium is denoted here as K D* (to keep same terminology as in Tummino and Copeland, 2008). K D* acts as a ‘macroscopic’/pseudo binding ‘affinity’ constant and, because both RL and R'L participate in the binding, this parameter is related to the microscopic rate constants in a different way from the earlier evoked K D, that is, K D* = k2.k4/(k1.(k3 + k4) (values are provided in Supporting Information Table S2). It is also shown in Supporting Information Figure S1 that the saturation binding curves of ligands A to D undergo a leftward shift and steepening with time. Yet, it is only after an exceedingly long incubation that the binding reaches equilibrium with a Hill coefficient, n H, close to unity and with [L] for half‐maximal binding close to K D*. Yet, to deal with shorter, more realistic time frames, we introduced the term app K D* to define [L] at which the observed binding is half‐maximal under non‐equilibrium conditions (values after 1 h incubation are provided in Supporting Information Table S2). While K D* is a genuine constant, app K D* is not because its value depends on the incubation time.
Important contribution of durable R'L
Durable R'L constitutes the centrepiece of the genuine induced‐fit model in the dedicated review articles (Tummino and Copeland, 2008; Zhang and Monsma, 2009; Lu and Tonge 2010). Ligands which, like C and D, are represented at the lower portion of the grid fit best with this paradigm because R'L furnishes an important contribution to the binding free energy/affinity and the ‘reverse isomerization’ between R'L and RL requires a high energy barrier to be overcome. Examples include sartans like candesartan and olmesartan (Van Liefde and Vauquelin, 2009) and CCR5 allosteric modulators such as maraviroc (Swinney et al., 2014).
Sartans constitute a family of AT1‐type angiotensin II (Ang II) receptor blockers that lower BP with few side effects (Michel et al., 2013). Indications of induced‐fit binding first emanated from ‘organ‐bath’‐type functional assays (Fierens et al., 1999a, 1999b). The assays involved pre‐incubating the tissues/cells with antagonist and then adding agonist (Leff and Martin, 1986). Earlier rabbit aortic strip contraction studies revealed that losartan and some other sartans shifted the Ang II concentration–contraction curves to the right (as shown in Figure 2a). Such a pattern is termed ‘surmountable’ and is observed with fast‐dissociating competitive antagonists; the response then reflects the emergence of new mass–action equilibrium. In contrast, other sartans decreased the maximal response as well, especially when their imidazole‐derived moiety carried a carboxyl group. Such a pattern is termed ‘insurmountable’ (Figure 2a) and can point to non‐competitive antagonism (Wienen et al., 1992; Christopoulos and Kenakin, 2002). Insurmountable behaviour may also be observed for competitive antagonists when they dissociate sufficiently slowly (Vauquelin et al., 2002a, 2002b; Kenakin et al., 2006).
The sartans exhibited the same insurmountable behaviour in assays with plated human AT1 receptor‐expressing CHO cells. In those assays, the production of inositol phosphates accounted for the response (Fierens et al., 1999a, 1999b; Verheijen et al., 2000; Vanderheyden et al., 2000a; Le et al., 2007). Non‐competitive antagonism was ruled‐out; all sartans only shifted the Ang II concentration–response curves to the right under co‐incubation conditions. Also, all the insurmountable sartans only produced a partial decline in the maximal response to Ang II (such as in Figure 2a), and the maximal extent of this decline differed between the sartans (Table 1).
Table 1.
Binding characteristics of different sartans to the human AT1 angiotensin II receptor stably expressed in recombinant CHO cells
| Sartan | COOH present | App. affinity versus candesartan | Dissociation t 1/2 (min) | Insurmountable (%) | Insurmountable if one step (%) |
|---|---|---|---|---|---|
| Candesartan | + | 1 | 120 | 95 | 98.5 |
| Olmesartan | + | 0.73 | 75 | 85 | 97.7 |
| EXP3174 | + | 0.45 | 30 | 70 | 94.4 |
| Telmisartan | − | 0.083 | 25 | 70 | 94.4 |
| Valsartan | + | 0.17 | 17 | 50 | 90.5 |
| Irbesartan | − | 0.15 | 7 | 30 | 78.9 |
| Losartan | − | 0.014 | <0.1* | 0 | <5.4 |
Adapted from Van Liefde and Vauquelin, 2009; data are from Fierens et al., 1999a, 1999b; Vanderheyden et al., 2000a, 2000b; Verheijen et al., 2000; and Le et al., 2007)
A carboxyl (COOH) group at the imidazole‐derived moiety contributes to the affinity and insurmountable behaviour of the sartans (e.g. compare the characteristics of EXP3174 with those of its precursor losartan where the carboxyl group is substituted by a hydroxyl group). Dissociation half‐lives were calculated from mono‐exponential dissociation curves of tritiated sartans and from functional experiments. Their affinities relative to candesartan are from [3H]‐valsartan competition‐binding experiments. Their degree of insurmountability refers to the largest decline in maximal Ang II‐mediated response (i.e. cumulated inositol triphosphate production during 5 min, in the absence of desensitization) after 30 min preincubation with sartan alone. These experimental data are compared with the degree of insurmountability that is expected for antagonists that bind according to a one‐step reversible bimolecular process and dissociate with the cited half‐lives.
Taking account of a potential 5% error on the degree of insurmountability of losartan, these latter calculations suggest that it dissociates with a half‐life of less than 0.1 min.
The response was not amplified (i.e. no ‘receptor reserve’) in either experimental system (Zhang et al., 1993; Le et al., 2005). In such a situation, the magnitude of the response is always proportional to the extent of receptor occupancy by the agonist. For the sake of simplicity, the simulations shown in Figure 2 also represent such situations. Mono‐exponential dissociation of the sartan–receptor complexes during the incubation with Ang II constitutes the simplest explanation for the observed partial insurmountability (Hall and Parsons, 2001; Lew and Ziogas, 2004). Yet it did not explain additional findings. While tritiated sartans dissociated mono‐exponentially under identical assay conditions (i.e. temperature, medium and the use of the same intact plated cells), this process was far too slow to account for the maximal extent of insurmountability in the functional assays (Table 1) (Fierens et al., 1999a, 1999b; Vanderheyden et al., 2000a; Verheijen et al., 2000; Le et al., 2007). Moreover, the responsiveness of Ang II was also shown to resume in a biphasic manner with time in functional washout experiments with olmesartan‐ and telmisartan‐pretreated cells. After an initial very rapid phase, the response recovered to the same rate as observed for the dissociation of the radiolabelled sartans (Le et al., 2007). These latter results support a two‐step induced‐fit binding model in which the formation of a highly unstable RL complex (accounting for surmountable inhibition) is followed by an isomerization into a more stable R'L complex (accounting for insurmountable inhibition) (Van Liefde and Vauquelin, 2009). A model was proposed in which the sartan's common biphenyltetrazole moiety binds first to form RL. The subsequent binding of their imidazole‐derived moiety (to yield R'L) then extends the residence time, especially when this moiety bears a carboxyl group (Table 1) (Van Liefde and Vauquelin, 2009; Ojima et al., 2011). Such structural elements may explain why the extent of insurmountability differs from one sartan to another (Van Liefde and Vauquelin, 2009). In this respect, site‐directed AT1 receptor mutagenesis and molecular modelling studies identified Lys199 as an important contributor to the stability of R'L (Fierens et al., 2000; Vauquelin et al., 2001b; Tuccinardi et al., 2006; Bhuiyan et al., 2009).
This example illustrates that the partial insurmountability of competitive antagonists may indicate an induced‐fit binding mechanism. Yet, as shown in Supporting Information Figure S3 for ligands A to D, the extent of insurmountability will gradually decline when the incubation with the agonist is prolonged. Ultimately, a new equilibrium between agonist, antagonist and receptor will be reached. The insurmountable nature of slow‐dissociating antagonists is therefore merely apparent and may only be observed when the challenge with the agonist is sufficiently brief. In contrast, surmountable antagonists dissociate so fast that such equilibrium is already reached within the time frame of the experiment. Finally, the duration of the pre‐incubation is also important. For ligands like C, RL is the most prominent species after a short pre‐incubation, but the contribution of R'L increases gradually when this preincubation is prolonged (Figure 2c). The binding of this antagonist will thus become increasingly insurmountable with time. Nonetheless, the pA2 values of the remaining surmountable component of C (Arunlakshana and Schild, 1959; plots are shown in Supporting Information Figure S4) do not vary irrespective of the pre‐incubation time (Figure 2c) and concentration of C (Figure 2b).
