

Abstract
Background
Acute exacerbations of idiopathic pulmonary fibrosis are major causes of morbidity and mortality among patients with idiopathic pulmonary fibrosis. However, acute exacerbations remain unpredictable. The aim of this study was to investigate risk factors for acute exacerbations of idiopathic pulmonary fibrosis.
Methods
We performed a retrospective cohort study of patients with idiopathic pulmonary fibrosis who visited our institutions from January 1999 to September 2014. We investigated risk factors for acute exacerbations in patients with idiopathic pulmonary fibrosis diagnosed retrospectively based on the official 2011 idiopathic pulmonary fibrosis ATS/ERS/JRS/ALAT Update Statement.
Results
The idiopathic pulmonary fibrosis study cohort included 65 subjects. The median follow-up period was 2.6 years. During follow-up, 24 patients (36.9 %) experienced acute exacerbations. A Kaplan-Meier curve demonstrated that the 1-year, 2-year, and 3-year incidences of acute exacerbation were 9.6, 19.2 and 31.0 %, respectively. Acute exacerbation exerted a significant impact on overall survival among those with the disease. A log-rank test showed that baseline cardiovascular diseases, higher GAP (gender, age, physiology) stage (≥II), higher serum lactate dehydrogenase level (≥180 U/L), higher serum surfactant protein-D level (≥194.7 ng/mL), higher neutrophil (≥1.77 %) and eosinophil (≥3.21 %) percentages in bronchoalveolar lavage fluid samples, and treatment with an immunosuppressive agent after diagnosis were associated with poor acute exacerbation-free probability. In the Cox analysis adjusted for treatment with an immunosuppressive agent, baseline cardiovascular diseases, higher GAP stage (≥II), and higher eosinophil percentage (≥3.21 %) in bronchoalveolar lavage fluid samples were predictors of an acute exacerbation of idiopathic pulmonary fibrosis.
Conclusions
This study demonstrated that baseline cardiovascular diseases, higher GAP stage (≥II), and higher eosinophil percentage (≥3.21 %) in bronchoalveolar lavage fluid samples were associated with the onset of an acute exacerbation of idiopathic pulmonary fibrosis.
Keywords: Bronchoalveolar lavage; Krebs von den Lungen-6; Surfactant protein-D; Lactate dehydrogenase; GAP (gender, age, Physiology) stage; Cardiovascular disease
Background
Idiopathic pulmonary fibrosis (IPF) is the most common form of idiopathic interstitial pneumonia [1, 2], and the median survival from the time of diagnosis is approximately 3 years [2]. The clinical course of individual patients with IPF is highly variable and unpredictable. IPF is not a uniform, clinically dynamic disease; there is a wide spectrum of disease courses, including stability or slow progression over a period of years, rapid deterioration, and even periods of relative stability punctuated by events causing rapid decline [3–5]. This acute worsening of the disease is sometimes attributed to identifiable conditions such as pneumonia and heart failure. However, many of these events occur without an identifiable cause, and are called acute exacerbations (AEs) of IPF [4]. AE-IPF is a major cause of morbidity and mortality in patients with IPF [3, 6–8]. In 553 Japanese patients with IPF, the most common cause of death was AE with a frequency of 40 % [8], which was similar to the 46 % value found in a previous study by Jeon and coworkers in Korea [6]. There is no proven treatment for AE-IPF. Patients with AEs have poor outcomes, with mortality exceeding 60 % during hospital admission; among those who survive, there is >90 % mortality within 6 months after discharge [3, 4]. Therefore, elucidating the pathogenesis of AE-IPF and predicting AE are important and challenging issues.
Kondoh et al. reported that a higher body mass index, higher modified Medical Research Council scale score, and decline in forced vital capacity (FVC) at 6 months after the diagnosis of IPF were independent risk factors for AE-IPF [7]. In one study, a comparison of clinical features between IPF patients with and without an AE-IPF showed no difference in age, smoking status, oxygenation, or lung function at the time of diagnosis [9]. Risk factors for AE-IPF reported by Song et al. [3] include a lower FVC at baseline as well as never having smoked. Pulmonary hypertension at baseline was also reported to be associated with a significant risk of a subsequent AE-IPF [10]. More recently, Kondoh et al. reported that a rapid decline in vital capacity percentage (≥10 % within 6 months after the diagnosis of IPF) and high baseline alveolar-arterial difference in oxygen (A-a DO2) value might be risk factors for an AE-IPF over the 52 weeks of the study [11]. However, studies regarding risk factors for AE-IPF are still limited, and AEs remain unpredictable. In addition, serum biomarkers and cellular analyses of bronchoalveolar lavage (BAL) fluid are still of unclear predictive value for AE-IPF. Furthermore, in most previous studies investigating risk factors for AE-IPF, the diagnosis of IPF was made based on diagnostic criteria developed before the official IPF ATS/ERS/JRS/ALAT Update Statement in 2011 [2].
The aim of this study was to investigate risk factors for AE-IPF, focusing on the significance of serum markers and cellular analyses of BAL fluid in patients with IPF who were diagnosed based on the official 2011 IPF ATS/ERS/JRS/ALAT Update Statement [2].
Methods
Study population
We performed a retrospective cohort study of patients with IPF who visited Nagasaki University Hospital, Nagasaki, Japan, or University Hospital of Occupational and Environmental Health, Kitakyushu, Japan, from January 1999 to September 2014. Diagnoses were made retrospectively according to the official ATS/ERS/JRS/ALAT statement [2]. Patients with underlying connective tissue disease, occupational or environmental exposure, or a histopathologic pattern on surgical lung biopsy other than usual interstitial pneumonia (UIP) were excluded from the study. At our institution, patients with suspected IPF routinely underwent bronchoscopy with BAL at the time of initial diagnosis, unless the patient’s status contraindicated to the need for bronchoscopy. Patients whose initial manifestation was an AE, patients without initial BAL fluid evaluations, and patients lost to follow-up within 90 days were excluded. Twelve of 65 (18.5 %) IPF diagnoses were confirmed pathologically in multiple lobes by open lung biopsy or video-assisted thoracoscopic surgery. AE-IPF was defined using the following previously reported criteria with a slight modification: (1) a previous diagnosis of IPF; (2) unexplained worsening of dyspnea within the past 30 days; (3) high-resolution computed tomography (HRCT) with new findings of bilateral ground-glass opacity or consolidation; and (4) the absence of an alternative explanation such as infection, pulmonary embolism, pneumothorax, or heart failure [2, 4]. The study protocol was approved by the Institutional Review Boards of Nagasaki University Hospital and University Hospital of Occupational and Environmental Health. The patients’ approval or informed consent was not required for a retrospective review of their records, pursuant to the ethical guidelines of the Japanese Ministry of Health, Labor, and Welfare.
