

Abstract
Chronic pain negatively impacts the quality of life in a variety of patient populations. The current therapeutic repertoire is inadequate in managing patient pain and warrants the development of new therapeutics. Adenosine and its four cognate receptors (A1, A2A, A2B and A3) have important roles in physiological and pathophysiological states, including chronic pain. Preclinical and clinical studies have revealed that while adenosine and agonists of the A1 and A2A receptors have antinociceptive properties, their therapeutic utility is limited by adverse cardiovascular side effects. In contrast, our understanding of the A3 receptor is only in its infancy, but exciting preclinical observations of A3 receptor antinociception, which have been bolstered by clinical trials of A3 receptor agonists in other disease states, suggest pain relief without cardiovascular side effects and with sufficient tolerability. Our goal herein is to briefly discuss adenosine and its receptors in the context of pathological pain and to consider the current data regarding A3 receptor‐mediated antinociception. We will highlight recent findings regarding the impact of the A3 receptor on pain pathways and examine the current state of selective A3 receptor agonists used for these studies. The adenosine‐to‐A3 receptor pathway represents an important endogenous system that can be targeted to provide safe, effective pain relief from chronic pain.
Abbreviations
- ADA
adenosine deaminase
- ADK
adenosine kinase
- CCI
chronic constriction injury
- CIPN
chemotherapy‐induced peripheral neuropathy
- CPP
conditioned place preference
- ENT
equilibrative nucleoside transporter
- PN
peroxynitrite
- RVM
rostral ventromedial medulla
- SO
superoxide
- TLR4
toll‐like receptor 4
Tables of Links
| TARGETS | |
|---|---|
| GPCRs a | Enzymes c |
| A1 receptor | ADA |
| A2A receptor | Adenylyl cyclase (AC) |
| A2B receptor | ADK |
| A3 receptor | Ecto‐5’‐nucleotidase |
| Catalytic receptors b | ERK1 |
| TLR4 | ERK2 |
| Transporters d | GAD65 |
| CNTs | p38 MAPK |
| ENT1 | PKA |
| ENT2 | S‐adenosylhomocysteine hydrolase |
| GAT1 | TrkB |
| GLT1 (EAAT1) | |
| KCC2 |
| LIGANDS | |
|---|---|
| ABT‐702 | IL‐1β |
| Adenosine | IL‐10 |
| ADP | Inosine |
| AMP | MRS1191 |
| ATP | MRS1220 |
| BDNF | MRS1523 |
| cAMP | MRS5698 |
| CCL2 | Nitric oxide (NO) |
| GABA | NMDA |
| IB‐MECA | TNF‐α |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,c,dAlexander et al., 2015a, 2015b, 2015c, 2015d).
Introduction
Chronic pain afflicts an estimated 10% of the world's adult population (Goldberg and McGee, 2011). The current therapeutic approaches for chronic pain include but are not limited to the use of nonsteroidal anti‐inflammatory drugs (NSAIDs), antidepressants, anticonvulsants and opioid pain relievers; however, these strategies are frequently either inadequate or are associated with side effects that reduce quality of life or result in the discontinuation of therapy (Goldberg and McGee, 2011; Pizzo and Clark, 2012). The search for new therapeutic targets is therefore of great importance. Adenosine and two of its associated adenosine receptor subtypes, the A1 and the A2A receptor, have been investigated in the field of pain with varying degrees of success; however, these agents lack a useful therapeutic index due to cardiovascular side effects. In response, the focus of research has turned to the previously overlooked A3 receptor, which displays both preclinical antinociceptive properties (Yoon et al., 2004; Janes et al., 2015, 2014b; Ford et al., 2015; Little et al., 2015) and, in trials for non‐pain conditions including psoriasis, hepatitis, rheumatoid arthritis, and glaucoma, offers a therapeutic index and tolerability that would be suitable for the treatment of chronic pain (Fishman et al., 2012). The aim of this review is to summarize the existing literature on adenosine and its receptors in the context of pain with a particular emphasis on the A3 receptor and its prospect as a novel solution to the problem of chronic pain management.
Adenosine production and metabolism
Adenosine is an endogenous purine nucleoside that regulates a number of physiological processes and is an important neuromodulator in the CNS (Fredholm et al., 2011). Concentrations of adenosine are generated by almost all cell types (Zimmermann, 2000), and accordingly, the physiological generation and neurobiology of adenosine has been thoroughly reviewed elsewhere (Hu et al., 2014). Here, we will provide a brief, contextual overview of adenosine homeostasis in nervous tissues.
In the context of pain, the most notable function of adenosine in the CNS is its role as a neuromodulator for neurotransmitter systems including glutamate, GABA, ACh and dopamine. In these systems, adenosine limits the extent of neuroexcitability and also regulates neuroplasticity (Sebastiao and Ribeiro, 1996). However, CNS adenosine is not restricted to neuronal synapses nor is it a directional signalling molecule., Adenosine plays a regulatory role in glial activation state and function, such that adenosine can be said to impact the entire synaptic unit (Cunha, 2008; Dias et al., 2013).
Concentrations of adenosine in CNS tissue are reported to persist at a basal level of 25–250 nM (Dunwiddie and Masino, 2001). Adenosine is generated as a metabolic intermediate in both the intracellular and extracellular spaces and passively exchanged along its concentration gradient via the ubiquitously expressed equilibrative nucleoside transporters (ENTs), ENT1 and ENT2 (Brundege and Dunwiddie, 1998; Peng et al., 2005) or through concentrative nucleoside transporters (CNTs) along the concentration gradient of sodium (Bonan, 2012; Choi and Berdis, 2012). The extracellular function of adenosine is regulated by (i) changes in the ratio of intracellular: extracellular adenosine generation and consequently its driving gradient across passive transport systems and (ii) the local expression profile of adenosine receptors mediating its response (Deussen et al., 1999; Zimmermann, 2000). Together, these two elements are modified in the pathological cellular environment in order to facilitate the action of adenosine as an anti‐inflammatory, inhibitory neuromodulator.
Intracellular adenosine generation in the CNS is predominantly a resultant from the dephosphorylation of AMP by soluble 5′‐nucleotidases (Latini and Pedata, 2001). Unlike other physiological tissues, adenosine generated from S‐adenosylhomocysteine hydrolysis does not significantly contribute to adenosine concentrations in the CNS. In the extracellular space, ATP is released as a co‐transmitter or in response to cellular insult (e.g. inflammation, cellular stress or excitotoxicity) (Ballarin et al., 1991; Engler, 1991; Latini and Pedata, 2001) and can be dephosphorylated by ectonucleoside triphosphate diphosphohydrolases (CD39 family) and either ecto‐5′‐nucleotidase (CD73) (Robson et al., 2006; Bonan, 2012) or by tissue‐nonspecific alkaline phosphatase (Sebastian‐Serrano et al., 2015) to form extracellular adenosine (Figure 1). Adenosine generation is therefore tightly coupled to the availability of ATP and ADP in the intracellular and extracellular spaces.
Figure 1.
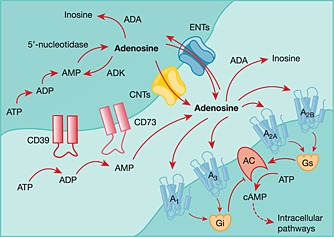
Adenosine synthesis and metabolism. ATP can be released from various cell types in response to cell excitation or insult. ATP can be deophosphorylated in sequence to form ADP, AMP and finally adenosine. In the extracellular space, ectonucleotidases (CD39 and CD73) facilitate formation of adenosine. Adenosine can act on its cognate receptors (A1, A2A, A2B and A3) or be removed from the extracellular space by metabolic enzymes (adenosine deaminase, ADA) or by transport back into the cell via equilibrative nucleoside transporters (ENTs) or concentrative nucleoside transporters (CNTs). In the intracellular space, adenosine can be converted to AMP (by adenosine kinase, ADK) which in turn is catalysed to ADP and then ATP. Intracellular adenosine can also be generated from AMP by 5′‐nucleotidase.
A second factor limiting the ratio of intracellular: extracellular adenosine is the metabolic inactivation of adenosine through adenosine kinase (ADK) phosphorylation (Spychala et al., 1996) or, to a lesser extent, the activity of adenosine deaminase (ADA) (Blackburn and Kellems, 1996). The function of these enzymes limits the physiological half‐life of adenosine to <1 s (Moser et al., 1989). Inhibition of ADK is the most effective strategy for increasing the extracellular concentration of adenosine and occurs by potentiating intracellular concentrations of adenosine and supporting an outward driving gradient through passive ENTs (Keil and DeLander, 1992; Zhang et al., 1993). Pharmacological blockade of ADK activity in the CNS results in adenosine‐mediated inhibition of spinal nociceptive transmission via the ENT‐dependent release of adenosine (Otsuguro et al., 2015), and inhibitors of ADK are efficacious in rodent experimental neuropathic pain models (Kowaluk et al., 2000; McGaraughty et al., 2005). It is important to note that ADK expression shifts from neurons to astrocytes during postnatal development, and accordingly, astrocytes are central to this aspect of adenosine homeostasis (Studer et al., 2006).
Adenosine receptors
As mentioned previously, the abundance of adenosine receptor subtypes is the second critical factor mediating the effects of extracellular adenosine, and much remains to be understood regarding the dynamics of adenosine receptor expression during cellular insult and pathological nociception. The extracellular actions of adenosine are mediated by its four cognate GPCRs: the A1, A2A, A2B and A3 receptor.
In the CNS, the A1 receptor is highly expressed both pre‐synaptically and post‐synaptically on neurons as well as on glia in the brain (cortex, cerebellum and hippocampus) and specifically in the superficial laminae of the spinal cord dorsal horn (Gessi et al., 2011). It is known that glial expression of the A1 receptor can be depressed in multiple sclerosis patients (Johnston et al., 2001), and expression of the A1 receptor in the lumbar dorsal horn is decreased in a model of post‐operative pain. However, expression of the A1 receptor is increased following sciatic nerve constriction (Yamaoka et al., 2013). These data suggest that the A1 receptor is responsive to neuroinflammatory and nociceptive pathology, and that A1 receptor antinociception may involve differential expression of the receptor post‐injury. However, in the context of off‐target effects, the presence of the A1 receptor in cardiovascular tissue—particularly the atrioventricular node—is responsible for high‐grade atrioventricular block mediated by A1 receptor agonists (Kiesman et al., 2009) and represents an unfortunate hurdle to the therapeutic exploitation of the A1 receptor
Expression of the A2A receptor in the brain is most notable in the striatum on post‐synaptic neurons, to a lesser extent pre‐synaptically in areas of the hippocampus and cerebral cortex and on glia (Svenningsson et al., 1997; Rebola et al., 2005). A2A receptor expression is known to increase in response to insults such as hypoxia, spinal cord injury, streptozotocin‐induced diabetes and in other circumstances such as chronic behavioural stress and ageing (Janes et al., 2014b). Of relevance to chronic pain, pro‐inflammatory mediators including IL‐1β and TNF‐α are also known to enhance expression of the A2A receptor in monocytes (Morello et al., 2006) such as microglia. Lastly, vasodilator A2A receptors expressed on the epithelium of coronary blood vessels is of note with respect to the cardiovascular side effects of A2A‐specific agents (Jacobson and Gao, 2006; Gao and Jacobson, 2007; Fredholm et al., 2011). The lower‐affinity A2B receptor is also expressed in the CNS—particularly on immune‐related cells—and in the cardiovascular system, but because of the low‐expression profile and micromolar affinity of adenosine for the A2B receptor, we will not extensively discuss the A2B receptor in this review (Fredholm et al., 2000; Aherne et al., 2011).