In traditional filtration‐based radioligand binding assays, RL and R'L will both contribute to bound radioligand provided that free radioligand molecules are removed faster than the dissociation of RL. However, only R'L may remain detectable with sartan‐like ligands C, E and F because of their high k2 value. The present simulations were based on this assumption. As shown in Figure 3, these radioligands will produce mono‐exponential association and dissociation curves (Figure 3a and b). All radiolabelled insurmountable sartans showed the same binding pattern in intact cell experiments (Fierens et al., 1999a, 1999b; Vanderheyden et al., 2000a; Verheijen et al., 2000; Le et al., 2007), and this can be attributed to the need to rinse the plated cells repeatedly with fresh medium before the measurement. Yet, evidence for an induced‐fit mechanism can still be obtained for such ligands by performing association experiments with multiple concentrations of radioligand and by plotting the kobs as a function of [L]. For a simple one‐step binding, such plots are linear with kobs = koff + kon.[L] (this also offers a convenient method to calculate kon and koff). However, when these plots are hyperbolic, such as for C (Figure 4a), they are more consistent with an induced‐fit mechanism. This pattern has been repeatedly observed in enzymology (e.g. Morrison, 1982; Harris Callan et al., 1997) and in radioligand binding studies (Järv et al., 1979; Toomela et al., 1994). The hyperbolic shape is due to the fact that the bimolecular binding and isomerization steps are rate limiting at low and high [L], respectively (Castro et al., 2005). Nonlinear regression analysis of such plots in terms of equation 5 in Figure 1b (Strickland et al. (1975) provides a value for the microscopic K D of the first binding step (i.e. k2/k1), k3 and k4. For C, this analysis provides a good estimate of k3 and the k2/k1 ratio, but a negative value was obtained for k4 (Table 2). Difficulty in obtaining k4 has also been experienced by others, especially when R'L is very stable (Toomela et al., 1994).
Figure 3.
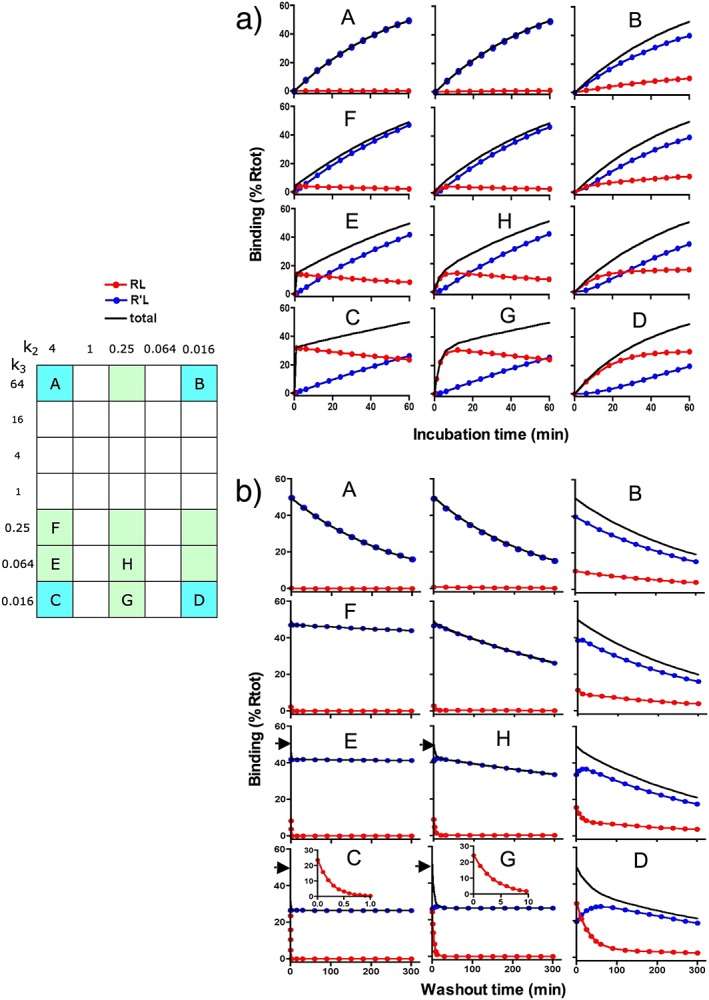
Simulated association and dissociation curves. Differential equations for the present simulations are given in the Supporting Information. Plots for representative ligands are shown in the same 2D configuration as those that are highlighted in the grid and an alphabetical character designates those that are subjected to closer scrutiny. Each of those plots shows values of RL, R'L and total binding (‘total’ = RL + R'L) individually. (a) Simulated association curves are shown for up to 60 min incubation and for a better comparison, they are preformed with a value of [L] that yields 50% receptor occupancy (i.e. ‘total’ = 50% Rtot) at the end. (b) Simulated dissociation curves are shown for up to 300 min washout during which remaining binding is recorded at different time intervals. The washout starts with ‘total’ = 50% Rtot and is triggered by setting [L] = 0 (which corresponds to replacing the ligand‐containing medium by naïve one). Arrows at the ordinate of D, E, G and H show the amount of initial total binding. Inserts in C and G show the decline in RL when the timescale is expanded. Values of R'L (for A, C, E and F) and of ‘total’ (for all other highlighted ligands) are analysed according to a one‐phase or two‐phase exponential association or dissociation paradigms where appropriate. Macroscopic rate constants are shown in Tables 2, 3 for the ligands that are designed by an alphabetical character.
Figure 4.
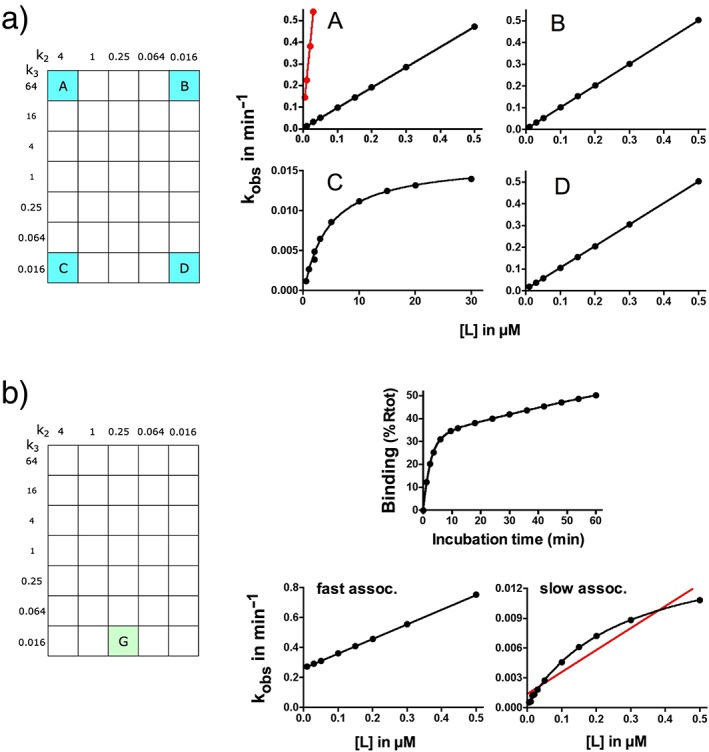
kobs versus [L] plots. (a) A to D: association curves for the highlighted ligands were simulated as in Figure 3a but with more ligand concentrations (abscissa) and 20 time points between 1 and 60 min. For ligands A and C, it is assumed that RL dissociates so fast that it is no longer detectable. Values of R'L (for A and C) and of total binding (for B and D) are analysed according to a monophasic exponential association paradigm, and kobs values are plotted against [L]. Plots are analysed by linear regression for A, B and D, and for C, by making use of the simplified Strickland equation (equation 5 in Figure 1b). The data points for ligand A coincide with those that are calculated from the original microscopic rate constants according to the extended Strickland equation (shown in black) but not with those that are calculated according to the simplified equation (shown in red). Results are presented in Table 2. (b) Association curves of ligand G were simulated as above, total binding is analysed according to a biphasic exponential association paradigm (an example for which ‘total’ = 50% Rtot after 60 min is shown at the top) and kobs values of the fast‐associating and slow‐associating components are plotted against [L]. The plot of the fast component (bottom left) is analysed by linear regression, and the plot of the slow component (bottom right) is analysed by making use of the simplified Strickland equation. For comparison, the red line was obtained via linear regression of the same data. Results are presented in Table 2.
Table 2.