Covariates
Data regarding patient characteristics were collected from clinical notes recorded at the time of the initial diagnosis of IPF and included age, sex, smoking history, partial pressure of oxygen in arterial blood (PaO2)/fraction of inspired oxygen (FiO2) ratio (P/F ratio), A-a DO2, hematologic data, pulmonary function tests, and BAL fluid cellular constituents. Data regarding serum concentrations of C-reactive protein (CRP), lactate dehydrogenase (LDH), Krebs von den Lungen-6 (KL-6), and surfactant protein (SP)-D were also collected from clinical notes recorded at the time of diagnosis. Gender-Age-Physiology (GAP) stage and composite physiologic index (CPI) were calculated as described previously [12, 13].
BAL and cell preparation
Bronchoscopy and BAL were performed as described previously [14].
Clinical and outcome assessments
In the AE analysis, subjects were censored if they (1) had not experienced an AE by September 30, 2014, or (2) were lost to follow-up. In the survival analysis, subjects were censored if they (1) were still alive on September 30, 2014, or (2) lost to follow-up. AE-free probability and survival probability were calculated from the time of the initial IPF diagnosis to censoring.
Statistical analysis
Baseline characteristics including demographics, baseline pulmonary function, respiratory parameters, hematologic data, serum markers, and BAL fluid findings were compared between patients who experienced an AE during the study period and those who did not. Data are described as frequencies for categorical variables and as the median and interquartile range (IQR) for quantitative variables. Associations between variables were assessed with Fisher’s exact test for categorical variables and Wilcoxon’s rank-sum test for quantitative variables. The cumulative AE incidence was estimated with the Kaplan-Meier method. The association between survival probability and AE was evaluated by the log-rank test. A log-rank test was used to evaluate associations among smoking status, cardiovascular diseases, GAP stage, biomarkers (LDH, KL-6, and SP-D), BAL fluid cellular profiles (percentages of lymphocytes, neutrophils, and eosinophils), treatment after diagnosis, and AE-free probability because the test is a simple method that did not require satisfying some assumptions. Cox’s proportional hazards regression models were used to evaluate hazard ratios (HRs) and adjust confounders. It was considered appropriate to determine a cut-off level instead of using continuous data because some factors did not satisfy the assumption that the log hazard increased linearly with the covariate. The optimum cut-off values for predicting AE incidence at the 3-year follow-up were determined by maximizing the Youden index [15] calculated from a time-dependent receiver operating characteristic (ROC) analysis with the R package timeROC [16]. We reported the results at the 3-year follow-up, as this was the interval with the most dramatic relationship between biomarkers, BAL fluid cellular profiles, and AE incidence. We evaluated HRs, 95 % confidence intervals (95 % CIs), and P values for each factor with an unadjusted Cox analysis. Because of the small number of AEs and deaths included in this study, it was considered inappropriate to adjust more than two factors in a multiple Cox analysis to evaluate the relative hazards of AE and death. Accordingly, we adjusted for treatment with an immunosuppressive agent in the multiple Cox analysis, which was found to be a significant risk factor for an AE-IPF in the log-rank test and unadjusted Cox analysis in this study. All tests were two-sided, and a P value less than 0.05 was considered statistically significant. All statistical analyses were performed using SAS version 9.4 (SAS Institute Inc., Cary, NC, USA) and R version 3.1.0.
Results
Subject characteristics
The IPF study cohort included 65 subjects. There were 51 men and 14 women with a median age of 69 years (IQR, 62 to 73 years). There were 13 never smokers, 42 former smokers, and 10 current smokers. The median follow-up period was 2.6 years (IQR, 1.3 to 4.2 years). The median survival time (MST) for all subjects was 4.5 years from the time of the initial visit.
AE incidence
During the follow-up period, 24 patients (36.9 %) experienced AEs. Eighteen patients (27.7 %) were lost to follow-up. The Kaplan-Meier curve demonstrated that the 1-year, 2-year, and 3-year incidences of AE-IPF were 9.6, 19.2 and 31.0 %, respectively (Fig. 1).
Fig. 1.
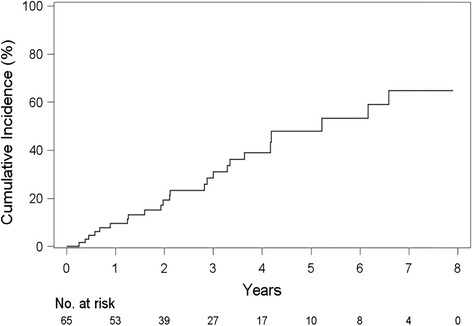
Incidence of acute exacerbation of idiopathic pulmonary fibrosis. The Kaplan-Meier curve demonstrates that the 1-year, 2-year, and 3-year incidences of AE-IPF were 9.6, 19.2 and 31.0 %, respectively
Demographic, physiological, gas exchange, and laboratory data and BAL fluid findings between patients with an AE (AE group) and those without an AE (Non-AE group)
No significant differences were observed in sex or age distributions between the groups. Patients who experienced an AE had a lower total lung capacity than those in the Non-AE group. Gas exchange and laboratory data and BAL fluid findings were similar between the groups (Table 1).
Table 1.