For many years, the A3 receptor was largely overlooked in CNS tissue due to a reported low profile of expression; however, it is now known that the A3 receptor can be found in high levels on many immune cell types, including glial cells (Abbracchio et al., 1997; Poulsen and Quinn, 1998; Ochaion et al., 2009), as well as on both peripheral (Ru et al., 2011) and central neurons (Jacobson et al., 1993; Lopes et al., 2003; Giannaccini et al., 2008; Zhang et al., 2010) in the brain and spinal cord (Borea et al., 2015; Haeusler et al., 2015). Both mRNA and protein for the A3 receptor have been documented in the lumbar spinal cord and in supraspinal areas including the rostral ventromedial medulla (RVM): indeed, A3 receptors at the level of the peripheral afferent, the spinal cord and the RVM are functionally relevant in pain as selective A3 receptor agonists administered via i.d., intrathecal (i.t.) or intra‐RVM routes dose‐dependently attenuate neuropathic pain behaviours (Little et al., 2015). It can therefore be concluded that the A3 receptor is functionally expressed at multiple levels of pain processing (Figure 2).
Figure 2.
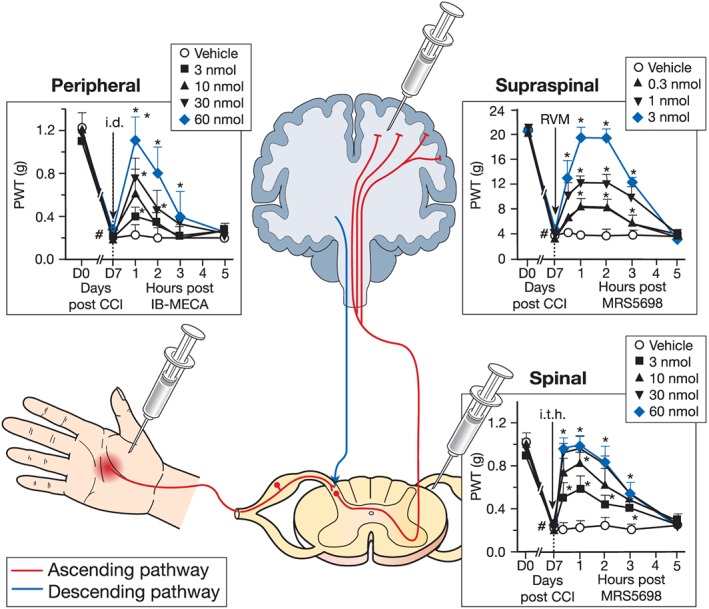
A3 receptor's multiple sites of action. Studies employing selective A3 receptor agonists have uncovered the efficacy of A3 receptor activation at several sites important for pain processing. In the ascending pathway (red), a stimulus is conducted from the periphery to the spinal cord where it is then transported to the brain to be interpreted. The brain is able to modulate these events via the descending pathway (green). The i.d. administration of IB‐MECA (3–60 nmol; Salvemini unpublished results) dose‐dependently attenuated the reduction in paw withdrawal thresholds (PWTs) associated with chronic constriction injury (CCI). These results were extended by MRS5698 administration via surgically implanted catheters placed into the intrathecal (i.t.; 3–60 nmol) space or rostral ventromedial medulla (RVM; 0.3–3 nmol). Data are mean ± SD for n = 5 animals per group and analysed by two‐way ANOVA with Bonferroni comparisons. #P < 0.05 vs. D0; *P < 0.05 vs. D7. The i.t. data are reprinted with permission from J Neuro (Ford et al., 2015) . Intra‐RVM data are reprinted with permission from Brain (Little et al., 2015).
The A3 receptor is unique among the adenosine receptors in that species disparities exist for both the receptor structure and distribution; the highest expression of the A3 receptor in rats exists in testis and mast cells, whereas the highest levels in human are found in the liver and lung (Borea et al., 2015). Importantly, while no direct evidence has been found for the existence of the A3 receptor in cardiomyocytes, several studies have demonstrated A3 receptor‐mediated cardioprotection from ischaemic injury (Tracey et al., 1997; Thourani et al., 1999a, 1999b; Cross et al., 2002; Harrison et al., 2002; Headrick and Peart, 2005) and doxorubicin‐induced cardiotoxicity (Shneyvays et al., 1998; Shneyvays et al., 2001). Additionally, high A3 receptor expression has been documented in the human coronary and carotid arteries (Hinze et al., 2012; Grandoch et al., 2013).
Given the distribution of adenosine receptors, it is also important to note the coupling mechanisms of these receptors. The A2A and A2B subtypes are Gs‐coupled GPCRs which stimulate AC and produce elevations in intracellular cAMP and associated signalling cascades (Fredholm et al., 2001). A1 and A3 receptors differ both in their coupling to the opposing Gi cascade which inhibits AC and in the ability to respond to inosine as a partial agonist (Boison et al., 2010; Fredholm et al., 2011). It is important to note that while these receptors have, at face value, opposing intracellular effects, the expression of these receptors on differing cell types can be responsible for commonalities in their activation in the CNS.
Adenosine and pain
Adenosine plays an integral role in CNS processing of pain through its regulation of excitatory neurotransmission, persistent neuronal signalling and regulation of glial activation and proliferation. Accordingly, the ability of adenosine and its analogues to inhibit pain behaviour has been documented in models of various aetiologies, including neuropathic pain associated with spinal cord injury, spinal nerve ligation and in the mustard oil, formalin and carrageenan pain models (Dickenson et al., 2000). Indeed, in the clinical setting, i.t. adenosine administration has been shown to provide sustained relief of chronic neuropathic pain lasting for several hours up to months in some patients (Hayashida et al., 2005). Adenosine therapy has also been employed for the prevention of post‐operative pain with mixed results: prophylactic i.v. administration of adenosine prior to surgical procedures conferred persistent pain relief in several studies (Hayashida et al., 2005; Gan and Habib, 2007), while another similar clinical trial did not observe a prophylactic effect (Habib et al., 2008). Unfortunately, the i.v. administration of adenosine is associated with undesirable cardiac side effects (Zylka, 2011) that limit its utility and route of administration (e.g. i.t.) in patients. Thus, isolating the antinociceptive qualities of adenosine from its cardiovascular side effects by evaluating receptor involvement has become an important focus for the development of adenosine‐based therapeutics in pain.
A1 and A2A receptors and pain
It is not our intent to discuss in detail the A1 and A2A receptors, because these targets have been the topic of many excellent reviews (Sawynok, 1998; Fredholm et al., 2011; Zylka, 2011; Chen et al., 2013), but these receptors have played an important role in the understanding of adenosine antinociception. The antinociceptive properties of adenosine were long attributed to the activation of the A1 and A2A receptor subtypes (Zylka, 2011; Sawynok, 2013). Genetic deletion of the A1 receptor produces thermal hypersensitivity and exacerbates neuropathic behavioural responses to cold and heat (Fedorova et al., 2003; Wu et al., 2005), and it is well documented in the literature that A1 receptor activation leads to antinociception in a range of pain models. A1 receptor activation produces beneficial outcomes in preclinical models of acute and chronic pain including nerve injury‐induced pain (Cui et al., 1997; Gong et al., 2010), peri‐operative pain (Gan and Habib, 2007), inflammatory pain (Sowa et al., 2010), central pain following spinal cord injury (Sjolund et al., 1998) and painful diabetic neuropathy (Vincenzi et al., 2014; Katz et al., 2015). Additionally, a spinally administered A1 receptor agonist reduces non‐evoked spontaneous pain behaviours resulting from a surgical model of pain (Zahn et al., 2007). The preclinical robustness of A1 receptor pain relief resulted in clinical trials for multiple A1 receptor agonists and an A1 receptor allosteric enhancer; however, these drug trials were discontinued due to limited efficacy, presumably driven by a low therapeutic index and dose limitation (Romagnoli et al., 2010; Gessi et al., 2011).
The findings regarding the A2A receptor have been controversial. Mice lacking the A2A receptor demonstrate depressed responses to acute pain stimuli as measured by the hot‐plate and tail‐flick tests (Ledent et al., 1997). Peripheral administration of an A2A receptor agonist is associated with nociceptive behaviours (Taiwo and Levine, 1990). However, very low doses of A2A receptor agonists administered spinally have been shown to promote the sustained reversal of nerve injury‐induced pain in rats for weeks after a single i.t. injection (Loram et al., 2009). In models of post‐surgical pain (Zahn et al., 2007) and inflammatory pain (Poon and Sawynok, 1998), i.t. administration of A2A receptor agonists had only transient antinociceptive effects that were minimal‐to‐negligible. However, an i.c.v. injection of an A2A‐targeted antibody with agonist‐like activity produces antinociceptive effects in naïve mice (By et al., 2011). At the clinical level, a phase II trial of an A2A receptor agonist BVT‐115959 in the treatment of diabetic neuropathy was completed in 2008, but the trial has been since abandoned (Swedish Orphan Biovitrum, 2014). The differing observations of A2A receptor agonists in pain highlight an apparent dichotomy of peripheral versus central A2A receptors in pain processing.
For the past decade, a narrow therapeutic focus on the A1 and A2A receptor has failed to harness adenosine antinociception effectively and without cardiovascular side effects (Zylka, 2011; Boison, 2013). In response, we anticipate that a greater emphasis on the A3 receptor is a better utilization of adenosine antinociception to provide safe, effective pain relief at the clinical level.
A3 receptors and pain
Our understanding of A3 receptor signalling in pain has evolved greatly since the human A3 receptor was first cloned in 1993 (Salvatore et al., 1993). Early investigations reached conclusions that were not necessarily correct due to the use of A3 receptor‐targeted compounds with poor specificity such as N 6‐benzyl–NECA (Sawynok et al., 1997; 1999) and the A3 receptor−/− mouse (Wu et al., 2002). A 1997 publication examining the contribution of the A3 receptor in pain reported that s.c. administration of N 6‐benzyl–NECA into the rodent hindpaw produced dose‐dependent flinching behaviour that was blocked by administration of a histamine H1 receptor antagonist or a 5‐HT2 receptor antagonist, but not by antagonists of A1 or A2 receptors (Sawynok et al., 1997). These studies lead to the incorrect hypothesis that the A3 receptor was likely to mediate the pro‐nociceptive, pro‐inflammatory effect of N 6‐benzyl–NECA via the induction of mast cell degranulation. A subsequent study clarified that N 6‐benzyl–NECA nociception was not influenced by an A3 receptor antagonist (MRS1191) but was in fact abolished by blockade of the A2B receptor, a subtype previously implicated in inflammation (Feoktistov and Biaggioni, 2011).