Association rate constants for ligands A to F that are obtained by analysing their simulated association profiles such as in Figure 3a, Figure 4 and Figure 5a
| Analysis of curve | kobs versus [L] | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| R 2 | Linear regression analysis | Analysis by simplified Strickland equation | ||||||||
| Ligand | [L] range (nM) | Monophasic | Biphasic | R 2 | kon M−1⋅min−1 | koff min−1 | R 2 | k3 (input) min−1 | k4 (input) min−1 | k2/k1 (input) nM |
| C (R'L) | 500–30 000 | 1 | 0.79 | 1 | 0.017 (0.016) | −6.5 × 10–4 (1.6 × 10–5) | 4.0 × 10–6 (4.0 × 10–6) | |||
| E (R'L) | 500–30 000 | 1 | 0.80 | 1 | 0.065 (0.064) | −5.5 × 10–4 (6.4 × 10–5) | 4.1 × 10–6 (4.0 × 10–6) | |||
| F (R'L) | 500–30 000 | 1 | 0.80 | 1 | 0.251 (0.250) | −2.8 × 10–4 (2.5 × 10–4) | 4.3 × 10–6 (4.0 × 10–6) | |||
| G (RL + RL) | 5–1000 | 0.93–0.98 | 1 | |||||||
| fast | 1 | 0.99 × 106 | 0.264 | |||||||
| slow | 0.82 | 1 | 0.016 (0.016) | 2.1 × 10–4 (2.6 × 10–4) | 2.6 × 10–7 (2.6 × 10–4) | |||||
| H (RL + R'L) | 5–500 | 0.92–0.98 | 1 | |||||||
| fast | 1 | 0.92 × 106 | 0.310 | |||||||
| slow | 0.99 | 1 | 0.068 (0.064) | −6.4 × 10–4 (1.0 × 10–3) | 3.2 × 10–7 (2.5 × 10–7) | |||||
| D (RL + R'L) | 5–500 | 1 | 1 | 0.99 × 106 | 9.39 × 10–3 | |||||
| A (R'L) | 5–500 | 1 | 1 | 0.94 × 106 | 3.95 × 10–3 | |||||
| B (RL + R'L) | 5–500 | 1 | 1 | 1.00 × 106 | 3.19 × 10–3 | |||||
Ligands are presented by order of appearance in the text. Bound species that are taken in consideration for the analysis are also mentioned. In this respect, it is assumed that RL dissociates so swiftly for ligands A, C, E and F that even at the earliest obtainable time points, it is no longer detectable when performing classical filtration radioligand binding assays. Data points are collected (≥10 per curve) and first analysed according to a monophasic exponential association model (Prism 4.0 by GraphPad Software Inc., San Diego, CA, USA). R 2 is represented as 1 in the table when above 0.999; if less curves are further analysed according to a biphasic exponential association model (in Prism 4.0 by GraphPad Software Inc.). All kobs versus [L] relationships are then checked for their linearity. When the plots are linear (i.e. R 2 = 1), they yield kon from the slope of the kobs versus [L] plot and koff from the extrapolated kobs for [L] = 0. When R 2 < 1, the plots are analysed according to the simplified Strickland equation (Prism 4.0 by GraphPad Software Inc. with equation 5 in Figure 1b as input) to yield k3, k4 and the k2/k1 ratio (Strickland et al., 1975). For comparison, the values of the microkinetic rate constants (or their ratio) that were utilized as input for the simulations are also given in brackets.
Figure 4a clearly shows that two‐step binding ligands like A, B and D yield linear rather than hyperbolic kobs versus [L] plots. Indeed, conditions need to be met for them to adopt a hyperbolic shape. Above all, these plots are based on values of R'L only (i.e. without interference from RL). RL and [L] should be in ‘instant equilibrium’ because the first binding step is merely represented by the k2/k1 ratio, k2 has to sufficiently exceed k3 (otherwise, a more elaborate version of the equation prevails), and the range of [L] is sufficiently large to attain (or even better to exceed) k2/k1 because the initial portion of a hyperbolic plot should closely approach a linear one. All these conditions are reasonably met for C and also for E and F in which k3 is higher (but still not high enough to infringe on the second condition) (Table 2). However, a further increase in k3 may cause its value to exceed that of k2, such as for A. In accordance with the elaborated version of the Strickland equation (Strickland et al. 1975), the kobs versus [L] plots are quasi‐linear in Figure 4a, even when [L] exceeds k2/k1 (data not shown).
Dissociation curves were simulated by pre‐incubating the receptors with ligand until 50% occupancy was achieved (Figure 3a), followed by setting [L] = 0 and measuring the remaining binding at different time intervals. This latter phase is often referred to as ‘wash‐out’, and it is usually carried out by adding an excess of unlabelled ligand to prevent new binding/rebinding events from taking place (Vauquelin, 2012). For A, C, E and F for which only R'L is measurable, these dissociation curves are mono‐exponential (Figure 3b), and koff values closely correspond to those that are calculated according to equation 6 in Figure 1b.
On the contrary, RL may become sufficiently stable to be detected in traditional radioligand binding studies when (starting from C and E) only k2 is lowered. For G and H, this results in biphasic association curves (Figure 3a and Figure 4b; Table 2). This pattern has been observed by Castro et al. (2005 when studying parathyroid hormone binding to its receptor in FRET experiments. The linear kobs versus [L] relationship for the rapid phase and the hyperbolic relationship for the slow phase endorsed an induced‐fit mechanism in which the slow phase corresponds to the switch from a resting to an active state of the receptor. Similar association characteristics were also very recently reported by Wilson et al. (2015 for the binding of the cancer drug Gleevec to c‐ABL kinase. Analysing the association curves of G and H according to a bi‐exponential association model yields a very similar outcome as well. The kobs versus [L] plots are linear for the fast component and provide a good estimation of k1 albeit only an approximate value of k2 (Figure 4b and Table 2). The plots are hyperbolic for the slow component and provide a good estimation of k3 and the k2/k1 ratio but only an approximation of k4. To deal with the subtle incongruities, it is of note that this analysis is in principle not applicable because, as initially reported by Galper et al. (1977 and also shown for G and H (Figure 3a), [R'L] displays an initial lag phase and [RL] even displays a bell‐shaped profile with time. The obvious biphasic profile of the dissociation curves of G (Figure 3b) may constitute additional evidence for an induced‐fit binding mechanism. Analysing these data according to a bi‐exponential dissociation model yields koff values that closely correspond to k2 for the rapid component and to the outcome of equation 5 (Figure 1b) for the slow component. Interestingly, because of the substantial difference between the rates of both components, almost identical koff values can also be obtained by analysing each component separately according to a mono‐exponential model (Table 3).
Table 3.
Macroscopic dissociation rate constants for ligands A to F that are obtained by analysing their dissociation profiles in simulated washout experiments such as in Figure 3b
| Ligand | k2 (min−1) | Theoretical koff for R'L (min−1) a | Analysis of the simulated dissociation data | ||
|---|---|---|---|---|---|
| koff (min−1) | R 2 | Washout time range | |||
| C | 4 | 1.59 × 10–5 | 1.59 × 10–5 | 1 | (0.5–24 h) |
| E | 4 | 6.30 × 10–5 | 6.29 × 10–5 | 1 | (0.5–24 h) |
| F | 4 | 2.35 × 10–4 | 2.35 × 10–4 | 1 | (0.5–24 h) |
| G (fast) | 0.25 | 0.265 | 1 (biphasic) | (0–12 min)b | |
| G (slow) | 2.40 × 10–4 | 2.41 × 10–4 | 1 (biphasic) | (0.5–24 h)b | |
| H (fast) | 0.25 | 0.293 | 1 (biphasic) | (0–12 min)b | |
| H (slow) | 8.13 × 10–4 | 8.15 × 10–4 | 1 (biphasic) | (0.5–24 h)b | |
| D (fast) c | 0.016 | 0.034 | 1 | (0–5 h) | |
| D (slow) c | 1.78 × 10–3 | 1.87 × 10–3 | |||
| A | 4 | 3.76 × 10–3 | 3.76 × 10–3 | 1 | (0–5 h) |
| B | 0.016 | 3.20 × 10–3 | 3.20 × 10–3 | 1 | (0–5 h) |
Ligands are presented by order of appearance in the text. In this respect, it is assumed that RL dissociates so swiftly for ligands A and C that even at the earliest obtainable time points, it is no longer detectable when performing classical filtration‐based radioligand binding assays.
k2 is the microscopic dissociation rate constant for RL, and the theoretical koff for the dissociation of R'L is calculated according to equation 6 in Figure 1b. Simulated dissociation data points are collected within the indicated time range (≥10 per curve) and, if not outspokenly biphasic, first analysed according to a mono‐exponential decay model with the nadir at 0% binding as constraint (in Prism 4.0 by GraphPad Software Inc.). A single value of koff is given when R 2 of this analysis exceeds 0.999 (represented as 1 in the table). R 2 was lower than 1 (not shown) for ligand D. Its dissociation curve was analysed according to a bi‐exponential decay model (same software) and so obtained koff values for the fast and slow components are given. The curves were more clearly biphasic for ligands G and H. The fast and slow components of the dissociation curves (time range of each component given) could be adequately (i.e. yielding R 2 = 1) analysed according to mono‐exponential decay model.
Corresponds to k2k4/(k2+ k3 + k4), that is, equation 6 in Figure 1
Similar results are obtained when the 0–12 min and 0.5–24 h data are combined and analysed according to a biphasic exponential decay model.
Similar analysis of the 0–5 h dissociation data shown in Figure 5b yields comparable koff values and reveals that prolonging the preincubation increases the relative contribution of the slow component, that is, 67%, 76%, 86% and 94% after 30 min, 1 h, 2 h and 5 h preincubation respectively.