Comparisons of baseline characteristics between patients with and without an acute exacerbation
| Non-AE | AE | P Value a | |||
|---|---|---|---|---|---|
| (N = 41) | (N = 24) | ||||
| Age, median (IQR), y | 69.0 | (62.0 to 73.0) | 68.0 | (62.0 to 73.5) | 0.995 |
| Sex, No. (%) | |||||
| Men | 31 | (75.6) | 20 | (83.3) | 1.000 |
| Women | 10 | (24.4) | 4 | (16.7) | |
| Smoking, No. (%) | |||||
| S | 7 | (17.1) | 3 | (12.5) | 1.000 |
| Ex | 25 | (61.0) | 17 | (70.8) | |
| N | 9 | (22.0) | 4 | (16.7) | |
| Smoking index, pack-years | 35.8 | (1.0 to 52.3) | 31.3 | (15.0 to 50.0) | 0.940 |
| Cardiovascular diseases, No. (%) | |||||
| Yes | 10 | (24.4) | 8 | (33.3) | 1.000 |
| No | 31 | (75.6) | 16 | (66.7) | |
| Atopic diseases, No. (%) | |||||
| Yes | 3 | (7.3) | 1 | (4.2) | 1.000 |
| No | 38 | (92.7) | 23 | (95.8) | |
| Baseline pulmonary function | |||||
| VC, median (IQR), % predicted | 81.0 | (64.1 to 95.0) | 69.2 | (59.5 to 83.0) | 0.074 |
| FVC, median (IQR), % predicted | 80.4 | (59.0 to 93.9) | 70.5 | (59.9 to 82.1) | 0.055 |
| TLC, median (IQR), % predicted | 75.9 | (63.0 to 90.0) | 66.1 | (54.0 to 77.0) | 0.020 |
| DLCO, median (IQR), % predicted | 58.0 | (46.1 to 72.5) | 46.2 | (41.0 to 72.3) | 0.276 |
| KCO, median (IQR), % predicted | 69.5 | (60.8 to 85.0) | 73.0 | (57.7 to 78.0) | 0.866 |
| GAP stage, No. (%) | |||||
| I | 25 | (69.4) | 10 | (45.5) | 1.000 |
| II | 9 | (25.0) | 12 | (54.5) | |
| III | 2 | (5.6) | 0 | (0.0) | |
| CPI | 37.3 | (22.2 to 47.6) | 47.7 | (30.5 to 54.9) | 0.190 |
| Respiratory parameters b | |||||
| PaO2, median (IQR), mm Hg | 80.7 | (75.3 to 91.0) | 81.0 | (75.2 to 94.5) | 0.810 |
| A-a DO2, median (IQR), mm Hg | 17.5 | (3.8 to 26.2) | 17.2 | (4.1 to 25.2) | 0.889 |
| Hematologic data | |||||
| WBC, median (IQR),/mm3 | 6700 | (5800 to 8100) | 7350 | (5750 to 9300) | 0.442 |
| Neutrophils, median (IQR), % | 62.8 | (57.0 to 69.4) | 63.9 | (58.0 to 69.0) | 0.897 |
| Eosinophils, median (IQR), % | 3.0 | (2.0 to 4.7) | 2.9 | (1.8 to 4.4) | 0.545 |
| Serum markers | |||||
| CRP, median (IQR), mg/dL | 0.31 | (0.06 to 0.69) | 0.29 | (0.13 to 0.64) | 0.688 |
| LDH, median (IQR), U/L | 235 | (187.0 to 255.0) | 247 | (223.5 to 296.0) | 0.082 |
| KL-6, median (IQR), U/mL | 912 | (595.0 to 1802.0) | 1258 | (744.5 to 2006.0) | 0.246 |
| SP-D, median (IQR), ng/mL | 203 | (131.0 to 342.0) | 269 | (178.6 to 343.0) | 0.285 |
| BAL fluid findings | |||||
| Total cell count, median (IQR), ×105/mL | 2.50 | (1.40 to 3.67) | 3.21 | (2.22 to 3.97) | 0.099 |
| Macrophages, median (IQR), % | 80.2 | (62.5 to 88.9) | 76.6 | (67.3 to 84.8) | 0.492 |
| Lymphocytes, median (IQR), % | 6.8 | (3.9 to 11.4) | 11.6 | (3.2 to 17.1) | 0.649 |
| Neutrophils, median (IQR), % | 6.7 | (2.9 to 13.3) | 7.0 | (3.2 to 10.5) | 0.807 |
| Eosinophils, median (IQR), % | 3.3 | (1.2 to 5.4) | 4.6 | (2.1 to 7.0) | 0.259 |
| CD4/8 ratio, median (IQR) | 1.90 | (0.86 to 3.70) | 1.10 | (0.60 to 1.90) | 0.065 |
| Treatment after the diagnosis | |||||
| Steroid, No. (%) | |||||
| yes | 7 | (17.1) | 8 | (33.3) | 1.000 |
| No | 34 | (82.9) | 16 | (66.7) | |
| Immunosuppressive agent c, No. (%) | |||||
| Yes | 3 | (7.3) | 6 | (25.0) | 1.000 |
| No | 38 | (92.7) | 18 | (75.0) | |
| Steroid with immunosuppressive agent, No. (%) | |||||
| Yes | 3 | (7.3) | 5 | (20.8) | 1.000 |
| No | 38 | (92.7) | 19 | (79.2) | |
| Pirfenidone, No. (%) | |||||
| Yes | 8 | (19.5) | 6 | (25.0) | 1.000 |
| No | 33 | (80.5) | 18 | (75.0) | |
| NAC, No. (%) | |||||
| Yes | 8 | (19.5) | 5 | (20.8) | 1.000 |
| No | 33 | (80.5) | 19 | (79.2) | |
Abbreviations: AE acute exacerbation, N number of patients, IQR interquartile range, S/Ex/N current smoker/ex-smoker/nonsmoker, VC vital capacity, FVC forced vital capacity, TLC total lung capacity, DLCO diffusing capacity for carbon monoxide, KCO carbon monoxide transfer coefficient, GAP gender-age-physiology, CPI composite physiologic index, P/F ratio PaO2/fraction of inspired oxygen ratio, A-a DO 2 alveolar-arterial difference in oxygen, WBC white blood cell count, CRP C-reactive protein, LDH lactate dehydrogenase, KL-6 Krebs von den Lungen-6, SP-D surfactant protein-D, BAL bronchoalveolar lavage, CD4/8 ratio CD4/CD8 lymphocyte subset ratio, NAC inhaled N-acetylcysteine
Data are presented as the median (interquartile range)
aWilcoxon’s rank-sum test was performed for continuous variables, and Fisher’s exact test was performed for categorical variables
bOne patient in the Non-AE group required oxygen (1 L/min O2 via a nasal cannula) at the time of sample collection. All other results were obtained at room air
cImmunosuppressive agents included cyclosporine (N = 7) and cyclophosphamide (N = 2)
Impact of AE on overall survival
AE exerted a significant impact on overall survival in IPF. There was a significant difference in survival between patients who experienced an AE (MST, 2.8 years) and those who did not (MST, not calculable) (log-rank test, P < 0.001) (Fig. 2).