The first definitive characterizations of the A3 receptor as antinociceptive resulted from the use of a more specific A3 receptor agonist, IB‐MECA (N 6‐(3‐iodobenzyl)‐adenosine‐5′‐N‐methyluronamide): IB‐MECA is 50‐fold selective for the A3 receptor over the A1 or A2A receptor, as compared with the 14‐fold selectivity of N 6‐benzyl–NECA (Gallo‐Rodriguez et al., 1994; Jacobson, 1998). In 2005, a study demonstrated that systemically administered IB‐MECA exerts a significant antinociceptive effect during the second phase of the formalin test without altering protective nociceptive responses (i.e. response to noxious thermal or mechanical stimuli) (Yoon et al., 2005). i.t. administration of an A3 receptor antagonist (MRS1220) prevented the antinociceptive actions of adenosine in the second phase of the formalin test, supporting a role for spinal A3 receptors in the effect of adenosine (Yoon et al., 2006).
Genetic work in the A3 receptor knockout mice (A3AR)−/− mouse has followed a similar pattern of evolution. In 2002, a study of carrageenan‐induced peripheral inflammatory pain in the A3AR−/− mouse noted a mild increase in the development of thermal hyperalgesia as compared with wild‐type controls (Wu et al., 2002). There was no alteration in the protective (i.e. non‐pathological) nociceptive responses of A3AR−/− animals to noxious heat and mechanical stimuli, implicating the A3 receptor in pathological rather than protective pain states. This report was partially contradicted by a later study that observed a decrease in the protective hot‐plate but not tail‐flick responses of A3AR−/− mice (Fedorova et al., 2003). Consequently, it was unclear whether the A3 receptor affected normal protective nociception or was solely implicated in pathological pain states.
There were no studies published between 2006 and 2012 that examined the contribution of A3 receptors in pain. In 2012, our laboratory revisited the A3 receptor hypothesis using the agents IB‐MECA and Cl‐IB‐MECA in neuropathic pain (Chen et al., 2012; Little et al., 2015). Importantly, we have observed no impact of these agents and others on baseline nociceptive thresholds, and we have therefore concluded that A3 receptor agents do not alter normal protective nociception. In neuropathic pain, both IB‐MECA and Cl‐IB‐MECA blocked the development of mechano‐allodynia following chronic constriction injury (CCI) in a manner prevented by an antagonist of the A3 receptor but not antagonists of the A1 or A2A receptor (Chen et al., 2012). Low doses of IB‐MECA given in combination with morphine, gabapentin or amitriptyline increased the potency of these agents as analgesics (Chen et al., 2012). The antinociceptive effects of IB‐MECA and Cl‐IB‐MECA have since been corroborated by better‐selective A3 agonists including MRS1898 [>100‐fold over A1 or A2A receptors (Gao et al., 2009)] and more recently MRS5698 [>10 000‐fold over A1 or A2A receptors (Tosh et al., 2012)] in rodent CCI, spared nerve injury and spinal nerve ligation models (Chen et al., 2012; Ford et al., 2015; Little et al., 2015). The specificity of a newer generation A3 receptor agonists has been corroborated by the attenuation of MRS5698 antinociception in the A3AR−/− mouse, and alternatively in the presence of a specific A3 receptor antagonist. (Little et al., 2015). In CCI, A3 receptor agonists administered via i.d. (ipsilateral paw to nerve injury) injection (IB‐MECA, 3–60 nmol), i.t. cannula (MRS5698, 3–60 nmol) or RVM cannula (MRS5698, 0.3–3 nmol) dose‐dependently attenuate mechanical allodynia (Little et al., 2015). Antinociception conferred via systemic administration of the CNS‐permeant MRS5698 is attenuated with i.t. or intra‐RVM delivery of an A3 receptor antagonist (Little et al., 2015). However, systemic administration of a peripherally restricted A3 receptor agonist also reverses CCI‐induced peak mechanical allodynia, and the effect is not reversed by administration of an i.t. A3 receptor antagonist (Paoletta et al., 2013). It can therefore be concluded that the A3 receptor produces antinociceptive input at central and peripheral levels, which is an important characteristic of successful analgesic agents (e.g. opioids). Further studies are warranted to explore the relationship between peripheral and central A3 receptors in pain.
A3 receptor agonists have also been validated in a number of cancer‐related pain states. In models of chemotherapy‐induced peripheral neuropathy (CIPN), IB‐MECA (Chen et al., 2012; Janes et al., 2014b) and MRS5698 (Janes et al., 2015; Little et al., 2015) blocked the development of neuropathic pain without interfering with the antitumour effects (Chen et al., 2012). In a rat model of breast cancer bone metastasis, treatment with Cl‐IB‐MECA reduced tumour growth and the related bone pain (Varani et al., 2013), and MRS5698 had similar antinociceptive effects in a mouse model of breast cancer bone metastasis (Little et al., 2015). High expression of the receptor is detected on many malignant cell types, and A3 receptor agonists have been shown to produce direct anticancer effects on their own and have been documented to enhance the actions of several widely used chemotherapeutics and attenuate the associated myelosuppression (Fishman et al., 2002; Fishman et al., 2009; 2012). Indeed, a successful phase I/II clinical trial of Cl‐IB‐MECA as an anticancer agent in hepatocellular carcinoma was recently completed by Can‐Fite BioPharma. Therefore, the use of A3 receptor agonists may provide dual benefits in the treatment of a variety of cancer‐related pain states.
Finally, it is important to note that because the antinociceptive effects of IB‐MECA and other selective A3 receptor agonists are not dependent upon endogenous opioid or endocannabinoid pathways (Ford et al., 2015; Little et al., 2015), studies have evaluated whether A3‐specific antinociception lacks classical tolerance and inherent reward properties. In preclinical studies, A3 receptor agonists are not subject to analgesic tolerance: in rats and mice, the effect of A3 receptor agonists persisted following six repeated daily injections (compared with morphine, which demonstrated tolerance following repeated injections for 6 days) or when administered as a continuous infusion for 7 days (Little et al., 2015). These findings are interesting as repeated or continuous exposure to adenosine receptor agonists usually results in diminished responses and receptor down‐regulation known as ‘desensitization phenomenon,’ a response that is characteristic for all adenosine receptor subtypes (Klaasse et al., 2008). However, similar findings have been reported in animal models of autoimmune disorders and cancer, wherein chronically administered A3 receptor agonists do not lose their anti‐inflammatory/anticancer effects in spite of A3 receptor protein down‐regulation (Madi et al., 2003). It was demonstrated that down‐regulation of the receptor is associated with downstream inhibition of key regulatory proteins involved in inflammation/tumour growth, such that receptor down‐regulation in fact represents receptor functionality in these cases (Fishman et al., 2006). It is possible that a similar mechanism exists for the action of IB‐MECA and other A3 receptor agonists in pain, but this hypothesis requires further investigation.
In order to evaluate the inherent reward and therefore abuse potential of A3 receptor agonists, the conditioned place preference (CPP) method was combined with the induction of nerve injury pain in rats to evaluate spontaneous pain behaviours. Results from these studies indicate that A3 receptor agonists such as MRS5698 produce CPP in nerve‐injured but not sham rats (unlike opioids and other drugs of abuse, which can elicit CPP from both naïve and injured animals), suggesting that A3 receptor activation attenuates spontaneous pain behaviours without inherent reward (Little et al., 2015). Taken together with the observation that A3 receptor agonists selectively modify pathological but not protective pain, it can be hypothesized that A3 receptor agonists may circumvent the classical complications of tolerance and abuse potential associated with opioid therapy.
Mechanisms of A3 receptor‐induced antinociception
Due to the recent emergence of the A3 receptor as a valid target for pain relief, much remains to be explored regarding the specific mechanisms of action downstream of receptor activation. To date, it is known that in the CCI model of neuropathic pain, the effects of A3 receptor agonists are mediated independently of the opioidergic and cannabinoid systems, but do act supraspinally to recruit the activation of 5‐hydroxytryptaminergic and noradrenergic bulbospinal circuits, and reduce the excitability of wide dynamic range spinal neurons (Little et al., 2015). Here, we will discuss the mechanisms by which A3 receptor activation in other disease states alters processes involved in the development of central sensitization and pain, including protein kinase activity, glutamatergic neurotransmission, ion conductance and neuroinflammation. A summary of this discussion can be found in Figure 3.
Figure 3.
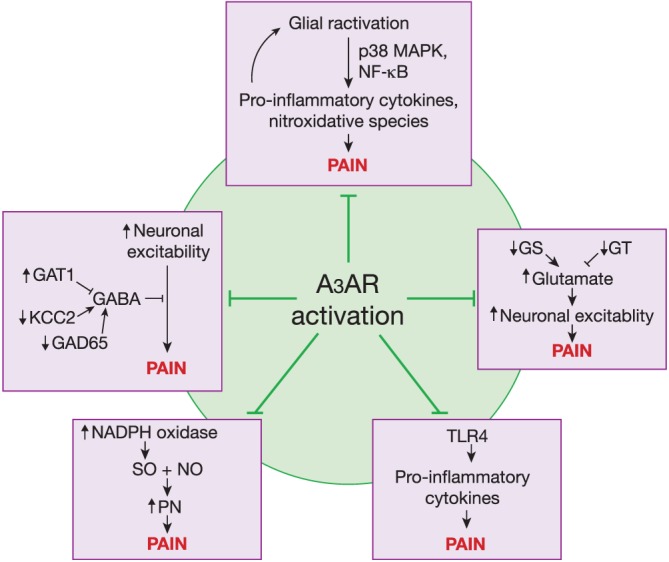
Potential mechanisms of A3 receptor (A3AR)‐mediated antinociception. Several pathways are known to be important in the establishment of pain states including impairment of GABAergic neurotransmission, enhanced neuroinflammation characterized by increased glial hyperactivation and TLR4 signalling, increased glutamatergic signalling and heightened production of nitroxidative species. The A3 receptor has been shown to modulate these pathways through the use of selective agonists, potentially explaining A3 receptor's antinociceptive actions.