Increasing k2 even more produces ligands like D whose association curves no longer suggest an induced‐fit mechanism. The association of D closely fits with a mono‐exponential model (Figure 3a), and the kobs versus [L] plot is linear (Figure 4a). Here again, [R'L] only starts to increase with time after an initial lag phase (Figure 5a). This may provide a potential indication in favour of an induced‐fit mechanism provided that R'L can be measured separately. However, the biphasic dissociation profile of D (Figure 3b) provides strong evidence for an induced‐fit mechanism. The slow component is consistent with the decline of [R'L] (Table 3), and the increased contribution of this component after a longer preincubation (Figure 5b) stems from the increased [R'L]/[RL] ratio at the onset of the washout. In contrast, the koff for the fast component was found to exceed k2. This phenomenon, as well as the initial modest but fast increase of [R'L] (Figure 3b), can be explained by the ability of RL to dissociate and to isomerize into R'L at an equal rate (Figure 5c). This phenomenon was also reported by Anderson and McConnell (1999).
Figure 5.
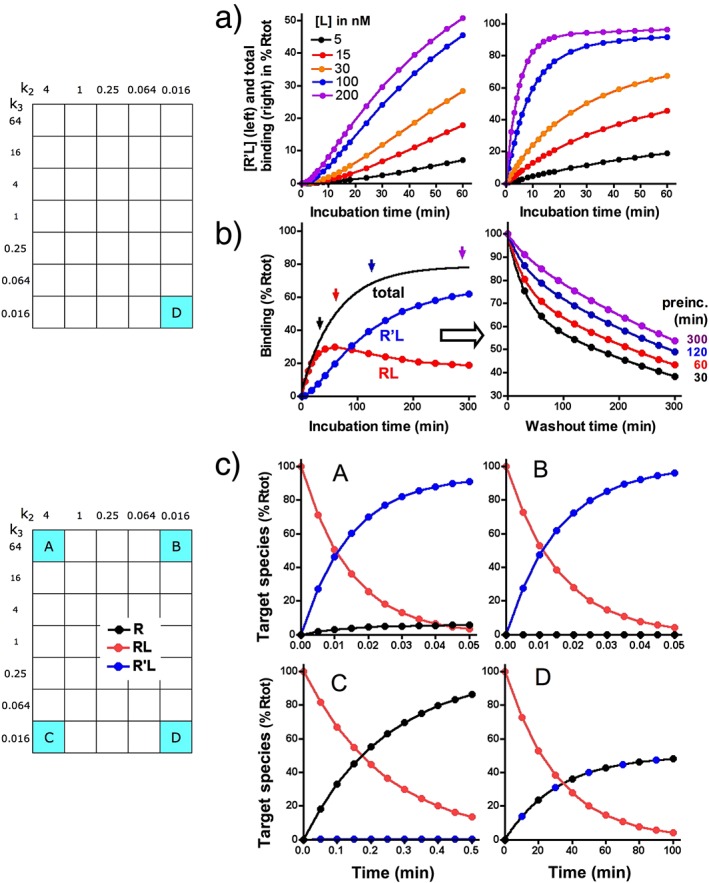
Deeper insight into (panels a and b) the association and dissociation profile of D and (panel c) the time‐dependent fate of the loosely bound RL complex for A to D. (a) Time‐dependent increase of R'L (left side) and total binding (right side) at different [L]. The simulated data for R'L show a distinct initial lag phase at all [L]. This precludes their analysis according to exponential paradigms. On the other hand, the total binding fits well with a monophasic exponential paradigm. (b) The relative contribution of the slow and fast dissociation components depends on the preincubation time. For the simulations, receptors are incubated with the same [L] as in Figure 3a but for 30 min, 1 h and 2 h and 5 h (arrows on top of the association binding curve at the left side). The washout is triggered by setting [L] = 0, and dissociation curves (right side) are shown for 5 h during which the remaining ‘total’ binding is recorded at different time intervals. For the sake of comparison, binding is expressed as % of binding at the onset of the washout. The dissociation data are analysed in terms of a biphasic exponential dissociation paradigm; the results are presented in Table 3. (c) To follow the time‐dependent fate of RL, its value is set at 100% of [Rtot] at the onset, and it is allowed to simultaneously dissociate with rate constant k2 or to evolve/convert into R'L with rate constant k3. Curves show the time‐dependent changes in concentration of the three species. Of note is that R'L is rapidly and almost exclusively formed for A to B, whereas at a much longer time scale, RL preferentially dissociates for C or dissociates with the same rate as its conversion into R'L for D.
Interestingly, a recent study dealing with the interaction between the tritiated allosteric modulator maraviroc and human chemokine receptors, CCR5, in membranes from recombinant CHO cells (Swinney et al., 2014) revealed that it is sometimes necessary to collect many data points in order to clearly perceive the biphasic pattern of dissociation curves. Indeed, curves that were initially generated with a rather restricted number of data points could hardly be distinguished from a mono‐exponential process. Yet, it was noticed that the koff value determined in these experiments was well below the koff that was obtained by analysing the association kobs versus [L] data. This constituted a preliminary indication of the occurrence of an induced‐fit mechanism, and additional investigations revealed that the maraviroc–CCR5 interaction presents the same overall kinetic binding profile as D. Interestingly, receptor mutation studies reveal that there is only a partial overlap between the amino acids that are necessary for obtaining R'L with maraviroc and other allosteric modulators such as vicriviroc and aplaviroc (Swinney et al., 2014). These drugs are all very efficacious in preventing the interaction between the GP‐120 domain of HIV‐1 and the second extracellular loop of the receptor (Maeda et al., 2006, 2008; Muniz‐Medina et al., 2009; Garcia‐Perez et al., 2011a, 2011b), and along with recent modelling data (Tan et al., 2013), the kinetic experiments suggest that they do so by triggering ligand‐specific conformational rearrangements of the receptor's extracellular domains rather than by steric hindrance. This interpretation concurs with the increasing awareness that GPCRs are flexible so that one binding site may give rise to multiple binding arrangements (Teague, 2003).
Repeated reformation of R'L
A stable R'L complex is not necessarily required for achieving a long residence time provided that the k3/k2 ratio is well above unity. This is demonstrated with ligands like A and B at the upper portion of the grid. Indeed, a high k3/k2 ratio will shift the balancing act between the two potential fates of RL, that is, to dissociate or to (re)evolve to R'L (Figure 5c) (Copeland, 2011; Vauquelin, 2013; Vauquelin et al., 2014). Because RL is more likely progress to R'L than fully dissociate, this species is thus likely to be formed repeatedly before A and B finally dissociate. This process will thus contribute to their long residence time.
As illustrated in Figure 2b and Figure 3a and b, the macroscopic binding patterns of A and B are nearly indiscernible from that of a single‐step, slow reversible bimolecular process. Also, as explained before, the high k3/k2 ratio also precludes the kobs versus [L] plot to adopt a hyperbolic shape for A and B (Figure 4a). For A, this is related to the fact that the intermediary RL species provides a negligible contribution to the binding even at the shortest time scales (not shown). For B, this species eludes detection as well in spite of its significant contribution to the binding. Yet, this contribution is likely to play an essential role in the action of salmeterol, a long‐acting synthetic β2‐adrenoceptor agonist and a well‐known bronchodilator (Coleman et al., 1996; Coleman, 2009), as well as of some other ligands. The extended lipophilic phenylalkoxyalkyl side chain of salmeterol is thought to bind tightly to an ‘exosite’ (equivalent to RL) that is present at the interface between the receptor and the lipid bilayer of the membrane. This allows the receptor‐activating saligenin head of this molecule to rapidly engage (to form R'L) and disengage in its binding pocket within the central cleft of the receptor according to a ‘charnière’ mechanism (Rocha e Silva, 1969). This could explain the quick disappearance of the salmeterol‐mediated relaxation of airway smooth muscle when an ‘orthosteric’ site‐binding antagonist is added, as well as the recovery of this effect after antagonist removal in in vitro studies (Bergendal et al., 1996). The simulations shown in Figure 6a and b show that this fast disappearance is related to the ability of such antagonists (denoted as M) to increase the apparent koff of ligands like A and B in particular. Moreover, simulations shown in Figure 6c also reveal that in competition‐binding experiments, ligands like M could also only partly depress the binding of B (and vice versa) provided that RL is sufficiently abundant and stable.
These latter simulations and also those that were based on the ‘charnière’‐like ‘two‐domain’ model by Hoare (2007) explain why some peptide agonists to class B GPCRs do not fully inhibit the specific binding of radiolabelled nonpeptides to the orthosteric site. However, due care should be exercised when interpreting experimental observations that are based on the interplay with two distinct ligands because increased radioligand dissociation such as shown in Figure 6 and incomplete competition binding (not shown) are also considered to constitute ‘hallmarks’ for allosteric interactions (Vauquelin and Van Liefde, 2012; Vauquelin et al., 2015). Further investigations are thus necessary to better define the interplay between these two ligands, such as comparing the ability of different unlabelled ligands to enhance the radioligand's affinity and/or residence time, focusing attention on receptor function and then examining the X‐ray crystal structure of receptors that are bound by one or two ligands (e.g. Kruse et al., 2013; Lane et al., 2013b).