Fig. 2.
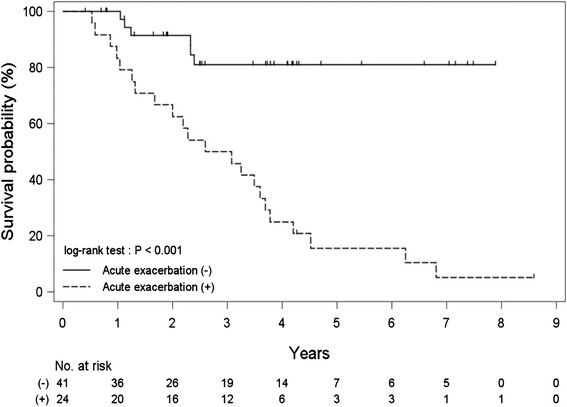
Impact of acute exacerbation on overall survival. There was a significant difference in survival between patients who experienced an acute exacerbation (median survival time, 2.8 years) and those who did not (median survival time, not calculable) (log-rank test, P < 0.001)
Risk factors for AE
According to the ROC curve analysis, the optimum cut-off levels to predict an AE-IPF were 180 U/L for LDH, 946 U/mL for KL-6, 194.7 ng/mL for SP-D; and 10.15 1.77 and 3.21 % for lymphocyte, neutrophil, and eosinophil percentages in BAL fluid samples, respectively. A log-rank test showed that baseline cardiovascular diseases, higher GAP stage (≥II), higher serum LDH level (≥180 U/L), higher serum SP-D level (≥194.7 ng/mL), higher neutrophil (≥1.77 %) and eosinophil (≥3.21 %) percentages in BAL fluid samples, and treatment with an immunosuppressive agent after diagnosis were associated with poor AE-free probability (P = 0.042, 0.002, 0.048, 0.014, 0.048, 0.009, and 0.009, respectively) (Table 2). In the unadjusted Cox analysis, treatment with an immunosuppressive agent, baseline cardiovascular diseases, higher GAP stage (≥II), higher serum SP-D level (≥194.7 ng/mL), and a higher eosinophil percentage in BAL fluid samples (≥3.21 %) were predictors of an AE-IPF (HR [95 % CI], 3.35 [1.27–8.84], 2.49 [1.00–6.19], 3.88 [1.54–9.80], 3.17 [1.21–8.31], and 3.27 [1.29–8.29], respectively). In the Cox analysis adjusted for treatment with an immunosuppressive agent, baseline cardiovascular diseases, higher GAP stage (≥II), and higher eosinophil percentage (≥3.21 %) in BAL fluid samples were predictors of an AE-IPF (HR [95 % CI], 3.21 [1.24–8.32], 3.23 [1.22–8.51], and 2.89 [1.11–7.51], respectively) (Table 3).
Table 2.
Log-rank test for risk factors for an acute exacerbation of idiopathic pulmonary fibrosis
| Non-AE (N = 41) | AE (N = 24) | log-rank P Value | |
|---|---|---|---|
| Smoking | |||
| S or Ex | 32 | 20 | 0.918 |
| N | 9 | 4 | |
| Cardiovascular diseases | |||
| Yes | 10 | 8 | 0.042 |
| No | 31 | 16 | |
| GAP stage | |||
| II or III | 11 | 12 | 0.002 |
| I | 25 | 10 | |
| Serum markers | |||
| LDH | |||
| ≥ 180 | 32 | 22 | 0.048 |
| < 180 | 9 | 2 | |
| KL-6 | |||
| ≥ 946 | 18 | 16 | 0.292 |
| < 946 | 21 | 8 | |
| SP-D | |||
| ≥ 194.7 | 22 | 18 | 0.014 |
| < 194.7 | 17 | 6 | |
| BAL fluid findings | |||
| Lymphocytes | |||
| ≥ 10.15 | 13 | 13 | 0.085 |
| < 10.15 | 28 | 11 | |
| Neutrophils | |||
| ≥ 1.77 | 34 | 22 | 0.048 |
| < 1.77 | 7 | 2 | |
| Eosinophils | |||
| ≥ 3.21 | 21 | 17 | 0.009 |
| < 3.21 | 20 | 7 | |
| Treatment after the diagnosis | |||
| Steroid | |||
| Yes | 7 | 8 | 0.443 |
| No | 34 | 16 | |
| Immunosuppressive agent | |||
| Yes | 3 | 6 | 0.009 |
| No | 38 | 18 | |
| Pirfenidone | |||
| Yes | 8 | 6 | 0.963 |
| No | 33 | 18 | |
| NAC | |||
| Yes | 8 | 5 | 0.547 |
| No | 33 | 19 | |
Abbreviations: AE acute exacerbation, N number of patients, S/Ex/N current smoker/ex-smoker/nonsmoker, GAP gender-age-physiology, LDH lactate dehydrogenase, KL-6 Krebs von den Lungen-6, SP-D surfactant protein-D, BAL bronchoalveolar lavage, NAC inhaled N-acetylcysteine
Table 3.