The GABAergic system is an important component regulating correct nociceptive transmission. Following the release of GABA from interneurons within the CNS, the neurotransmitter GABA can act upon its receptors to dampen neuronal excitability and reduce nociceptive signalling. During pathological pain, dysfunction of the GABAergic interneuron system liberates excitatory neuronal signalling and ultimately results in a state of system hyperexcitability (Zeilhofer et al., 2012). An abnormality in GABA signalling can occur through a variety of mechanisms, including reduced GABA synthesis by the enzyme GAD65 (Stiller et al., 1996; Eaton et al., 1998), enhanced expression of the GABA reuptake transporter GAT‐1 (Eaton et al., 1998; Moore et al., 2002) and impairment of the K+‐Cl− cotransporter KCC2, which maintains the anion gradient required for the inhibitory action of Cl− through GABAA channels (Coull et al., 2003; Price et al., 2005). It was recently demonstrated that the A3 receptor agonist MRS5698 reverses CCI‐induced pain via a GABA‐mediated mechanism, wherein the CCI‐related dephosphorylation of GAD65 and GAT‐1 and the phosphorylation of KCC2 are ameliorated by treatment with an A3 receptor agonist (Ford et al., 2015). These results indicate that restoration of the GABAergic inhibitory system contributes to the reversal of neuropathic pain following A3 receptor activation. In addition to modifying phosphorylation of these enzymes, the A3 receptor may also exert its effects on GABA‐mediated inhibition indirectly through the attenuation of brain‐derived neurotrophic factor (BDNF) signalling. Glial‐derived BDNF–TrkB signalling has been shown to reduce GABAergic signalling in the CCI model of neuropathic pain (Biggs et al., 2010; Ferrini and De, 2013; Smith, 2014). A3 receptor activation is associated with the attenuation of astrocyte reactivity, neuroinflammatory response (Janes et al., 2015) and reactive microglial chemotaxis (Choi et al., 2011), such that A3 receptor agonists may reduce BDNF associated with glial hyperactivation and free the GABAergic system to function properly.
A3 receptor agonists have demonstrated neuroprotection in animal models that is potentially mediated through the induction of pro‐survival RhoA‐PLD signalling pathways (Jacobson, 1998; Fredholm et al., 2011). For example, A3 receptor agonists prevent the decrease in PLD activity that occurs in response to prolonged reactive oxygen species exposure during apoptosis in cardiomyoctytes (Lee et al., 2001; Asemu et al., 2005). Accordingly, the protection of proper PLD function can then go on to increase the production of choline, which leads to activation of α7 nicotinic ACh receptors (Lee et al., 1993). This effect is known to be both neuroprotective and antinociceptive in chronic neuropathic pain (Feuerbach et al., 2009).
Many studies implicate spinal glia in the development and maintenance of chronic pain, and a variety of pain states can be prevented or attenuated with agents that disrupt the stimulation of glial cells (Milligan and Watkins, 2009; Gwak et al., 2012; Old et al., 2015). During an enhanced response state, glial cells can release pro‐inflammatory cytokines and nitroxidative species, which can then sensitize the neurons in the dorsal horn leading to pain (Cao and Zhang, 2008; Milligan and Watkins, 2009). These glial‐derived pro‐inflammatory mediators act not only on neurons but also on glial cells leading to an amplification loop that is potentially responsible for the long‐lasting hypersensitivity underpinning some chronic pain states (Bradesi et al., 2001). A3 receptor agonists have been shown to impart their beneficial actions at least partly through modulation of spinal neuroinflammatory processes, as IB‐MECA treatment results in a reduction of hyperactive astrocytes and reduced production of pro‐inflammatory/neuroexcitatory cytokines in models of CIPN (Janes et al., 2014a; Janes et al., 2015). Interestingly, A3 receptor activation also enhances formation of the anti‐inflammatory cytokine IL‐10 (Hasko et al., 1996; Janes et al., 2014a; Janes et al., 2015) and the production of glial‐derived neuroprotective substances including CCL2 (Wittendorp et al., 2004). Both in vitro and in vivo studies have revealed that A3 receptors produce these effects by inhibiting the p38 MAPK and NF‐κB signalling pathways (Madi et al., 2007; Varani et al., 2010; Varani et al., 2011; Janes et al., 2014a). A better understanding of whether this mechanism occurs in A3 receptor‐mediated antineuroinflammatory pain relief is critical to the development of an A3 therapeutic strategy in pain.
Increased formation of nitroxidative species including superoxide, NO and their highly pro‐nociceptive reaction product peroxynitrite (Salvemini and Neumann, 2010) have been shown to play an important role in the development and maintenance of pain of several aetiologies including acute and chronic inflammation (Ndengele et al., 2008), orofacial pain (Yeo et al., 2008), opiate‐induced hyperalgesia and antinociceptive tolerance (Muscoli et al., 2007), nerve injury‐induced pain (Rausaria et al., 2011) and CIPN (Doyle et al., 2012; Janes et al., 2013). In a model of CIPN, the A3 receptor agonist IB‐MECA attenuated the spinal activation of NADPH oxidase, a source of superoxide as a precursor to peroxynitrite formation (Poderoso et al., 1996; Janes et al., 2014a). In prostate cancer cells, IB‐MECA treatment has been shown to inhibit NADPH oxidase activation through inhibition of intracellular cAMP/PKA (Jajoo et al., 2009) and by reducing the expression of NADPH oxidase subunits through inhibition of ERK1/2 activity (Jajoo et al., 2009). As such, the downstream effects of the A3 receptor on PKA activation and ERK phosphorylation could underlie the observed effect of IB‐MECA on NADPH oxidase in the CNS during pain.
Given the role of adenosine in limiting excitatory neurotransmission, it is unsurprising that A3 receptor activation impacts glutamatergic signalling. A3 receptor agonists protect against the neurotoxic P2X7 receptor‐mediated (Zhang et al., 2006) or the glutamate‐ and NMDA‐mediated rises in intracellular Ca2 + and neuronal excitability in vitro (Zhang et al., 2010). Alterations in glutamatergic neurotransmission and increased neuronal excitability are widely observed in models of chronic pain (Hansson and Ronnback, 2004; Latremoliere and Woolf, 2009; Gwak et al., 2012). A3 receptor activation is associated with the inhibition of the post‐translational nitration and inactivation of the glutamate transporter GLT‐1 and glutamate synthase (Janes et al., 2014a), together largely responsible for maintaining proper glutamatergic signalling and termination (Mao et al., 2002). In addition, A3 receptor agonists in neurons inhibit signalling through presynaptic metabotropic glutamate receptors (normally involved in reducing neurotransmission at glutamatergic synapses) (Macek et al., 1998), which have been shown to be involved in the induction of several pain states (Fundytus, 2001).
Finally, activation of the innate immune receptor toll‐like receptor 4 (TLR4) expressed on glial cells has been implicated in the development of neuropathic pain (Watkins et al., 2009; Li et al., 2014). Stimulation of the A3 receptor with IB‐MECA has been documented to decrease the TLR4‐induced release of pro‐inflammatory mediators including TNF and macrophage inflammatory protein‐1α as well as to increase the production of the anti‐inflammatory IL‐10 (Hasko et al., 1996; Sajjadi et al., 1996; Szabo et al., 1998). Suppression of pro‐inflammatory mediators following TLR stimulation is lost in A3 receptor knockout mice, suggesting that the A3 receptor has a critical role in suppressing TLR4 responses (Salvatore et al., 2000).
A3 receptor‐specific tools for the study of pain
For the benefit of future investigations of the A3 receptor, we have provided a table of preclinically useful adenosine and A3AR modulators, their pharmacological characteristics (Table 1) and their structures (Figure 4). Adenosine itself is a native, nonselective adenosine receptor agonist, while its metabolite inosine, generated following the action of ADA, weakly activates the A3 receptor (Gao et al., 2011). Preclinically successful A3 receptor agonists (compounds 1–6) have been generated through substitutions at the C2, N6 and 5′ positions, most favorably including: N 6‐benzyl (compounds 1–6) or small alkyl (compound 6); C2‐arylethynyl (compounds 4–6) or aryltriazolyl substitutions. IB‐MECA (compound 1) and Cl‐IB‐MECA (compound 2) are widely used in pharmacological probes of nM affinity with CI‐IB‐MECA being more selective for the A3 receptor (K D 2.4 nM versus 1.5 nM respectively), and both of these agents have been employed in clinical trials, such that they represent clinically available avenues for targeting the A3 receptor. Compounds 3–6 contain a conformationally constrained bicyclic (N‐methanocarba) ring in place of ribose, which adds to the A3 receptor selectivity (Tosh et al., 2012; 2014; 2015), and these agents are specific for the A3 receptor with a selectivity of 0.7–3.5 nM. MRS5481 (compound 4) is a peripherally restricted agonist that is highly A3‐selective (K D 1.9 nM) (Paoletta et al., 2013). MRS5698 (compound 5) is balanced in affinity at the human and mouse A3 receptors (K D 3 nM) with insignificant activity at A1 and A2A receptors at 10 μM. Compound 6 displays a long duration of action in vivo when administered p.o.
Table 1.
A3 receptor‐selective agents for use in the study of A3‐mediated antinociception
| Compound | K i or K D, nM (or % inhibition at 10 μM) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Human | Mouse | Rat | |||||||
| A1 | A2A | A3 | A1 | A2A | A3 | A1 | A2A | A3 | |
| Agonists | |||||||||
| 1 – IB‐MECA | 700 ± 270 | 6200 ± 100 | 2.4 ± 0.5 | 5.9 | ~700 | 0.087 | 54 | 56 | 1.1 |
| 2 – Cl‐IB‐MECA | 220 ± 20 | 5400 ± 2500 | 1.5 ± 0.2 | 35 | ~10 000 | 0.18 | 820 | 470 | 0.33 |
| 3 – MRS1898 | 136 ± 22 | 784 ± 97 | 1.51 ± 0.23 | 83.9 ± 10.3 | 1660 ± 260 | 0.17 ± 0.04 | |||
| 4 – MRS5841 | 16% | 7% | 1.90 ± 0.03 | 15% | 1% | 11.3 ± 1.9 | |||
| 5 – MRS5698 | 6% | 41% | 3.49 ± 1.84 | 16% | 27% | 3.08 ± 0.23 | |||
| 6 – MRS5980 | 6% | 24% | 0.70 ± 0.11 | 38% | 7% | 36.1 ± 4.7 | |||
| Antagonists | |||||||||
| 7 – MRS1523 | >10 000 | 3660 ± 930 | 18.9 | 8000 | >10 000 | 731 | 15 600 | 2050 | 113 |
| 8 – MRS1191 | >10 000 | >10 000 | 31.4 | 40 100 | >10 000 | 1850 | |||
Figure 4.
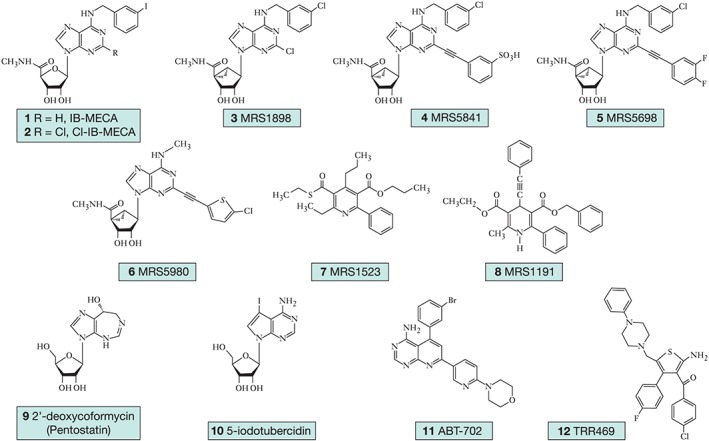
Pharmacological agents useful for the study of A3 receptor‐mediated antinociception. Preclinically useful selective A3 receptor agonists (1–6), A3 receptor antagonists (7–8) and adenosine modulators (9–12).