Equations that are mathematically similar to those that apply to the induced‐fit model (i.e. equations 5 to 7 in Figure 1b) are also able to describe the binding properties of bivalent ligands without the necessity for a conformational change to be involved. In this respect, bi/multivalency is well known to increase the residence time and affinity of antibodies and other biologically important polyvalent interactions (Mammen et al., 1998). This principle is also well established for biological therapeutics (Hudson and Kortt, 1999; Rudnick Adams, 2009; Holliger and Hudson, 2005) and constitutes a rationale for developing pharmaceuticals by covalently linking small peptides or synthetic molecules (often denoted as ‘pharmacophores’) by a spacer arm (Todorovska et al., 2001; Mohr et al., 2010; Kroll et al., 2013). In the prevailing models, both pharmacophores bind to their respective receptor sites according to a one‐step bimolecular mechanism. Yet, the pharmacophore that binds first will bring the second one in close proximity to its receptor site and thereby substantially increase its local concentration (Kramer and Karpen 1998; Vauquelin and Charlton, 2013). This second association can now be described by a k3‐like composite first‐order rate constant which, because of the high local concentration of the pharmacophore involved is likely to exceed the k2‐like dissociation rate constant of the already bound one (Vauquelin, 2013; Vauquelin et al., 2014). Simulations that are based on such models revealed that the binding properties of such constructs are similar to those of A and B, including accelerated dissociation in the presence of a monovalent ligand that binds exclusively to one of the receptor sites (because the bivalent ligand can no longer benefit from the high k3/k2 ratio) and incomplete inhibition in competition‐binding experiments (Vauquelin and Van Liefde, 2012; Vauquelin et al., 2015).
Technical considerations
The present simulations indicate that partial insurmountability (Figure 2), hyperbolic kobs versus [L] plots (Figure 4), biphasic association and dissociation curves (Figure 3), lags in the formation and elimination of R'L (Figure 3) and accelerated dissociation (Figure 6) and partial competition already provide useful indications in favour of an induced‐fit (or related) mechanism. Such observations require radioligand binding and/or functional assays and, as clearly illustrated for AT1 receptor antagonists (Le et al., 2007), comparing experimental data from both approaches may be useful. Also, compared with the earlier and still widespread use of membrane suspensions, studies with intact recombinant cells are easier to interpret: they are more likely to express higher levels of the receptor (subtype) of interest; binding and/or functional assays can be carried out under identical conditions and the information is also more relevant from the physiological perspective (Kenakin, 2009; Vauquelin and Charlton, 2010). In this respect, it is of note that receptors in membrane preparations have lost part of their natural microenvironment and this may affect the residence time of some ligands (Fierens et al., 2002; Verheijen et al., 2004). Even more compelling is that while the candesartan–AT1 receptor interaction was essentially enthalpy‐driven in intact cells, entropy also contributed significantly in membrane preparations thereof (Fierens et al., 2002).
The present simulations were based on differential equations (shown in Supporting Information Table S2) and carried out with a custom‐written Microsoft Excel™ programme (Vauquelin et al., 2001a). Similar simulations have been carried out by others (Walkup et al., 2015) using commercially available software like Mathematics (Wolfram, Champaign, Il). The simulated data do not account for experimental variance/error that will show up in real‐life situations. Yet, even so, analysing ‘biphasic’ association and dissociation curves according to the common practice of splitting them into their fast and slow components (Castro et al., 2005; Wilson et al., 2015) does not yield the correct microscopic rate constants. As already reported (Galper et al., 1977; Bellamy et al., 2002) and shown in Figure 3, this discrepancy can be linked to the more complex evolution of [RL] and [R'L] with time. These caveats could be overcome by fitting data with ‘explicit’ kinetic equations (obtained by integrating the differential equations) and by nonlinear regression analysis using a graphical package such as Prism® by GraphPad software. Such equations have already been published by Schreiber et al. (1985 for direct binding kinetics and competition kinetics), and by Bellamy et al. (2002 for dissociation kinetics.
With regard to the generation of experimental data, the use of ‘homogeneous’ binding assay techniques are an alternative to the classical binding experiments that require the physical separation between free and bound radioligand before the measurement. To be of the most value, these techniques should be adaptable to intact cells and allow the collection of more data (for a more reliable analysis) and also at shorter time scales. Potential challenges to collecting reliable kinetic data with homogeneous assays include the rate of mixing for adherent/plated cells. To overcome this caveat, assays could be carried out on cell suspensions in plate readers with built‐in dispensers (Schiele et al., 2015). Another technical challenge is the limited ‘mass transport’ of ligands to the metal surface in plasmon resonance biosensors. This is a well‐known limiting factor when the flow is slow and/or when the receptor density and/or association rate is high (Christensen, 1997). ‘Homogeneous’ assay techniques have already been reviewed elsewhere (e.g. Kenakin, 2009; Cusack et al., 2015), and we will only briefly focus on those that are most promising with respect to induced‐fit‐type binding. As illustrated in Table 4 and further commented upon in the legend, these assays belong to three categories: ‘homogeneous’ radioligand binding, fluorometry based on spectroscopic labels and label‐free assays.
Table 4.
Alternative, homogenous binding techniques to detect and study induced‐fit binding
| Category | 1 | 2 | 2 | 3 |
|---|---|---|---|---|
| Technique | SPA | FRET | BRET | SPR |
| Label requirement | + radioactive | + fluorescent | + fluorescent | − |
| Many data points on single sample | + | + | + | + |
| Time frame of data collection | minute–hours | second–minutes | second–minutes | second–minutes |
| Whole‐cell assay | + | + | + | − |
Techniques belong to three categories. For categories 1 and 2, detection relies on the transfer of energy between a radioactive, fluorescent or bioluminescent donor and luminescent/fluorescent acceptor and only requires the ligand and the receptor to be in close proximity. This could potentially give rise to a high background at high concentrations of ligand. For category 3, detection relies on the actual binding.
- ‘Homogeneous’ radioligand binding: the scintillation proximity assay (SPA) in which a radiolabelled ligand triggers the luminescence of receptor‐bearing imaging beads allows recordings to be made for up to 24 h with membrane preparations (Swinney et al., 2014) and can be carried out with intact cells.
- Fluorimetry based on spectroscopic labels: for the FRET technique, fluorescently labelled receptors are excited by an external light source and act as energy donor. Compared with using the classical fluorescent protein–receptor hybrids, the more promising time‐resolved FRET approach is based on labelling receptors (e.g. at the N‐terminal end of GPCRs) with much smaller lanthanide chelates. In this respect, terbium merits special attention because of its great emission strength, stability and (because of its well‐separated emission peaks) compatibility with a wide spectrum of acceptors that can be covalently linked to the ligand (Schiele et al., 2015). The related bioluminiscence resonance energy transfer (BRET) technique, in which the energy donor is a receptor‐associated enzyme that emits light upon the catalytic degradation of a suitable substrate, has recently been improved by introducing Nanoluc (Nlluc) a small‐sized luciferase with high emission intensity as energy donor. The large spectral resolution with red light‐emitting acceptors greatly improves the detection sensitivity (Machleidt et al., 2015). Constructs in which Nlluc was linked to the N‐terminal end of GPCRs were recently shown to translocate to the cell surface and allowed ligand binding to be measured in living cells (Stoddart et al., 2015). Photobleaching of the acceptor may still be a matter of concern for FRET and BRET, but their replacement by self‐healing fluorophores (Tinnefeld and Cordes, 2012) could further enhance their performance. Also, it should be checked whether or not the covalent coupling of an acceptor to the ligand affects its pharmacological properties (Cusack et al., 2015).
- Label‐free assays: surface plasmon resonance (SPR) measures changes in the refractive index of the medium nearby the sensor's thin metal surface when a ligand or receptor in solution increases the mass at that surface by binding to its immobilized counterpart (Patching, 2014). SPR has become a leading approach for characterizing the kinetics of molecular interactions, but for membrane‐associated receptors, a major challenge is that they have to be solubilized and purified in order to be being immobilized. Although they can then be reconstituted in lipid bilayers and retain some of their original characteristics, they are no longer in their native environment (Patching, 2014). It is only very recently that the binding of antibody and lectins to their targets on immobilized intact cells could be recorded (Wang et al., 2012), and it is to be hoped that these pioneering studies will pave the way to the detection of small ligand binding as well.
Final considerations
In this work, we have shown that a number of experimental approaches can be used to identify induced‐fit mechanisms including partial insurmountability (Figure 2), hyperbolic kobs versus [L] plots (Figure 4), biphasic association and dissociation curves (Figure 3), lags in the formation and elimination of R'L (Figure 3) and accelerated dissociation and partial competition (Figure 6).
‘All models are wrong but some are useful’ (Box and Draper, 1969). This statement is highly pertinent when ascribed to the intricate mechanistic aspects of ligand/drug–receptor/target interactions. While a one‐step reversible binding still constitutes a favourite mechanism, it has become increasingly evident that freshly formed ‘encounter complexes’ may go through multiple states via small conformational adjustments and that GPCRs are sufficiently flexible for the binding site to adopt multiple binding arrangements (Teague, 2003; Garvey, 2010; Copeland, 2011; Dror et al., 2011). Yet, such situations are customarily reduced to a two‐step process. While a thermodynamic cycle that includes the induced‐fit and conformational selection mechanisms (Figure 1c) offers the most complete description of binding events that go along with conformational changes (Copeland, 2011), it is usually even further reduced to the induced‐fit mechanism. This mechanism, in which a loose initial RL complex is subsequently converted via a conformational change into a more stable R'L complex, (Swinney, 2004; Tummino and Copeland, 2008) is considered to account for the binding of a significant portion of ligands with high affinity and clinical efficacy (Copeland, 2010, 2011).