Cox analysis for risk factors for an acute exacerbation of idiopathic pulmonary fibrosis
| HR | (95 % CI) | P Value | |
|---|---|---|---|
| Treatment after the diagnosis | |||
| Immunosuppressive agent | |||
| Unadjusted | 3.35 | (1.27 to 8.84) | 0.014 |
| Cardiovascular diseases | |||
| Unadjusted | 2.49 | (1.00 to 6.19) | 0.049 |
| Adjusteda | 3.21 | (1.24 to 8.32) | 0.016 |
| GAP stage (≥II) | |||
| Unadjusted | 3.88 | (1.54 to 9.80) | 0.004 |
| Adjusteda | 3.23 | (1.22 to 8.51) | 0.018 |
| Serum markers | |||
| LDH, U/L, Cut off point = 180 | |||
| Unadjusted | 4.00 | (0.92 to 17.46) | 0.065 |
| Adjusteda | 3.49 | (0.78 to 15.57) | 0.101 |
| KL-6, U/mL, Cut off point = 946 | |||
| Unadjusted | 1.57 | (0.67 to 3.68) | 0.296 |
| Adjusteda | 1.53 | (0.65 to 3.59) | 0.326 |
| SP-D, ng/mL, Cut off point = 194.7 | |||
| Unadjusted | 3.17 | (1.21 to 8.31) | 0.019 |
| Adjusteda | 2.69 | (0.98 to 7.42) | 0.056 |
| BAL fluid findings | |||
| Lymphocytes, %, Cut off point = 10.15 | |||
| Unadjusted | 2.01 | (0.89 to 4.52) | 0.091 |
| Adjusteda | 2.03 | (0.90 to 4.57) | 0.087 |
| Neutrophils, %, Cut off point = 1.77 | |||
| Unadjusted | 4.04 | (0.92 to 17.79) | 0.065 |
| Adjusteda | 3.54 | (0.79 to 15.94) | 0.100 |
| Eosinophils, %, Cut off point = 3.21 | |||
| Unadjusted | 3.27 | (1.29 to 8.29) | 0.013 |
| Adjusteda | 2.89 | (1.11 to 7.51) | 0.029 |
Abbreviations: HR hazard ratio, CI confidence interval, GAP gender-age-physiology, LDH lactate dehydrogenase, KL-6 Krebs von den Lungen-6, SP-D surfactant protein-D, BAL bronchoalveolar lavage
aAdjusted for immunosuppressive agent therapy
Prognostic factors for overall survival from the initial IPF diagnosis
In the unadjusted Cox analysis, higher baseline GAP stage (≥II), higher serum SP-D level (≥194.7 ng/mL), and higher eosinophil percentage (≥3.21 %) in BAL fluid samples were significant prognostic factors for IPF (HR [95 % CI], 4.02 [1.68–9.64], 3.49 [1.36–8.94], and 2.42 [1.03–5.68], respectively). In the Cox analysis adjusted for treatment with an immunosuppressive agent, baseline cardiovascular diseases, higher GAP stage (≥II), and higher serum SP-D level (≥194.7 ng/mL) were significant prognostic factors for IPF (HR [95 % CI], 2.37 [1.03–5.44], 3.63 [1.47–8.93], and 3.21 [1.21–8.48]) (Table 4).
Table 4.
Cox analysis for prognostic factors for overall survival from the idiopathic pulmonary fibrosis initial diagnosis
| HR | (95 % CI) | P Value | |
|---|---|---|---|
| Treatment after diagnosis | |||
| Immunosuppressive agent | |||
| Unadjusted | 2.40 | (0.96 to 6.02) | 0.062 |
| Cardiovascular diseases | |||
| Unadjusted | 2.08 | (0.91 to 4.54) | 0.086 |
| Adjusteda | 2.37 | (1.03 to 5.44) | 0.042 |
| GAP stage (≥II) | |||
| Unadjusted | 4.02 | (1.68 to 9.64) | 0.002 |
| Adjusteda | 3.63 | (1.47 to 8.93) | 0.005 |
| Serum markers | |||
| LDH, U/L, Cut off point = 180 | |||
| Unadjusted | 2.49 | (0.74 to 8.37) | 0.139 |
| Adjusteda | 2.24 | (0.66 to 7.63) | 0.198 |
| KL-6, U/mL, Cut off point = 946 | |||
| Unadjusted | 1.46 | (0.67 to 3.18) | 0.340 |
| Adjusteda | 1.45 | (0.67 to 3.17) | 0.345 |
| SP-D, ng/mL, Cut off point = 194.7 | |||
| Unadjusted | 3.49 | (1.36 to 8.94) | 0.009 |
| Adjusteda | 3.21 | (1.21 to 8.48) | 0.019 |
| BAL fluid findings | |||
| Lymphocytes, %, Cut off point = 10.15 | |||
| Unadjusted | 1.96 | (0.93 to 4.14) | 0.077 |
| Adjusteda | 1.99 | (0.94 to 4.21) | 0.072 |
| Neutrophils, %, Cut off point = 1.77 | |||
| Unadjusted | 2.48 | (0.73 to 8.45) | 0.147 |
| Adjusteda | 2.21 | (0.63 to 7.69) | 0.213 |
| Eosinophils, %, Cut off point = 3.21 | |||
| Unadjusted | 2.42 | (1.03 to 5.68) | 0.043 |
| Adjusteda | 2.20 | (0.92 to 5.25) | 0.075 |
Abbreviations: HR hazard ratio, CI confidence interval, GAP gender-age-physiology, LDH lactate dehydrogenase, KL-6 Krebs von den Lungen-6, SP-D surfactant protein-D, BAL bronchoalveolar lavage
aAdjusted for immunosuppressive agent therapy
Discussion
This study examined associations between patient characteristics, serum markers, and BAL fluid cellular constituents and the incidence of AE-IPF and survival in a cohort of IPF patients who were diagnosed based on the official 2011 IPF ATS/ERS/JRS/ALAT Update Statement [2]. A log-rank test showed that treatment with an immunosuppressive agent after diagnosis, baseline cardiovascular diseases, higher GAP stage (≥II), higher serum LDH level (≥180 U/L), higher serum SP-D level (≥194.7 ng/mL), and higher neutrophil (≥1.77 %) and eosinophil (≥3.21 %) percentages in BAL fluid samples were associated with poor AE-free probability. In the Cox analysis adjusted for treatment with an immunosuppressive agent, baseline cardiovascular diseases, higher GAP stage (≥II), and a higher eosinophil percentage (≥3.21 %) in BAL fluid samples were predictors of an AE-IPF.