An alternative strategy for the study of the A3 receptor is the use of compounds that enhance extracellular adenosine concentrations such as inhibitors of ADA (e.g. compound 9, pentostatin) and inhibitors of adenosine kinase (e.g. compound 10 5‐iodotubercidin and compound 11 ABT‐702) used in tandem with selective A3 receptor antagonists to elucidate A3‐mediated effects (Figure 4). As an ADK inhibitor, compound 11 decreases both chronic and acute pain with peripheral or central administration (Kowaluk et al., 2000), and compound 12 (TRR496) is a selective A1 receptor allosteric enhancer that suppresses pain in a manner comparable and additive with morphine in formalin and writhing tests and has anti‐allodynic effects in the streptozotocin‐induced model of diabetic neuropathic pain (Vincenzi et al., 2014). An effective and moderately selective A3 receptor antagonist for use in mouse and rat is compound 8 (MRS1523) (Li et al., 1998), although species differences in affinity should be taken into consideration when using selective adenosine receptor ligands, as heterocyclic antagonists of the A3 receptor often have much higher affinity at the human than the murine A3 receptor.
Concluding remarks
Although early studies investigating the A3 receptor presumed a peripheral pro‐nociceptive role in pain—in large part due to the use of nonselective agonists—the development of selective pharmacological tools targeting the A3 receptor has now uncovered the robust antinociceptive properties of A3 receptor agonists in a variety of pathological pain states. Emerging evidence suggests that harnessing the endogenous antinociceptive A3 receptor pathway yields effective pain relief without altering normal protective nociception and without producing reward effects associated with abuse potential. Noteworthy, the cardiovascular side effects marring the usefulness of adenosine‐targeted therapies is reduced in A3 receptor‐mediated strategies, and A3 receptor agonists do not display complications in on‐going phase II/III clinical trials for non‐pain conditions. We propose that A3 receptor activation may be a safe and successful strategy for exploiting the potent analgesic actions of adenosine to provide a breakthrough non‐opioid treatment for patients suffering from chronic pain.
Author contributions
All authors contributed to writing and reviewing sections of the manuscript and approved the final version.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
We would like to thank Amanda Ford for her contributions to the experiments outlined in Figure 2. D. S. is funded by the Leukemia and Lymphoma Society Translational Research Program, the National Cancer Institute (RO1CA16519) and the Mayday Fund with additional support from the Saint Louis Cancer Center. K. A. J. acknowledges funding from the NIDDK Intramural Research Program.
Janes, K. , Symons‐Liguori, A. , Jacobson, K. A. , and Salvemini, D. (2016) Identification of A3 adenosine receptor agonists as novel non‐narcotic analgesics. British Journal of Pharmacology, 173: 1253–1267. doi: 10.1111/bph.13446.
References
- Abbracchio MP, Rainaldi G, Giammarioli AM, Ceruti S, Brambilla R, Cattabeni F, et al. (1997). The A3 adenosine receptor mediates cell spreading, reorganization of actin cytoskeleton, and distribution of Bcl‐XL: studies in human astroglioma cells. Biochem Biophys Res Commun 241: 297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aherne CM, Kewley EM, Eltzschig HK (2011). The resurgence of A2B adenosine receptor signaling. Biochim Biophys Acta 1808: 1329–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E, et al. (2015d). The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asemu G, Dent MR, Singal T, Dhalla NS, Tappia PS (2005). Differential changes in phospholipase D and phosphatidate phosphohydrolase activities in ischemia–reperfusion of rat heart. Arch Biochem Biophys 436: 136–144. [DOI] [PubMed] [Google Scholar]
- Ballarin M, Fredholm BB, Ambrosio S, Mahy N (1991). Extracellular levels of adenosine and its metabolites in the striatum of awake rats: inhibition of uptake and metabolism. Acta Physiol Scand 142: 97–103. [DOI] [PubMed] [Google Scholar]
- Biggs JE, Lu VB, Stebbing MJ, Balasubramanyan S, Smith PA (2010). Is BDNF sufficient for information transfer between microglia and dorsal horn neurons during the onset of central sensitization? Mol Pain 6: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn MR, Kellems RE (1996). Regulation and function of adenosine deaminase in mice. Prog Nucleic Acid Res Mol Biol 55: 195–226. [DOI] [PubMed] [Google Scholar]
- Boison D (2013). Adenosine kinase: exploitation for therapeutic gain. Pharmacol Rev 65: 906–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, Chen JF, Fredholm BB (2010). Adenosine signaling and function in glial cells. Cell Death Differ 17: 1071–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonan CD (2012). Ectonucleotidases and nucleotide/nucleoside transporters as pharmacological targets for neurological disorders. CNS Neurol Disord Drug Targets 11: 739–750. [DOI] [PubMed] [Google Scholar]
- Borea PA, Varani K, Vincenzi F, Baraldi PG, Tabrizi MA, Merighi S, et al. (2015). The A3 adenosine receptor: history and perspectives. Pharmacol Rev 67: 74–102. [DOI] [PubMed] [Google Scholar]
- Bradesi S, Eutamene H, Theodorou V, Fioramonti J, Bueno L (2001). Effect of ovarian hormones on intestinal mast cell reactivity to substance P. Life Sci 68: 1047–1056. [DOI] [PubMed] [Google Scholar]
- Brundege JM, Dunwiddie TV (1998). Metabolic regulation of endogenous adenosine release from single neurons. Neuroreport 9: 3007–3011. [DOI] [PubMed] [Google Scholar]
- By Y, Condo J, Durand‐Gorde JM, Lejeune PJ, Mallet B, Guieu R, et al. (2011). Intracerebroventricular injection of an agonist‐like monoclonal antibody to adenosine A(2 A) receptor has antinociceptive effects in mice. J Neuroimmunol 230: 178–182. [DOI] [PubMed] [Google Scholar]
- Cao H, Zhang YQ (2008). Spinal glial activation contributes to pathological pain states. Neurosci Biobehav Rev 32: 972–983. [DOI] [PubMed] [Google Scholar]
- Chen JF, Eltzschig HK, Fredholm BB (2013). Adenosine receptors as drug targets–what are the challenges? Nat Rev Drug Discov 12: 265–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Janes K, Chen C, Doyle T, Bryant L, Tosh DK, et al. (2012). Controlling murine and rat chronic pain through A3 adenosine receptor activation. FASEB J 26: 1855–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi IY, Lee JC, Ju C, Hwang S, Cho GS, Lee HW, et al. (2011). A3 adenosine receptor agonist reduces brain ischemic injury and inhibits inflammatory cell migration in rats. Am J Pathol 179: 2042–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JS, Berdis AJ (2012). Nucleoside transporters: biological insights and therapeutic applications. Future Med Chem 4: 1461–1478. [DOI] [PubMed] [Google Scholar]
- Coull JA, Boudreau D, Bachand K, Prescott SA, Nault F, Sik A, et al. (2003). Trans‐synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature 424: 938–942. [DOI] [PubMed] [Google Scholar]
- Cross HR, Murphy E, Black RG, Auchampach J, Steenbergen C (2002). Overexpression of A(3) adenosine receptors decreases heart rate, preserves energetics, and protects ischemic hearts. Am J Physiol Heart Circ Physiol 283: H1562–H1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui JG, Sollevi A, Linderoth B, Meyerson BA (1997). Adenosine receptor activation suppresses tactile hypersensitivity and potentiates spinal cord stimulation in mononeuropathic rats. Neurosci Lett 223: 173–176. [DOI] [PubMed] [Google Scholar]
- Cunha RA (2008). Different cellular sources and different roles of adenosine: A1 receptor‐mediated inhibition through astrocytic‐driven volume transmission and synapse‐restricted A2A receptor‐mediated facilitation of plasticity. Neurochem Int 52: 65–72. [DOI] [PubMed] [Google Scholar]
- Deussen A, Stappert M, Schafer S, Kelm M (1999). Quantification of extracellular and intracellular adenosine production: understanding the transmembranous concentration gradient. Circulation 99: 2041–2047. [DOI] [PubMed] [Google Scholar]
- Dias RB, Rombo DM, Ribeiro JA, Henley JM, Sebastiao AM (2013). Adenosine: setting the stage for plasticity. Trends Neurosci 36: 248–257. [DOI] [PubMed] [Google Scholar]
- Dickenson AH, Suzuki R, Reeve AJ (2000). Adenosine as a potential analgesic target in Inflammatory and neuropathic pains. CNS Drugs 13: 77–85. [Google Scholar]
- Doyle T, Chen Z, Muscoli C, Bryant L, Esposito E, Cuzzocrea S, et al. (2012). Targeting the overproduction of peroxynitrite for the prevention and reversal of paclitaxel‐induced neuropathic pain. J Neurosci 32: 6149–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA (2001). The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci 24: 31–55. [DOI] [PubMed] [Google Scholar]
- Eaton MJ, Plunkett JA, Karmally S, Martinez MA, Montanez K (1998). Changes in GAD‐ and GABA‐ immunoreactivity in the spinal dorsal horn after peripheral nerve injury and promotion of recovery by lumbar transplant of immortalized serotonergic precursors. J Chem Neuroanat 16: 57–72. [DOI] [PubMed] [Google Scholar]
- Engler RL (1991). Adenosine. The signal of life? Circulation 84: 951–954. [DOI] [PubMed] [Google Scholar]
- Fedorova IM, Jacobson MA, Basile A, Jacobson KA (2003). Behavioral characterization of mice lacking the A3 adenosine receptor: sensitivity to hypoxic neurodegeneration. Cell Mol Neurobiol 23: 431–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feoktistov I, Biaggioni I (2011). Role of adenosine A(2B) receptors in inflammation. Adv Pharmacol 61: 115–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrini F, De Koninck Y (2013). Microglia Control Neuronal Network Excitability via BDNF Signalling, Neural Plasticity, Article ID 429815, 11 pages. doi:10.1155/2013/429815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerbach D, Lingenhoehl K, Olpe HR, Vassout A, Gentsch C, Chaperon F, et al. (2009). The selective nicotinic acetylcholine receptor alpha7 agonist JN403 is active in animal models of cognition, sensory gating, epilepsy and pain. Neuropharmacology 56: 254–263. [DOI] [PubMed] [Google Scholar]
- Fishman P, Bar‐Yehuda S, Madi L, Cohn I (2002). A3 adenosine receptor as a target for cancer therapy. Anticancer Drugs 13: 437–443. [DOI] [PubMed] [Google Scholar]