The induced‐fit mechanism focuses on conformational changes. However, the binding of additional moieties of the ligand may also give rise to a stable R'L complex and, in practice, it is not always possible to make a clear‐cut distinction between either mechanism. For example, the long residence time of insurmountable sartans was attributed to the formation of a stable R'L complex by the binding of their imidazole‐derived moiety (Van Liefde and Vauquelin, 2009), but conformational changes could also be involved. Also, bi/multivalency and conformational changes may go hand in hand (Valant et al., 2012; Lane et al., 2013a, 2013b; Vauquelin et al., 2014). For example, AT1 receptor mutation studies have indicated that the initial binding of the N‐terminal end of angiotensin II already triggers partial receptor activation, while the subsequent binding of the C‐terminal end results in full receptor activation (Le et al., 2002).
Author contributions
G.V. contributed to the study design and writing and performed the simulations. I.V.L. contributed to the writing and performed bibliographic search and artwork. D.C.S. contributed to the study design and writing.
Conflict of interest
The authors declare no conflicts of interest.
Supporting information
Table S1 Binding models differential equations that are relevant to the present article.
Table S2 Definitions and values of binding parameters of the investigated drugs.
Figure S1 Saturation binding curves: effect of the incubation time.
Figure S2 Time‐wise decline of the contribution of [RL] to the total binding of Ligand C.
Figure S3 Effect of the duration of agonist‐ exposure on the insurmountable behaviour of antagonists A to D.
Figure S4 Agonist EC50 values after pre‐treatment with antagonist C: effect of different experimental conditions.
Supporting info item
Acknowledgements
We are very much indebted to the reviewers for their pertinent and highly instructive comments.
Vauquelin, G. , Van Liefde, I. , and Swinney, D. C. (2016) On the different experimental manifestations of two‐state ‘induced‐fit’ binding of drugs to their cellular targets. British Journal of Pharmacology, 173: 1268–1285. doi: 10.1111/bph.13445.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE, et al (2015). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson TG, McConnell HM (1999). Interpretation of biphasic dissociation kinetics for isomeric class II major histocomptability complex‐peptide complexes. Biophys J 77: 2451–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arunlakshana O, Schild HO (1959). Some quantitative uses of drug antagonists. Br J Pharmacol Chemother 14: 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellamy TC, Wood J, Garthwaite J (2002). On the activation of soluble guanylyl cyclase by nitric oxide. Proc Natl Acad Sci U S A 99: 507–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergendal A, Lindén A, Skoogh B‐E, Gerspacher M, Anderson GP, Lolfdahl C‐G (1996). Salmeterol mediated reassertion of relaxation persists in guinea‐pig trachea pretreated with aliphatic side chain structural analogues. Br J Pharmacol 117: 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhuiyan MA, Ishiguro M, Hossain M, Nakamura T, Ozakic M, Miurad S, et al (2009). Binding sites of valsartan, candesartan and losartan with angiotensin II receptor 1 subtype by molecular modeling. Life Sci 85: 136–140. [DOI] [PubMed] [Google Scholar]
- Black JW, Leff P (1983). Operational models of pharmacological agonism. Proc R Soc Lond B Biol Sci 220: 141–162. [DOI] [PubMed] [Google Scholar]
- Box G, Draper N (1969). Evolutionary operation: a statistical model for process improvement. John Wiley and Sons: New York. [Google Scholar]
- Castro M, Nikolaev VO, Palm D, Lohse MJ, Vilardaga J‐P (2005). Turn‐on switch in parathyroid hormone receptor by a two‐step parathyroid hormone binding mechanism. Proc Natl Acad Sci U S A 102: 16084–16089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen LLH (1997). Theoretical analysis of protein concentration determination using biosensor technology under conditions of partial mass transport limitation. Anal Biochem 249: 153–164. [DOI] [PubMed] [Google Scholar]
- Christopoulos A, Kenakin TG (2002). Protein‐coupled receptor allosterism and complexing. Pharmacol Rev 54: 323–374. [DOI] [PubMed] [Google Scholar]
- Coleman RA (2009). On the mechanism of the persistent action of salmeterol: what is the current position? Br J Pharmacol 158: 180–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RA, Johnson M, Nials AT, Vardey CJ (1996). Exosites: their current status, and their relevance to the duration of action of long‐acting beta 2‐adrenoceptor agonists. Trends Pharmacol Sci 17: 324–330. [PubMed] [Google Scholar]
- Copeland RA, Pompliano DL, Meek TD (2006). Drug‐target residence time and its implications for lead optimization. Nat Rev Drug Discov 5: 730–739. [DOI] [PubMed] [Google Scholar]
- Copeland RA (2010). The dynamics of drug–target interactions: drug–target residence time and its impact on efficacy and safety. Expert Opin Drug Discov 5: 305–310. [DOI] [PubMed] [Google Scholar]
- Copeland RA (2011). Conformational adaptation in drug–target interactions and residence time. Future Med Chem 3: 1491–1501. [DOI] [PubMed] [Google Scholar]
- Cusack KP, Wang Y, Hoemann M, Marjanovic J, Heym RG, Vasudevan A (2015). Design strategies to address kinetics of drug binding and residence time. Bioorg Med Chem Lett . doi:10.1016/j.bmcl.2015.02.027. [DOI] [PubMed] [Google Scholar]
- Dahl G, Akerud T (2013). Pharmacokinetics and the drug–target residence time concept. Drug Discov Today 18: 697–707. [DOI] [PubMed] [Google Scholar]
- Del Castillo J, Katz B (1957). Interaction at end‐plate receptors between different choline derivatives. Proc R Soc Lond Ser B Biol Sci 146: 369–381. [DOI] [PubMed] [Google Scholar]
- Dror RO, Pan AC, Arlow DH, Borhani DW, Maragakis P, Shan Y, et al (2011). Pathway and mechanism of drug binding to G‐protein‐coupled receptors. Proc Natl Acad Sci U S A 108: 13118–13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierens FLP, Vanderheyden PML, De Backer J‐P, Vauquelin G (1999a). Binding of the antagonist [3H]candesartan to angiotensin II AT1 receptor‐transfected Chinese hamster ovary cells. Eur J Pharmacol 367: 413–422. [DOI] [PubMed] [Google Scholar]
- Fierens FLP, Vanderheyden PML, De Backer J‐P, Vauquelin G (1999b). Insurmountable angiotensin II AT1 receptor antagonists: the role of tight antagonist binding. Eur J Pharmacol 372: 199–206. [DOI] [PubMed] [Google Scholar]
- Fierens FLP, Vanderheyden PML, Gaborik Z, Le Minh T, De Backer JP, Hunyady L, et al (2000). Lys199 Mutation of the human angiotensin II AT1 receptor differently affects the binding of surmountable and insurmountable non‐peptide antagonists. J Renin Angiotensin Aldosterone Syst 1: 283–288. [DOI] [PubMed] [Google Scholar]
- Fierens F, Vanderheyden PML, Roggeman C, Vande Gucht P, De Backer J‐P, Vauquelin G (2002). Distinct binding properties of the AT1 receptor antagonist [3H]candesartan to intact cells and membrane preparations. Biochem Pharmacol 63: 1273–1279. [DOI] [PubMed] [Google Scholar]
- Galper JB, Klein W, Catterall WA (1977). Muscarinic acetylcholine receptors in developing chick heart. J Biol Chem 252: 8692–8699. [PubMed] [Google Scholar]
- Galper JB, Dziekan LC, O'Hara DS, Smith TW (1982). The biphasic response of muscarinic cholinergic receptors in cultured heart cells to agonists. Effects of receptor number and affinity in intact cells and homogenates. J Biol Chem 257: 10344–10356. [PubMed] [Google Scholar]
- Garcia‐Perez J, Rueda P, Staropoli I, Kellenberger E, Alcami J, Arenzana‐Seisdedos F, et al (2011a). New insights into the mechanisms whereby low molecular weight CCR5 ligands inhibit HIV‐1 infection. J Biol Chem 286: 4978–4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Perez J, Rueda P, Alcami J, Rognan D, Arenzana‐Seisdedos F, Lagane B, et al (2011b). Allosteric model of maraviroc binding to CC chemokine receptor 5 (CCR5). J Biol Chem 286: 33409–33421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garvey EP (2010). Structural mechanism of slow‐onset, two‐step enzyme inhibition. Curr Chem Biol 4: 64–73. [Google Scholar]
- Hall D, Parsons S (2001). Non‐surmountable antagonism: a general drawback of pre‐steady state measurement? Trends Pharmacol Sci 22: 63–65. [DOI] [PubMed] [Google Scholar]