It was reported that the human lung microbiome and any changes in its numbers and composition play important roles in the pathogenesis and progression of lung diseases, including IPF [17, 18]. In addition, it was recently reported that patients with IPF had a higher bacterial load in BAL fluid compared with control subjects and that an increased bacterial load at the time of diagnosis identified patients with more rapidly progressive IPF and those at a higher risk of mortality [19]. The multicenter PANTHER trial found that combination therapy (prednisone, azathioprine, and N-acetylcysteine) was associated with more AEs than a placebo. [20]. The present study also demonstrated that treatment with an immunosuppressive agent was associated with an AE-IPF in the log-rank test and unadjusted Cox analysis. In addition, Papiris et al. recently reported that immunosuppression adversely affected the outcomes of AE-IPFs [21]. These findings imply that treatment with an immunosuppressive agent may increase the bacterial load in the lungs of patients with IPF and lead to an AE-IPF. Further studies are needed to elucidate the association between bacterial load and AE-IPFs. However, baseline cardiovascular diseases, higher GAP stage (≥II), and higher eosinophil percentage (≥3.21 %) in BAL fluid samples were predictors of an AE-IPF, even after adjustment for treatment with an immunosuppressive agent in the Cox analysis. These risk factors were thought to be independent of treatment with an immunosuppressive agent.
BAL fluid analyses in IPF patients typically show an increase in total cell count, neutrophils (>5 %), and eosinophils (>5 %) [22]. In the present study, baseline median neutrophil and eosinophil percentages in the BAL fluid of patients who experienced an AE were 7.0 % (IQR: 3.2–10.5 %) and 4.6 % (IQR: 2.1–7.0 %), respectively. These BAL fluid findings were consistent with those previously reported in patients with IPF [22]. This study showed that higher neutrophil (≥1.77 %) and eosinophil (≥3.21 %) percentages in BAL fluid samples were associated with poor AE-free probability in the log-rank test. In the adjusted Cox analysis, a higher baseline eosinophil percentage (≥3.21 %) in BAL fluid samples was a predictor of an AE-IPF. Relative increases in eosinophil and neutrophil percentages in BAL fluid may identify a subset of patients with disease that is more “active” or in an accelerated phase of tissue damage that may predispose them to an AE. Although inflammation is never a prominent histopathological finding in UIP [2], occult sustained eosinophil and neutrophil accumulations in the alveolar space may be key immune effector cells driving the inflammatory response and may play a role in the occurrence of an AE-IPF. Findings of recent studies of high BAL fluid eosinophil and neutrophil percentages in patients with IPF during AEs lend support to this possibility [3, 9]. Tabuena et al. [23] found that BAL fluid neutrophil and lymphocyte counts predicted mortality in current smokers in a study of 81 patients with IPF. Kinder et al. recently reported that each doubling of baseline BAL fluid neutrophil percentage was associated with a 30 % increase in mortality risk in a series of 156 patients with biopsy-proven IPF [24]. These observations also suggest the potential contribution of inflammation in the pathogenesis of IPF. Although the possible role of a chronic inflammatory response has been largely neglected because of the inefficacy of corticosteroids and immunosuppressants in the treatment of IPF [2, 20], further studies focusing on the potential role of inflammation in IPF pathogenesis and AE-IPF occurrence are needed. Current practice and consensus guidelines [2] do not recommend performing BAL for a determination of cellular constituents in BAL fluid at the time of IPF diagnosis. However, the indication for BAL with a cellular analysis may become clearer when effective therapies to prevent an AE-IPF are discovered.
This study showed that a baseline higher GAP stage (≥II) was one of the predictors of an AE-IPF and a significant prognostic factor for IPF in the adjusted Cox analysis. This GAP model consists of four baseline variables: gender (G), age (A), and two lung physiology variables (P) (FVC and diffusing capacity for carbon monoxide) [12]. It has been proposed that the GAP model was useful in the prediction of mortality at baseline in patients with IPF [12]. In addition, it was reported that GAP stage III IPF patients tended to have a higher incidence of AEs than stage II and stage I patients [25]. The present findings along with those obtained in previous studies suggest that the GAP staging system could be used as a quick and simple screening method for predicting AE-IPF.
Cardiovascular disease has been reported to be one of the most frequently observed comorbidities in patients with IPF and associated with shortened survival time [26, 27]. However, the impact on AE-IPF has remained unknown. This study showed that baseline cardiovascular disease was not only a prognostic factor for IPF, but also a risk factor for an AE-IPF in the adjusted Cox analysis. Further studies are needed to elucidate the mechanisms of why cardiovascular disease increases the risk of AE-IPF.
SP-D is a lipoprotein complex synthesized mainly by alveolar type II epithelial cells and is secreted into a liquid layer lining the lung epithelium. Increased serum SP-D levels obtained at the time of IPF diagnosis were reported to be associated with increased mortality [28–30]. In the present study, serum SP-D levels obtained at the time of initial diagnosis were associated with AE-free probability in a log-rank test and overall survival in the adjusted Cox analysis. Two mechanisms may contribute to elevated SP-D serum levels in pulmonary fibrosis: (1) an absolute increase in alveolar type II epithelial cells due to diffuse hyperplasia increases at the source of pulmonary SP-D; and (2) epithelial injury and basal membrane leakage may cause spillover into the circulation [28]. Evidence indicates that repetitive injuries to alveolar epithelial cells trigger an exaggerated wound healing response resulting in extensive scar formation, and finally, pulmonary fibrosis [31, 32]. Together with previous observations, the present findings suggest that injuries to alveolar epithelial cells may be one of the important triggers for IPF progression.