- Fishman P, Bar‐Yehuda S, Liang BT, Jacobson KA (2012). Pharmacological and therapeutic effects of A3 adenosine receptor agonists. Drug Discov Today 17: 359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman P, Bar‐Yehuda S, Madi L, Rath‐Wolfson L, Ochaion A, Cohen S, et al. (2006). The PI3K‐NF‐kappaB signal transduction pathway is involved in mediating the anti‐inflammatory effect of IB‐MECA in adjuvant‐induced arthritis. Arthritic Res Ther 8 (R33): 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman P, Bar‐Yehuda S, Synowitz M, Powell JD, Klotz KN, Gessi S, et al. (2009). Adenosine receptors and cancer. Handb Exp Pharmacol 193: 399–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford A, Castonguay A, Cottet M, Little JW, Chen Z, Ligouri A, et al. (2015). Engagement of the GABA to KCC2 signaling pathway contributes to the analgesic effects of A3AR agonists in neuropathic pain. J Neurosci 35: 6057–6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchetti P, Cappellacci L, Vita P, Petrelli R, Lavecchia A, Kachler S, et al. (2009). N6‐Cycloalkyl‐ and N6‐bicycloalkyl‐C5'(C2')‐modified adenosine derivatives as high‐affinity and selective agonists at the human A1 adenosine receptor with antinociceptive effects in mice. J Med Chem 52: 2393–2406. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, IJzerman AP, J KA, K KN, Linden J (2001). International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev 53: 527–552. [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, IJzerman AP, J KA, Linden J, M CE (2011). International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors–an update. Pharmacol Rev 63: 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, Arslan G, Halldner L, Kull B, Schulte G, Wasserman W (2000). Structure and function of adenosine receptors and their genes. Naunyn Schmiedebergs Arch Pharmacol 362: 364–374. [DOI] [PubMed] [Google Scholar]
- Fundytus ME (2001). Glutamate receptors and nociception: implications for the drug treatment of pain. CNS Drugs 15: 29–58. [DOI] [PubMed] [Google Scholar]
- Gallo‐Rodriguez C, Ji XD, Melman N, Siegman BD, Sanders LH, Orlina J, et al. (1994). Structure–activity relationships of N6‐benzyladenosine‐5'‐uronamides as A3‐selective adenosine agonists. J Med Chem 37: 636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan TJ, Habib AS (2007). Adenosine as a non‐opioid analgesic in the perioperative setting. Anesth Analg 105: 487–494. [DOI] [PubMed] [Google Scholar]
- Gao ZG, Jacobson KA (2007). Emerging adenosine receptor agonists. Expert Opin Emerg Drugs 12: 479–492. [DOI] [PubMed] [Google Scholar]
- Gao ZG, Jacobson KA (2011). Emerging adenosine receptor agonists: an update. Expert Opin Emerg Drugs 16: 597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao ZG, Teng B, Wu H, Joshi BV, Griffiths GL, Jacobson KA (2009). Synthesis and pharmacological characterization of [(125)I]MRS1898, a high‐affinity, selective radioligand for the rat A(3) adenosine receptor. Purinergic Signalling 5: 31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao ZG, Verzijl D, Zweemer A, Ye K, Goblyos A, Ijzerman AP, et al. (2011). Functionally biased modulation of A(3) adenosine receptor agonist efficacy and potency by imidazoquinolinamine allosteric enhancers. Biochem Pharmacol 82: 658–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge ZD, Peart JN, Kreckler LM, Wan TC, Jacobson MA, Gross GJ, et al. (2006). Cl‐IB‐MECA [2‐chloro‐N6‐(3‐iodobenzyl)adenosine‐5'‐N‐methylcarboxamide] reduces ischemia/reperfusion injury in mice by activating the A3 adenosine receptor. J Pharmacol Exp Ther 319: 1200–1210. [DOI] [PubMed] [Google Scholar]
- Gessi S, Merighi S, Varani K, Borea PA (2011). Adenosine receptors in health and disease. Adv Pharmacol 61: 41–75. [DOI] [PubMed] [Google Scholar]
- Giannaccini G, Betti L, Palego L, Fabbrini L, Schmid L, Castagna M, et al. (2008). Species comparison of adenosine receptor subtypes in brain and testis. Neurochem Res 33: 852–860. [DOI] [PubMed] [Google Scholar]
- Goldberg DS, McGee SJ (2011). Pain as a global public health priority. BMC Public Health 11: 770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong QJ, Li YY, Xin WJ, Wei XH, Cui Y, Wang J, et al. (2010). Differential effects of adenosine A1 receptor on pain‐related behavior in normal and nerve‐injured rats. Brain Res 1361: 23–30. [DOI] [PubMed] [Google Scholar]
- Grandoch M, Hoffmann J, Rock K, Wenzel F, Oberhuber A, Schelzig H, et al. (2013). Novel effects of adenosine receptors on pericellular hyaluronan matrix: implications for human smooth muscle cell phenotype and interactions with monocytes during atherosclerosis. Basic Res Cardiol 108: 340. [DOI] [PubMed] [Google Scholar]
- Gwak YS, Kang J, Unabia GC, Hulsebosch CE (2012). Spatial and temporal activation of spinal glial cells: role of gliopathy in central neuropathic pain following spinal cord injury in rats. Exp Neurol 234: 362–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib AS, Minkowitz H, Osborn T, Ogunnaike B, Candiotti K, Viscusi E, et al. (2008). Phase 2, double‐blind, placebo‐controlled, dose–response trial of intravenous adenosine for perioperative analgesia. Anesthesiology 109: 1085–1091. [DOI] [PubMed] [Google Scholar]
- Haeusler D, Grassinger L, Fuchshuber F, Horleinsberger WJ, Hoftberger R, Leisser I, et al. (2015). Hide and seek: a comparative autoradiographic in vitro investigation of the adenosine A3 receptor. Eur J Nucl Med Mol Imag 42: 928–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson E, Ronnback L (2004). Altered neuronal‐glial signaling in glutamatergic transmission as a unifying mechanism in chronic pain and mental fatigue. Neurochem Res 29: 989–996. [DOI] [PubMed] [Google Scholar]
- Harrison GJ, Cerniway RJ, Peart J, Berr SS, Ashton K, Regan S, et al. (2002). Effects of A(3) adenosine receptor activation and gene knock‐out in ischemic‐reperfused mouse heart. Cardiovasc Res 53: 147–155. [DOI] [PubMed] [Google Scholar]
- Hasko G, Szabo C, Nemeth ZH, Kvetan V, Pastores SM, Vizi ES (1996). Adenosine receptor agonists differentially regulate IL‐10, TNF‐alpha, and nitric oxide production in RAW 264.7 macrophages and in endotoxemic mice. J Immunol 157: 4634–4640. [PubMed] [Google Scholar]
- Hayashida M, Fukuda K, Fukunaga A (2005). Clinical application of adenosine and ATP for pain control. J Anesth 19: 225–235. [DOI] [PubMed] [Google Scholar]
- Headrick JP, Peart J (2005). A3 adenosine receptor‐mediated protection of the ischemic heart. Vascul Pharmacol 42: 271–279. [DOI] [PubMed] [Google Scholar]
- Hinze AV, Mayer P, Harst A, von Kugelgen I (2012). Adenosine A(3) receptor‐induced proliferation of primary human coronary smooth muscle cells involving the induction of early growth response genes. J Mol Cell Cardiol 53: 639–645. [DOI] [PubMed] [Google Scholar]
- Hu X, Liou AK, Leak RK, Xu M, An C, Suenaga J, et al. (2014). Neurobiology of microglial action in CNS injuries: receptor‐mediated signaling mechanisms and functional roles. Prog Neurobiol 119‐120: 60–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA (1998). Adenosine A3 receptors: novel ligands and paradoxical effects. Trends Pharmacol Sci 19: 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Gao ZG (2006). Adenosine receptors as therapeutic targets. Nat Rev Drug Discov 5: 247–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Nikodijevic O, Shi D, Gallo‐Rodriguez C, Olah ME, Stiles GL, et al. (1993). A role for central A3‐adenosine receptors. Mediation of behavioral depressant effects. FEBS Lett 336: 57–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Nikodijevic O, Ji XD, Berkich DA, Eveleth D, Dean RL, et al. (1992). Synthesis and biological activity of N6‐(p‐sulfophenyl)alkyl and N6‐sulfoalkyl derivatives of adenosine: water‐soluble and peripherally selective adenosine agonists. J Med Chem 35: 4143–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jajoo S, Mukherjea D, Watabe K, Ramkumar V (2009). Adenosine A(3) receptor suppresses prostate cancer metastasis by inhibiting NADPH oxidase activity. Neoplasia 11: 1132–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes K, Esposito E, Doyle T, Cuzzocrea S, Tosh DK, Jacobson KA, et al. (2014a). A3 adenosine receptor agonist prevents the development of paclitaxel‐induced neuropathic pain by modulating spinal glial‐restricted redox‐dependent signaling pathways. Pain 155: 2560–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes K, Wahlman C, Little JW, Doyle T, Tosh DK, Jacobson KA, et al. (2015). Spinal neuroimmmune activation is independent of T‐cell infiltration and attenuated by A3 adenosine receptor agonists in a model of oxaliplatin‐induced peripheral neuropathy. Brain Behav Immun 44: 91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes K, Doyle T, Bryant L, Esposito E, Cuzzocrea S, Ryerse J, et al. (2013). Bioenergetic deficits in peripheral nerve sensory axons during chemotherapy‐induced neuropathic pain resulting from peroxynitrite‐mediated post‐translational nitration of mitochondrial superoxide dismutase. Pain 154: 2432–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes K, Little JW, Li C, Bryant L, Chen C, Chen Z, et al. (2014b). The development and maintenance of paclitaxel‐induced neuropathic pain require activation of the sphingosine 1‐phosphate receptor subtype 1. J Biol Chem 289: 21082–21097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JB, Silva C, Gonzalez G, Holden J, Warren KG, Metz LM, et al. (2001). Diminished adenosine A1 receptor expression on macrophages in brain and blood of patients with multiple sclerosis. Ann Neurol 49: 650–658. [PubMed] [Google Scholar]
- Katz NK, Ryals JM, Wright DE (2015). Central or peripheral delivery of an adenosine A1 receptor agonist improves mechanical allodynia in a mouse model of painful diabetic neuropathy. Neuroscience 285: 312–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keil GJ II, DeLander GE (1992). Spinally‐mediated antinociception is induced in mice by an adenosine kinase‐, but not by an adenosine deaminase‐, inhibitor. Life Sci 51: PL171–PL176. [DOI] [PubMed] [Google Scholar]
- Kiesman WF, Elzein E, Zablocki J (2009). A1 adenosine receptor antagonists, agonists, and allosteric enhancers. Handbook Exp Pharmacol 193: 25–58. [DOI] [PubMed] [Google Scholar]