- Harris Callan O, So O‐Y, Swinney D (1997). The kinetic factors that determine the affinity and selectivity for slow binding inhibition of human prostaglandin H synthase 1 and 2 by indomethacin and fubriprofen. J Biol Chem 271: 3548–3554. [DOI] [PubMed] [Google Scholar]
- Hirschberg BT, Schimerlik MI (1994). A kinetic model for oxotremorine M binding to recombinant porcine M2 muscarinic receptors expressed in Chinese hamster ovary cells. J Biol Chem 269: 26127–26135. [PubMed] [Google Scholar]
- Hoare SRJ (2007). Allosteric modulators of class B G‐protein‐coupled receptors. Curr Neuropharmacol 5: 168–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliger P, Hudson PJ (2005). Engineered antibody fragments and the rise of single domains. Nat Biotechnol 23: 1126–1136. [DOI] [PubMed] [Google Scholar]
- Hudson PJ, Kortt AA (1999). High avidity scFv multimers; diabodies and triabodies. J Immunol Methods 23: 177–189. [DOI] [PubMed] [Google Scholar]
- Hulme EC, Trevethick MA (2010). Ligand binding assays at equilibrium: validation and interpretation. Br J Pharmacol 161: 1219–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Järv J, Hedlund B, Bartfai T (1979). Isomerisation of the muscarinic receptor‐antagonist complex. J Biol Chem 254: 5595–5598. [PubMed] [Google Scholar]
- Kenakin TP (2009). Cellular assays as portals to seven‐transmembrane receptor‐based drug discovery. Nat Rev Drug Discov 8: 617–626. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Jenkinson S, Watson C (2006). Determining the potency and molecular mechanism of action of insurmountable antagonists. J Pharmacol Exp Ther 319: 710–723. [DOI] [PubMed] [Google Scholar]
- Kloog Y, Sokolovsky M (1977). Studies on muscarinic acetylcholine receptors from mouse brain, characterization of the interaction with antagonists. Brain Res 144: 31–48. [DOI] [PubMed] [Google Scholar]
- Kramer RH, Karpen JW (1998). Spanning biding sites on allosteric proteins with polymer‐linked ligand dimers. Nature 395: 710–713. [DOI] [PubMed] [Google Scholar]
- Kroll C, Mansi R, Braun F, Dobitz S, Maecke HR, Wennemers H (2013). Hybrid bombesin analogues: combining an agonist and antagonist in defined distances for optimized tumor targeting. J Am Chem Soc 135: 16793–16796. [DOI] [PubMed] [Google Scholar]
- Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, et al (2013). Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504: 101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane JR, Sexton PM, Christopoulos A (2013a). Bridging the gap: bitopic ligands of G‐protein‐coupled receptors. Trends Pharmacol Sci 34: 59–65. [DOI] [PubMed] [Google Scholar]
- Lane JR, Chubukov P, Liu W, Canals M, Cherezov V, Abagyan R, et al (2013b). Structure based ligand discovery targeting orthosteric and allosteric pockets of dopamine receptors. Mol Pharmacol 84: 794–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le MT, Vanderheyden PML, Szaszák M, Hunyady L, Vauquelin G (2002). Angiotensin IV is a potent agonist for constitutive active human AT1 receptors. Distinct roles of the N‐ and C‐terminal amino acid residues of angiotensin II in human AT1 receptor activation. J Biol Chem 277: 23107–23110. [DOI] [PubMed] [Google Scholar]
- Le MT, De Backer J‐P, Hunyady L, Vanderheyden PML, Vauquelin G (2005). Comparison of ligand binding and functional properties of human AT1 receptors transiently and stably expressed in CHO‐K1 cells. Eur J Pharmacol 513: 35–45. [DOI] [PubMed] [Google Scholar]
- Le MT, Pugsley M, Vauquelin G, Van Liefde I (2007). Molecular characterisation of the interactions between olmesartan and telmisartan and the human angiotensin II AT1 receptor. Br J Pharmacol 151: 952–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leff P (1995). The two‐state model of receptor activation. Trends Pharmacol Sci 16: 89–97. [DOI] [PubMed] [Google Scholar]
- Leff P, Martin GR (1986). Peripheral 5‐HT2‐like receptors. Can they be classified with the available antagonists? Br J Pharmacol 88: 585–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew MJ, Ziogas J (2004). The two‐state model of antagonist–AT1 receptor interaction: an hypothesis defended but not tested. Biochem Pharmacol 67: 397–399. [DOI] [PubMed] [Google Scholar]
- Lu H, Tonge PJ (2010). Drug–target residence time: critical information for lead optimization. Curr Opin Chem Biol 14: 467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machleidt T, Woodroofe CC, Schwinn MK, Méndez J, Robeers MB, et al (2015). NanoBRET: a novel BRET platform for the analysis of protein–protein interactions. ACS Chem Biol . doi:10.1021/acschembio.5b00143. [DOI] [PubMed] [Google Scholar]
- Maeda K, Das D, Ogata‐Aoki H, Nakata H, Miyakawa T, et al (2006). Structural and molecular interactions of CCR5 inhibitors with CCR5. J Biol Chem 281: 12688–12698. [DOI] [PubMed] [Google Scholar]
- Maeda K, Das D, Yin PD, Tsuchiya K, Ogata‐Aoki H (2008). Involvement of the second extracellular loop and transmembrane residues of CCR5 in inhibitor binding and HIV‐1 fusion: insights into the mechanism of allosteric inhibition. J Mol Biol 381: 956–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammen M, Choi S‐K, Whitesides GM (1998). Polyvalent interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angew Chem Int Ed 37: 2754–2794. [DOI] [PubMed] [Google Scholar]
- Michel MC, Foster C, Brunner HR, Liu L (2013). A systematic comparison of the properties of clinically used angiotensin II type 1 receptor antagonists. Pharmacol Rev 65: 809–848. [DOI] [PubMed] [Google Scholar]
- Mohr K, Tränkle C, Kostenis E, Barocelli E, De Amici M, Holzgrabe U (2010). Rational design of dualsteric GPCR ligands: quests and promise. Br J Pharmacol 159: 997–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JF (1982). The slow‐biding and slow, tight‐binding inhibition of enzyme‐catalysed reactions. Trends Biochem Sci 7: 102–105. [Google Scholar]
- Muniz‐Medina VM, Jones S, Maglich JM, Galardi C, Hollingsworth RE, et al (2009). The relative activity of “function sparing” HIV‐1 entry inhibitors on viral entry and CCR5 internalization: is allosteric functional selectivity a valuable therapeutic property? Mol Pharmacol 75: 490–501. [DOI] [PubMed] [Google Scholar]
- Neubig R, Spedding M, Kenakin T, Christopoulos A (2003). International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on terms and symbols in quantitative pharmacology. Pharmacol Rev 55: 597–606. [DOI] [PubMed] [Google Scholar]
- Núñez S, Venhorst J, Kruse CG (2012). Target–drug interactions: first principles and their application to drug discovery. Drug Discov Today 17: 10–22. [DOI] [PubMed] [Google Scholar]
- Ojima M, Igata H, Tanaka M, Sakamoto H, Kuroita T, Kohora Y, et al (2011). In vitro antagonistic properties of a new angiotensin type 1 receptor blocker, azilsartan, in receptor binding and function studies. J Pharmacol Exp Ther 336: 801–808. [DOI] [PubMed] [Google Scholar]
- Patching SG (2014). Surface plasmon resonance spectroscopy for characterization of membrane protein–ligand interactions and its potential fro drug discovery. Biochim Biophys Acta Biomembr 1838: 43–55. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al (2014). The IUPHAR/BPS guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucleic Acids Res 42: D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha e Silva M (1969). A thermodynamic approach to problems of drug antagonism I. The “Charnière theory”. Eur J Pharmacol 6: 294–302. [DOI] [PubMed] [Google Scholar]
- Rudnick SI, Adams GP (2009). Affinity and avidity an antibody‐based tumor targeting. Cancer Biother Radiopharm 24: 155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheibe S, Bennett DB, Spain JW, Roth BL, Cosica CJ (1984). Kinetic evidence for differential agonist and antagonist binding to bovine hippocampical synaptic membrane opioid receptors. J Biol Chem 259: 13298–13303. [PubMed] [Google Scholar]
- Schiele F, Ayaz P, Fernández‐Montalván A (2015). A universal homogenous assay for high‐throughput determination of binding kinetics. Anal Biochem 468: 42–49. [DOI] [PubMed] [Google Scholar]
- Schreiber G, Henis YI, Sokolovsky M (1985). Rate constants of agonist binding to muscarinic receptors in rat brain medulla. Evaluation by competition kinetics. J Biol Chem 260: 8789–8794 and 8795–8802. [PubMed] [Google Scholar]
- Stoddart LA, Johnstone EKM, Wheal AJ, Goulding J, Robers MB, et al (2015). Application of BRET to monitor ligand binding to GPCRs. Nat Methods 12: 661–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland S, Palmer G, Massey V (1975). Determination of dissociation constants and specific rate constants of enzyme–substrate (or protein–ligand) interactions from rapid reaction kinetic data. J Biol Chem 250: 4048–4052. [PubMed] [Google Scholar]