In a randomized trial of pirfenidone, a positive treatment effect was demonstrated, including fewer AE-IPF episodes (14 % versus none) [33]. However, a previous study did not reproduce the inhibitory effect of pirfenidone on AE-IPF [34]. In the present study, neither treatment with pirfenidone nor N-acetylcysteine was associated with AE-free probability in a log-rank test. However, this might be due to the small number of patients treated with pirfenidone or NAC in this study. Whether new antifibrotic agents have preventive effects against AE-IPF is an interesting clinical question. Further studies are needed to elucidate this question.
Previously reported conflicting results regarding risk factors for an AE-IPF were probably partly related to the use of multiple AE-IPF definitions. According to previously reported AE-IPF criteria, endotracheal aspirate or BAL fluid should be examined to exclude pulmonary infection, and patients who do not undergo these procedures should be termed as having a “suspected AE” [4]. However, these procedures are often not feasible given the significant hypoxemia typical in an AE-IPF. There is a frequent inability to exclude underlying infection results confidently in a large number of “suspected” AE-IPF cases that cannot be confirmed. Therefore, the definition of AE-IPF in the present study did not include the performance of endotracheal aspirate collection or BAL as an essential component. Collard et al. recently reported that suspected AE-IPF cases were clinically indistinguishable from definite AE-IPF cases, and were associated with similarly high risks of disease progression and short-term mortality in IPF [35]. Ryerson et al. recently proposed expanding the AE-IPF diagnostic criteria by allowing the diagnostic criteria to be met without the performance of invasive procedures (e.g. bronchoscopy) [36]. In fact, the definition of AE-IPF used in recent clinical trials of nintedanib did not include the performance of endotracheal aspirate collection or BAL as essential components, and infection was excluded in accordance with routine clinical practice and microbiologic studies [37]. Therefore, the results obtained in the present study are thought to be practical for the management of patients with IPF in daily clinical settings.
There are some limitations to this study. First, this was a retrospective study with a small number of subjects. Because of the small numbers of AEs and deaths, risk factors and prognostic factors were adjusted only by treatment with an immunosuppressive agent in the multiple Cox analysis. Because the associations among several factors seem to be complex, further study in a larger patient cohort is needed to analyze independent risk factors for AE-IPF and prognostic factors for IPF. Second, although the present study suggests that alveolar inflammation may be one of the key pathways activated prior to the development of an AE-IPF, the precise biological mechanisms of AE-IPF remain unknown. Third, this study included only Japanese patients. Therefore, further studies are needed to ascertain whether these results can be applied equally to other ethnic groups. The relative rarity of AE-IPF suggests that these research questions should be answered by multicenter collaborations.
Conclusions
In conclusion, this study demonstrated that baseline cardiovascular diseases, higher GAP stage (≥II), and higher eosinophil percentage (≥3.21 %) in BAL fluid samples were predictors of an AE-IPF. Further studies are needed to elucidate the association between these markers and mechanisms of AE occurrence.
Abbreviations
A-a DO2, alveolar-arterial difference in oxygen; AE, acute exacerbation; BAL, bronchoalveolar lavage; CI, confidence interval; CPI, composite physiologic index; CRP, C-reactive protein; DLCO, diffusing capacity for carbon monoxide; FiO2, fraction of inspired oxygen; FVC, forced vital capacity; GAP, gender-age-physiology; HR, hazard ratio; HRCT, high-resolution computed tomography; IPF, idiopathic pulmonary fibrosis; IQR, interquartile range; KCO, carbon monoxide transfer coefficient; KL-6, Krebs von den Lungen-6; LDH, lactate dehydrogenase; MST, median survival time; NAC, N-acetylcysteine; P/F ratio, partial pressure of oxygen in arterial blood (PaO2)/fraction of inspired oxygen (FiO2) ratio; PaO2, partial pressure of oxygen in arterial blood; ROC, receiver operating characteristic; SP-D, surfactant protein-D; UIP, usual interstitial pneumonia
Acknowledgments
The authors thank Dr. M. Kitaichi (Department of Laboratory Medicine and Pathology, NHO Kinki-chuo Chest Medical Center, Sakai, Japan) for valuable advice regarding pathological diagnosis, and Mr. A. Yokoyama and Mr. A. Nishigaki for excellent technical support. We would like to thank Editage (www.editage.jp) for English language editing.
Funding
This study was supported in part by a research grant from Takeda Science Foundation and Grants-in-Aid for Scientific Research of Japan Society for the Promotion of Science (JSPS KAKENHI), grant numbers 25860649 and 15 K09223.
Availability of data and material
The data will not be shared with participant confidentiality.
Authors’ contributions
TK made substantial contributions to the study conception and design. SS participated in the design of the study and performed the statistical analysis. TK, NS, HU, TH, SN, AH, KO, HI, KY, YI, and YO collected the clinical data from clinical notes and retrospectively made diagnoses of idiopathic pulmonary fibrosis. TK drafted the article. SK and HM critically revised the article for important intellectual content. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The study protocol was approved by the Institutional Review Boards of Nagasaki University Hospital and University Hospital of Occupational and Environmental Health. The patients’ approval or informed consent was not required for a retrospective review of their records, pursuant to the ethical guidelines of the Japanese Ministry of Health, Labor, and Welfare.
Contributor Information
Tomoyuki Kakugawa, Phone: +81-95-819-7273, Email: kakugawa@nagasaki-u.ac.jp.
Noriho Sakamoto, Email: nsakamot@nagasaki-u.ac.jp.
Shuntaro Sato, Email: shuntarosato@nagasaki-u.ac.jp.
Hirokazu Yura, Email: h_yura.5994@me.com.
Tatsuhiko Harada, Email: harada-nagasaki@umin.ac.jp.
Shota Nakashima, Email: shonaka@nagasaki-u.ac.jp.
Atsuko Hara, Email: a-hara@nagasaki-u.ac.jp.