- Klaasse EC, Ijzerman AP, de Grip WJ, Beukers MW (2008). Internalization and desensitization of adenosine receptors. Purinergic signalling 4: 21–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowaluk EA, Mikusa J, Wismer CT, Zhu CZ, Schweitzer E, Lynch JJ, et al. (2000). ABT‐702 (4‐amino‐5‐(3‐bromophenyl)‐7‐(6‐morpholino‐pyridin‐ 3‐yl)pyrido[2,3‐d]pyrimidine), a novel orally effective adenosine kinase inhibitor with analgesic and anti‐inflammatory properties. II In vivo characterization in the rat. J Pharmacol Exp Therapeut 295: 1165–1174. [PubMed] [Google Scholar]
- Latini S, Pedata F (2001). Adenosine in the central nervous system: release mechanisms and extracellular concentrations. J Neurochem 79: 463–484. [DOI] [PubMed] [Google Scholar]
- Latremoliere A, Woolf CJ (2009). Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain 10: 895–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledent C, Vaugeois JM, Schiffmann SN, Pedrazzini T, El Yacoubi M, Vanderhaeghen JJ, et al. (1997). Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2A receptor. Nature 388: 674–678. [DOI] [PubMed] [Google Scholar]
- Lee HC, Fellenz‐Maloney MP, Liscovitch M, Blusztajn JK (1993). Phospholipase D‐catalyzed hydrolysis of phosphatidylcholine provides the choline precursor for acetylcholine synthesis in a human neuronal cell line. Proc Natl Acad Sci U S A 90: 10086–10090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Bokoch G, Liang BT (2001). A novel cardioprotective role of RhoA: new signaling mechanism for adenosine. FASEB Journal 15: 1886–1894. [DOI] [PubMed] [Google Scholar]
- Li AH, Moro S, Melman N, Ji XD, Jacobson KA (1998). Structure–activity relationships and molecular modeling of 3, 5‐diacyl‐2,4‐dialkylpyridine derivatives as selective A3 adenosine receptor antagonists. J Med Chem 41: 3186–3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhang H, Kosturakis AK, Jawad AB, Dougherty PM (2014). Toll‐like receptor 4 signaling contributes to paclitaxel‐induced peripheral neuropathy. J Pain 15: 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang BT, Urso M, Zambraski E, Jacobson KA (2010). Adenosine A3 receptors in muscle protection In: Adenosine Receptors from Cell Biology to Pharmacology and Therapeutics. Borea PA, Springer Netherlands: Dordrecht, Netherlands, pp. 257–280. [Google Scholar]
- Little JW, Ford A, Symons‐Liguori AM, Chen Z, Janes K, Doyle T, et al. (2015). Endogenous adenosine A3 receptor activation selectively alleviates persistent pain states. Brain 138: 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes LV, Rebola N, Pinheiro PC, Richardson PJ, Oliveira CR, Cunha RA (2003). Adenosine A3 receptors are located in neurons of the rat hippocampus. Neuroreport 14: 1645–1648. [DOI] [PubMed] [Google Scholar]
- Loram LC, Harrison JA, Sloane EM, Hutchinson MR, Sholar P, Taylor FR, et al. (2009). Enduring reversal of neuropathic pain by a single intrathecal injection of adenosine 2 A receptor agonists: a novel therapy for neuropathic pain. J Neurosci 29: 14015–14025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macek TA, Schaffhauser H, Conn PJ (1998). Protein kinase C and A3 adenosine receptor activation inhibit presynaptic metabotropic glutamate receptor (mGluR) function and uncouple mGluRs from GTP‐binding proteins. J Neurosci 18: 6138–6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madi L, Bar‐Yehuda S, Barer F, Ardon E, Ochaion A, Fishman P (2003). A3 adenosine receptor activation in melanoma cells: association between receptor fate and tumor growth inhibition. J Biol Chem 278: 42121–42130. [DOI] [PubMed] [Google Scholar]
- Madi L, Cohen S, Ochayin A, Bar‐Yehuda S, Barer F, Fishman P (2007). Overexpression of A3 adenosine receptor in peripheral blood mononuclear cells in rheumatoid arthritis: involvement of nuclear factor‐kappaB in mediating receptor level. J Rheumatol 34: 20–26. [PubMed] [Google Scholar]
- Mao J, Sung B, Ji RR, Lim G (2002). Chronic morphine induces downregulation of spinal glutamate transporters: implications in morphine tolerance and abnormal pain sensitivity. J Neurosci 22: 8312–8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaraughty S, Cowart M, Jarvis MF, Berman RF (2005). Anticonvulsant and antinociceptive actions of novel adenosine kinase inhibitors. Curr Top Med Chem 5: 43–58. [DOI] [PubMed] [Google Scholar]
- Melman A, Gao ZG, Kumar D, Wan TC, Gizewski E, Auchampach JA, et al. (2008). Design of (N)‐methanocarba adenosine 5'‐uronamides as species‐independent A3 receptor‐selective agonists. Bioorg Med Chem Lett 18: 2813–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Watkins LR (2009). Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci 10: 23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KA, Kohno T, Karchewski LA, Scholz J, Baba H, Woolf CJ (2002). Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. J Neurosci 22: 6724–6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morello S, Ito K, Yamamura S, Lee KY, Jazrawi E, Desouza P, et al. (2006). IL‐1 beta and TNF‐alpha regulation of the adenosine receptor (A2A) expression: differential requirement for NF‐kappa B binding to the proximal promoter. J Immunol 177: 7173–7183. [DOI] [PubMed] [Google Scholar]
- Moser GH, Schrader J, Deussen A (1989). Turnover of adenosine in plasma of human and dog blood. Am J Physiol 256: C799–C806. [DOI] [PubMed] [Google Scholar]
- Muscoli C, Cuzzocrea S, Ndengele MM, Mollace V, Porreca F, Fabrizi F, et al. (2007). Therapeutic manipulation of peroxynitrite attenuates the development of opiate‐induced antinociceptive tolerance in mice. J Clin Invest 117: 3530–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndengele MM, Cuzzocrea S, Esposito E, Mazzon E, Di Paola R, Matuschak GM, et al. (2008). Cyclooxygenases 1 and 2 contribute to peroxynitrite‐mediated inflammatory pain hypersensitivity. FASEB J 22: 3154–3164. [DOI] [PubMed] [Google Scholar]
- Ochaion A, Bar‐Yehuda S, Cohen S, Barer F, Patoka R, Amital H, et al. (2009). The anti‐inflammatory target A(3) adenosine receptor is over‐expressed in rheumatoid arthritis, psoriasis and Crohn's disease . Cell Immunol 258: 115–122. [DOI] [PubMed] [Google Scholar]
- Old EA, Clark AK, Malcangio M (2015). The role of glia in the spinal cord in neuropathic and inflammatory pain. Handb Exp Pharmacol 227: 145–170. [DOI] [PubMed] [Google Scholar]
- Otsuguro KI, Tomonari Y, Otsuka S, Yamaguchi S, Kon Y, Ito S (2015). An adenosine kinase inhibitor, ABT‐702, inhibits spinal nociceptive transmission by adenosine release via equilibrative nucleoside transporters in rat. Neuropharmacology 97: 160–170. [DOI] [PubMed] [Google Scholar]
- Paoletta S, Tosh DK, Finley A, Gizewski ET, Moss SM, Gao ZG, et al. (2013). Rational design of sulfonated A3 adenosine receptor‐selective nucleosides as pharmacological tools to study chronic neuropathic pain. J Med Chem 56: 5949–5963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. (2014). The IUPHAR/BPS guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucleic Acids Res 42: D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng L, Huang R, Yu AC, Fung KY, Rathbone MP, Hertz L (2005). Nucleoside transporter expression and function in cultured mouse astrocytes. Glia 52: 25–35. [DOI] [PubMed] [Google Scholar]
- Pizzo PA, Clark NM (2012). Alleviating suffering 101–pain relief in the United States . N Engl J Med 366: 197–199. [DOI] [PubMed] [Google Scholar]
- Poderoso JJ, Carreras MC, Lisdero C, Riobo N, Schopfer F, Boveris A (1996). Nitric oxide inhibits electron transfer and increases superoxide radical production in rat heart mitochondria and submitochondrial particles. Arch Biochem Biophys 328: 85–92. [DOI] [PubMed] [Google Scholar]
- Poon A, Sawynok J (1998). Antinociception by adenosine analogs and inhibitors of adenosine metabolism in an inflammatory thermal hyperalgesia model in the rat. Pain 74: 235–245. [DOI] [PubMed] [Google Scholar]
- Poulsen SA, Quinn RJ (1998). Adenosine receptors: new opportunities for future drugs. Bioorg Med Chem 6: 619–641. [DOI] [PubMed] [Google Scholar]
- Price TJ, Cervero F, de Koninck Y (2005). Role of cation‐chloride‐cotransporters (CCC) in pain and hyperalgesia. Curr Top Med Chem 5: 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausaria S, Ghaffari MM, Kamadulski A, Rodgers K, Bryant L, Chen Z, et al. (2011). Retooling manganese(III) porphyrin‐based peroxynitrite decomposition catalysts for selectivity and oral activity: a potential new strategy for treating chronic pain. J Med Chem 54: 8658–8669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebola N, Canas PM, Oliveira CR, Cunha RA (2005). Different synaptic and subsynaptic localization of adenosine A2A receptors in the hippocampus and striatum of the rat. Neuroscience 132: 893–903. [DOI] [PubMed] [Google Scholar]
- Robson SC, Sevigny J, Zimmermann H (2006). The E‐NTPDase family of ectonucleotidases: Structure function relationships and pathophysiological significance. Purinergic Signalling 2: 409–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagnoli R, Baraldi PG, Tabrizi MA, Gessi S, Borea PA, Merighi S (2010). Allosteric enhancers of A1 adenosine receptors: state of the art and new horizons for drug development. Curr Med Chem 17: 3488–3502. [DOI] [PubMed] [Google Scholar]
- Ru F, Surdenikova L, Brozmanova M, Kollarik M (2011). Adenosine‐induced activation of esophageal nociceptors. Am J Physiol Gastrointest Liver Physiol 300: G485–G493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajjadi FG, Takabayashi K, Foster AC, Domingo RC, Firestein GS (1996). Inhibition of TNF‐alpha expression by adenosine: role of A3 adenosine receptors. J Immunol 156: 3435–3442. [PubMed] [Google Scholar]
- Salvatore CA, Jacobson MA, Taylor HE, Linden J, Johnson RG (1993). Molecular cloning and characterization of the human A3 adenosine receptor. Proc Natl Acad Sci U S A 90: 10365–10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvatore CA, Tilley SL, Latour AM, Fletcher DS, Koller BH, Jacobson MA (2000). Disruption of the A(3) adenosine receptor gene in mice and its effect on stimulated inflammatory cells. J Biol Chem 275: 4429–4434. [DOI] [PubMed] [Google Scholar]