- Swinney DC (2004). Biochemical mechanisms of drug action: what does it take for success? Nat Rev Drug Discov 3: 801–808. [DOI] [PubMed] [Google Scholar]
- Swinney DC (2006). Can binding kinetics translate to a clinically differentiated drug? From theory to practice. Lett Drug Des Discov 3: 569–574. [Google Scholar]
- Swinney DC, Beavis P, Chuang K‐T, Zheng Y, Lee I, Gee P, et al (2014). A study into the molecular mechanism of binding kinetics and long residence times of human CCR5 receptor antagonists. Br J Pharmacol 171: 3364–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Q, Zhu Y, Li J, Chen Z, Han GW, et al (2013). Structure of the CCR5 chemokine receptor–HIV entry inhibitor maraviroc complex. Science 341: 1387–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teague SJ (2003). Implications of protein flexibility for drug discovery. Nat Rev Drug Discov 2: 527–541. [DOI] [PubMed] [Google Scholar]
- Tinnefeld P, Cordes J (2012). “Self‐healing” dyes: intramolecular stabilization of organic fluorophores. Nat Methods 9: 426–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorovska A, Roovers RC, Dolezal O, Kortt AA, Hoogenboom HR, Hudson PJ (2001). Design and application of diabodies, triabodies and tetrabodies for cancer targeting. J Immunol Methods 248: 47–66. [DOI] [PubMed] [Google Scholar]
- Toomela T, Jolkkonen M, Rinken A, Järv J, Karlsson E (1994). Two‐step binding of green mamba toxin to muscarinic acetylcholine receptor. FEBS Lett 352: 95–97. [DOI] [PubMed] [Google Scholar]
- Tuccinardi T, Calderone V, Rapposelli S, Martinelli A (2006). Proposal of a new binding orientation for non‐peptide AT1 antagonists: homology modeling, docking and three‐dimensional quantitative structure–activity relationship analysis. J Med Chem 49: 4305–4316. [DOI] [PubMed] [Google Scholar]
- Tummino PJ, Copeland RA (2008). Residence time of receptor–ligand complexes and its effect on biological function. Biochemistry 47: 5481–5492. [DOI] [PubMed] [Google Scholar]
- Valant C, Lane JR, Sexton PM, Christopoulos A (2012). The best of two worlds? Bitopic orthosteric/allosteric ligands of G protein‐coupled receptors. Annu Rev Pharmacol Toxicol 52: 153–178. [DOI] [PubMed] [Google Scholar]
- van Giersbergen PLM, Shatzer SA, Henderson AK, Lai J, Nakanishi S, Yamamura HI, et al (1991). Characterization of a tachikinin peptide NK2 receptor transfected in murine fibroblast B82 cells. Proc Natl Acad Sci U S A 88: 1661–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Liefde I, Vauquelin G (2009). Sartan–AT1 receptor interactions: in vitro evidence for insurmountable antagonism and inverse agonism. Mol Cell Endocrinol 302: 237–243. [DOI] [PubMed] [Google Scholar]
- Vanderheyden PML, Fierens FLP, Verheijen I, De Backer J‐P, Vauquelin G (2000a). Binding characteristics of [3H]irbesartan to human recombinant angiotensin II type 1 receptors. J Renin Angiotensin Aldosterone Syst 1: 159–165. [DOI] [PubMed] [Google Scholar]
- Vanderheyden PML, Fierens FLP, De Backer J‐P, Vauquelin G (2000b). Reversible and syntopic interaction between angiotensin II AT1 receptor antagonists and human AT1 receptors expressed in CHO‐K1 cells. Biochem Pharmacol 59: 927–935. [DOI] [PubMed] [Google Scholar]
- Vauquelin G (2012). Determination of drug–receptor residence times by radioligand binding and functional assays: experimental strategies and physiological relevance. Med Chem Commun 3: 645–651. [Google Scholar]
- Vauquelin G (2013). Simplified models for heterobivalent ligand binding: when are they applicable and which are the factors that affect their target residence time. Naunyn Schmiedebergs Arch Pharmacol 386: 949–962. [DOI] [PubMed] [Google Scholar]
- Vauquelin G, Charlton S (2010). Long‐lasting target binding and rebinding as mechanisms to prolong in vivo drug action. Br J Pharmacol 161: 488–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vauquelin G, Van Liefde I (2012). Radioligand dissociation measurements: potential interference of rebinding and allosteric mechanisms and physiological relevance of different model systems. Expert Opin Drug Discov 7: 583–595. [DOI] [PubMed] [Google Scholar]
- Vauquelin G, Charlton S (2013). Exploring avidity, understanding the potential gains in functional affinity and target residence time of bivalent and heterobivalent ligands. Br J Pharmacol 168: 1771–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vauquelin G, Morsing P, Fierens FLP, De Backer J‐P, Vanderheyden PML (2001a). A two‐state receptor model for the interaction between angiotensin II AT1 receptors and their non‐peptide antagonists. Biochem Pharmacol 61: 277–284. [DOI] [PubMed] [Google Scholar]
- Vauquelin G, Fierens FLP, Gáborik Z, Le Minh T, De Backer J‐P, Hunyady L, et al (2001b). Role of basic amino acids of the human angiotensin type1 receptor in the binding of the non‐peptide antagonist candesartan. J Renin Angiotensin Aldosterone Syst 2: S32–S36. [DOI] [PubMed] [Google Scholar]
- Vauquelin G, Van Liefde I, Vanderheyden P (2002a). Models and methods for studying insurmountable antagonism. Trends Pharmacol Sci 23: 514–518. [DOI] [PubMed] [Google Scholar]
- Vauquelin G, Van Liefde I, Birzbier BB, Vanderheyden PML (2002b). New insights in insurmountable antagonism. Fundam Clin Pharmacol 16: 263–272. [DOI] [PubMed] [Google Scholar]
- Vauquelin G, Bricca G, Van Liefde I (2014). Avidity and positive allosteric modulation may act hand in hand to increase the residence time of divalent receptor ligands. Fundam Clin Pharmacol 28: 530–543. [DOI] [PubMed] [Google Scholar]
- Vauquelin G, Hall D, Charlton P (2015). “Partial” competition of heterobivalent ligand binding may be mistaken for allosteric interactions: a comparison of different target interaction models. Br J Pharmacol 172: 2300–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheijen I, Tourlousse D, Vanderheyden PML, De Backer J‐P, Vauquelin G (2004). Effect of saponin and filipin on antagonist binding to AT1 receptors in intact cells. Biochem Pharmacol 67: 601–606. [DOI] [PubMed] [Google Scholar]
- Verheijen I, Fierens FLP, De Backer J‐P, Vauquelin G, Vanderheyden PML (2000). Interaction between the partially insurmountable antagonist valsartan and human recombinant angiotensin II type 1 receptors. Fundam Clin Pharmacol 14: 577–585. [DOI] [PubMed] [Google Scholar]
- Walkup GK, You Z, Ross PL, Allen EKH, et al (2015). Translating slow‐binding inhibition kinetics into cellular and in vivo effects. Nat Chem Biol 4: 16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Yang Y, Wang S, Nagaraj VJ, Liu Q, Wu N (2012). Label‐free measuring of binding kinetics of membrane proteins in single living cells. Nat Chem 4: 846–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienen W, Mauz ABM, Van Meel JCA, Entzeroth M (1992). Different types of receptor interaction of peptide and nonpeptide angiotensin II antagonists revealed by receptor binding and functional studies. Mol Pharmacol 41: 1081–1088. [PubMed] [Google Scholar]
- Wilson C, Agafonov RV, Hoemberger M, Kutter S, Zorba A, Halpin J, et al (2015). Using ancient protein kinases to unravel a modern cancer drug's mechanism. Science 347: 882–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin N, Pei J, Lai L (2013). A comprehensive analysis of the influence of drug binding kinetics on drug action at the molecular and systemic levels. Mol Biosyst 9: 1381–1389. [DOI] [PubMed] [Google Scholar]
- Zhang R, Monsma F (2009). The importance of drug–target residence time. Curr Opin Drug Discov Devel 12: 488–496. [PubMed] [Google Scholar]
- Zhang JC, Van Meel A, Pfaffendorf M, Van Zwieten P (1993). Different types of angiotensin II receptor antagonism induced by BIBS 222 in the rat portal vein and rabbit aorta; the influence of receptor reserve. J Pharmacol Exp Ther 269: 509–514. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Binding models differential equations that are relevant to the present article.
Table S2 Definitions and values of binding parameters of the investigated drugs.
Figure S1 Saturation binding curves: effect of the incubation time.
Figure S2 Time‐wise decline of the contribution of [RL] to the total binding of Ligand C.
Figure S3 Effect of the duration of agonist‐ exposure on the insurmountable behaviour of antagonists A to D.
Figure S4 Agonist EC50 values after pre‐treatment with antagonist C: effect of different experimental conditions.
Supporting info item