- Salvemini D, Neumann W (2010). Targeting peroxynitrite driven nitroxidative stress with synzymes: a novel therapeutic approach in chronic pain management. Life Sci 86: 604–614. [DOI] [PubMed] [Google Scholar]
- Sawynok J (1998). Adenosine receptor activation and nociception. Eur J Pharmacol 347: 1–11. [DOI] [PubMed] [Google Scholar]
- Sawynok J (2013). Adenosine and Pain In: Adenosine: A Key Link between Metabolism and Brain Activity. Masino S. and Boison D., Springer‐Verlag New York: New York, pp. 343–360. [Google Scholar]
- Sawynok J, Reid A, Liu XJ (1999). Acute paw oedema induced by local injection of adenosine A(1), A(2) and A(3) receptor agonists. Eur J Pharmacol 386: 253–261. [DOI] [PubMed] [Google Scholar]
- Sawynok J, Zarrindast MR, Reid AR, Doak GJ (1997). Adenosine A3 receptor activation produces nociceptive behaviour and edema by release of histamine and 5‐hydroxytryptamine. Eur J Pharmacol 333: 1–7. [DOI] [PubMed] [Google Scholar]
- Sebastian‐Serrano A, de Diego‐Garcia L, Martinez‐Frailes C, Avila J, Zimmermann H, Millan JL, et al. (2015). Tissue‐nonspecific alkaline phosphatase regulates purinergic transmission in the central nervous system during development and disease. Comput Struct Biotechnol J 13: 95–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastiao AM, Ribeiro JA (1996). Adenosine A2 receptor‐mediated excitatory actions on the nervous system. Prog Neurobiol 48: 167–189. [DOI] [PubMed] [Google Scholar]
- Shneyvays V, Nawrath H, Jacobson KA, Shainberg A (1998). Induction of apoptosis in cardiac myocytes by an A3 adenosine receptor agonist. Exp Cell Res 243: 383–397. [DOI] [PubMed] [Google Scholar]
- Shneyvays V, Mamedova L, Zinman T, Jacobson K, Shainberg A (2001). Activation of A(3)adenosine receptor protects against doxorubicin‐induced cardiotoxicity. J Mol Cell Cardiol 33: 1249–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjolund KF, von Heijne M, Hao JX, Xu XJ, Sollevi A, Wiesenfeld‐Hallin Z (1998). Intrathecal administration of the adenosine A1 receptor agonist R‐phenylisopropyl adenosine reduces presumed pain behaviour in a rat model of central pain. Neurosci Lett 243: 89–92. [DOI] [PubMed] [Google Scholar]
- Smith PA (2014). BDNF: No gain without pain? Neuroscience 283C: 107–123. [DOI] [PubMed] [Google Scholar]
- Sowa NA, Street SE, Vihko P, Zylka MJ (2010). Prostatic acid phosphatase reduces thermal sensitivity and chronic pain sensitization by depleting phosphatidylinositol 4,5‐bisphosphate. J Neurosci 30: 10282–10293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spychala J, Datta NS, Takabayashi K, Datta M, Fox IH, Gribbin T, et al. (1996). Cloning of human adenosine kinase cDNA: sequence similarity to microbial ribokinases and fructokinases. Proc Natl Acad Sci U S A 93: 1232–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiller CO, Cui JG, O'Connor WT, Brodin E, Meyerson BA, Linderoth B (1996). Release of gamma‐aminobutyric acid in the dorsal horn and suppression of tactile allodynia by spinal cord stimulation in mononeuropathic rats. Neurosurgery 39: 367–374 discussion 374‐365. [DOI] [PubMed] [Google Scholar]
- Studer FE, Fedele DE, Marowsky A, Schwerdel C, Wernli K, Vogt K, et al. (2006). Shift of adenosine kinase expression from neurons to astrocytes during postnatal development suggests dual functionality of the enzyme. Neuroscience 142: 125–137. [DOI] [PubMed] [Google Scholar]
- Svenningsson P, Hall H, Sedvall G, Fredholm BB (1997). Distribution of adenosine receptors in the postmortem human brain: an extended autoradiographic study. Synapse 27: 322–335. [DOI] [PubMed] [Google Scholar]
- Swedish Orphan Biovitrum (2014). Efficacy and Tolerability of Novel A2A Agonist in Treatment of Diabetic Neuropathic Pain. NCT00452777. [Online] Available from ClinicalTrials.gov. [Google Scholar]
- Szabo C, Scott GS, Virag L, Egnaczyk G, Salzman AL, Shanley TP, et al. (1998). Suppression of macrophage inflammatory protein (MIP)‐1alpha production and collagen‐induced arthritis by adenosine receptor agonists. Br J Pharmacol 125: 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taiwo YO, Levine JD (1990). Direct cutaneous hyperalgesia induced by adenosine. Neuroscience 38: 757–762. [DOI] [PubMed] [Google Scholar]
- Thourani VH, Ronson RS, Jordan JE, Guyton RA, Vinten‐Johansen J (1999a). Adenosine A3 pretreatment before cardioplegic arrest attenuates postischemic cardiac dysfunction. Ann Thorac Surg 67: 1732–1737. [DOI] [PubMed] [Google Scholar]
- Thourani VH, Nakamura M, Ronson RS, Jordan JE, Zhao ZQ, Levy JH, et al. (1999b). Adenosine A(3)‐receptor stimulation attenuates postischemic dysfunction through K(ATP) channels. Am J Physiol 277: H228–H235. [DOI] [PubMed] [Google Scholar]
- Tosh DK, Paoletta S, Chen Z, Crane S, Lloyd J, Gao ZG (2015). Structure‐based design, synthesis by click chemistry and in vivo activity of highly selective A3 adenosine receptor agonists. [DOI] [PMC free article] [PubMed]
- Tosh DK, Deflorian F, Phan K, Gao ZG, Wan TC, Gizewski E, et al. (2012). Structure‐guided design of A(3) adenosine receptor‐selective nucleosides: combination of 2‐arylethynyl and bicyclo[3.1.0]hexane substitutions. J Med Chem 55: 4847–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosh DK, Finley A, Paoletta S, Moss SM, Gao ZG, Gizewski ET, et al. (2014). In vivo phenotypic screening for treating chronic neuropathic pain: modification of c2‐arylethynyl group of conformationally constrained A3 adenosine receptor agonists. J Med Chem 57: 9901–9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey WR, Magee W, Masamune H, Kennedy SP, Knight DR, Buchholz RA, et al. (1997). Selective adenosine A3 receptor stimulation reduces ischemic myocardial injury in the rabbit heart. Cardiovasc Res 33: 410–415. [DOI] [PubMed] [Google Scholar]
- Varani K, Padovan M, Vincenzi F, Targa M, Trotta F, Govoni M, et al. (2011). A2A and A3 adenosine receptor expression in rheumatoid arthritis: upregulation, inverse correlation with disease activity score and suppression of inflammatory cytokine and metalloproteinase release. Arthritis Res Ther 13(R197): 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varani K, Vincenzi F, Targa M, Paradiso B, Parrilli A, Fini M, et al. (2013). The stimulation of A(3) adenosine receptors reduces bone‐residing breast cancer in a rat preclinical model. Eur J Cancer 49: 482–491. [DOI] [PubMed] [Google Scholar]
- Varani K, Vincenzi F, Tosi A, Targa M, Masieri FF, Ongaro A, et al. (2010). Expression and functional role of adenosine receptors in regulating inflammatory responses in human synoviocytes. Br J Pharmacol 160: 101–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincenzi F, Targa M, Romagnoli R, Merighi S, Gessi S, Baraldi PG, et al. (2014). TRR469, a potent A(1) adenosine receptor allosteric modulator, exhibits anti‐nociceptive properties in acute and neuropathic pain models in mice. Neuropharmacology 81: 6–14. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Rice KC, Maier SF (2009). The “toll” of opioid‐induced glial activation: improving the clinical efficacy of opioids by targeting glia. Trends Pharmacol Sci 30: 581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittendorp MC, Boddeke HW, Biber K (2004). Adenosine A3 receptor‐induced CCL2 synthesis in cultured mouse astrocytes. Glia 46: 410–418. [DOI] [PubMed] [Google Scholar]
- Wu WP, Hao JX, Halldner‐Henriksson L, Xu XJ, Jacobson MA, Wiesenfeld‐Hallin Z, et al. (2002). Decreased inflammatory pain due to reduced carrageenan‐induced inflammation in mice lacking adenosine A3 receptors. Neuroscience 114: 523–527. [DOI] [PubMed] [Google Scholar]
- Wu WP, Hao JX, Halldner L, Lovdahl C, DeLander GE, Wiesenfeld‐Hallin Z, et al. (2005). Increased nociceptive response in mice lacking the adenosine A1 receptor. Pain 113: 395–404. [DOI] [PubMed] [Google Scholar]
- Yamaoka G, Horiuchi H, Morino T, Miura H, Ogata T (2013). Different analgesic effects of adenosine between postoperative and neuropathic pain. J Orthop Sci 18: 130–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo JF, Ling SF, Tang N, Ong WY (2008). Antinociceptive effect of CNS peroxynitrite scavenger in a mouse model of orofacial pain. Exp Brain Res 184: 435–438. [DOI] [PubMed] [Google Scholar]
- Yoon MH, Bae HB, Choi JI (2005). Antinociception of intrathecal adenosine receptor subtype agonists in rat formalin test. Anesth Analg 101: 1417–1421. [DOI] [PubMed] [Google Scholar]
- Yoon MH, Choi JI, Park HC, Bae HB (2004). Interaction between intrathecal gabapentin and adenosine in the formalin test of rats. J Korean Med Sci 19: 581–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon MH, Bae HB, Choi JI, Kim SJ, Chung ST, Kim CM (2006). Roles of adenosine receptor subtypes in the antinociceptive effect of intrathecal adenosine in a rat formalin test. Pharmacology 78: 21–26. [DOI] [PubMed] [Google Scholar]
- Zahn PK, Straub H, Wenk M, Pogatzki‐Zahn EM (2007). Adenosine A1 but not A2A receptor agonist reduces hyperalgesia caused by a surgical incision in rats: a pertussis toxin‐sensitive G protein‐dependent process. Anesthesiology 107: 797–806. [DOI] [PubMed] [Google Scholar]
- Zeilhofer HU, Wildner H, Yevenes GE (2012). Fast synaptic inhibition in spinal sensory processing and pain control. Physiol Rev 92: 193–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Franklin PH, Murray TF (1993). Manipulation of endogenous adenosine in the rat prepiriform cortex modulates seizure susceptibility. J Pharmacol Exp Ther 264: 1415–1424. [PubMed] [Google Scholar]
- Zhang M, Hu H, Zhang X, Lu W, Lim J, Eysteinsson T, et al. (2010). The A3 adenosine receptor attenuates the calcium rise triggered by NMDA receptors in retinal ganglion cells. Neurochem Int 56: 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhang M, Laties AM, Mitchell CH (2006). Balance of purines may determine life or death of retinal ganglion cells as A3 adenosine receptors prevent loss following P2X7 receptor stimulation. J Neurochem 98: 566–575. [DOI] [PubMed] [Google Scholar]
- Zimmermann H (2000). Extracellular metabolism of ATP and other nucleotides. Naunyn Schmiedebergs Arch Pharmacol 362: 299–309. [DOI] [PubMed] [Google Scholar]
- Zylka MJ (2011). Pain‐relieving prospects for adenosine receptors and ectonucleotidases. Trends Mol Med 17: 188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]