Keishi Oda, Email: oda-keishi@med.uoeh-u.ac.jp.
Hiroshi Ishimoto, Email: h-ishimoto@nagasaki-u.ac.jp.
Kazuhiro Yatera, Email: yatera@med.uoeh-u.ac.jp.
Yuji Ishimatsu, Email: yuji-i@nagasaki-u.ac.jp.
Yasushi Obase, Email: obaseya@nagasaki-u.ac.jp.
Shigeru Kohno, Email: s-kohno@nagasaki-u.ac.jp.
Hiroshi Mukae, Email: hmukae@nagasaki-u.ac.jp.
References
- 1.Travis WD, Costabel U, Hansell DM, King TE, Jr, Lynch DA, Nicholson AG, Ryerson CJ, Ryu JH, Selman M, Wells AU, et al. An official American thoracic society/european respiratory society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188:733–748. doi: 10.1164/rccm.201308-1483ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Song JW, Hong SB, Lim CM, Koh Y, Kim DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J. 2011;37:356–363. doi: 10.1183/09031936.00159709. [DOI] [PubMed] [Google Scholar]
- 4.Collard HR, Moore BB, Flaherty KR, Brown KK, Kaner RJ, King TE, Jr, Lasky JA, Loyd JE, Noth I, Olman MA, et al. Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2007;176:636–643. doi: 10.1164/rccm.200703-463PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim DS, Collard HR, King TE., Jr Classification and natural history of the idiopathic interstitial pneumonias. Proc Am Thorac Soc. 2006;3:285–292. doi: 10.1513/pats.200601-005TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jeon K, Chung MP, Lee KS, Chung MJ, Han J, Koh WJ, Suh GY, Kim H, Kwon OJ. Prognostic factors and causes of death in Korean patients with idiopathic pulmonary fibrosis. Respir Med. 2006;100:451–457. doi: 10.1016/j.rmed.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 7.Kondoh Y, Taniguchi H, Katsuta T, Kataoka K, Kimura T, Nishiyama O, Sakamoto K, Johkoh T, Nishimura M, Ono K, Kitaichi M. Risk factors of acute exacerbation of idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. 2010;27:103–110. [PubMed] [Google Scholar]
- 8.Natsuizaka M, Chiba H, Kuronuma K, Otsuka M, Kudo K, Mori M, Bando M, Sugiyama Y, Takahashi H. Epidemiologic survey of Japanese patients with idiopathic pulmonary fibrosis and investigation of ethnic differences. Am J Respir Crit Care Med. 2014;190:773–779. doi: 10.1164/rccm.201403-0566OC. [DOI] [PubMed] [Google Scholar]
- 9.Kim DS, Park JH, Park BK, Lee JS, Nicholson AG, Colby T. Acute exacerbation of idiopathic pulmonary fibrosis: frequency and clinical features. Eur Respir J. 2006;27:143–150. doi: 10.1183/09031936.06.00114004. [DOI] [PubMed] [Google Scholar]
- 10.Judge EP, Fabre A, Adamali HI, Egan JJ. Acute exacerbations and pulmonary hypertension in advanced idiopathic pulmonary fibrosis. Eur Respir J. 2012;40:93–100. doi: 10.1183/09031936.00115511. [DOI] [PubMed] [Google Scholar]
- 11.Kondoh Y, Taniguchi H, Ebina M, Azuma A, Ogura T, Taguchi Y, Suga M, Takahashi H, Nakata K, Sugiyama Y, et al. Risk factors for acute exacerbation of idiopathic pulmonary fibrosis - Extended analysis of pirfenidone trial in Japan. Respir Investig. 2015;53:271–278. doi: 10.1016/j.resinv.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 12.Ley B, Ryerson CJ, Vittinghoff E, Ryu JH, Tomassetti S, Lee JS, Poletti V, Buccioli M, Elicker BM, Jones KD, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156:684–691. doi: 10.7326/0003-4819-156-10-201205150-00004. [DOI] [PubMed] [Google Scholar]
- 13.Wells AU, Desai SR, Rubens MB, Goh NS, Cramer D, Nicholson AG, Colby TV, du Bois RM, Hansell DM. Idiopathic pulmonary fibrosis: a composite physiologic index derived from disease extent observed by computed tomography. Am J Respir Crit Care Med. 2003;167:962–969. doi: 10.1164/rccm.2111053. [DOI] [PubMed] [Google Scholar]
- 14.Sakamoto N, Mukae H, Fujii T, Kakugawa T, Kaida H, Kadota J, Kohno S. Soluble form of Fas and Fas ligand in serum and bronchoalveolar lavage fluid of individuals infected with human T-lymphotropic virus type 1. Respir Med. 2004;98:213–219. doi: 10.1016/j.rmed.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 15.Youden WJ. Index for rating diagnostic tests. Cancer. 1950;3:32–35. doi: 10.1002/1097-0142(1950)3:1<32::AID-CNCR2820030106>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 16.Heagerty PJ, Lumley T, Pepe MS. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics. 2000;56:337–344. doi: 10.1111/j.0006-341X.2000.00337.x. [DOI] [PubMed] [Google Scholar]
- 17.Human Microbiome Project C Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han MK, Zhou Y, Murray S, Tayob N, Noth I, Lama VN, Moore BB, White ES, Flaherty KR, Huffnagle GB, et al. Lung microbiome and disease progression in idiopathic pulmonary fibrosis: an analysis of the COMET study. Lancet Respir Med. 2014;2:548–556. doi: 10.1016/S2213-2600(14)70069-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Molyneaux PL, Cox MJ, Willis-Owen SA, Mallia P, Russell KE, Russell AM, Murphy E, Johnston SL, Schwartz DA, Wells AU, et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2014;190(8):906–13. doi: 10.1164/rccm.201403-0541OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Idiopathic Pulmonary Fibrosis Clinical Research N. Raghu G, Anstrom KJ, King TE, Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366:1968–1977. doi: 10.1056/NEJMoa1113354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Papiris SA, Kagouridis K, Kolilekas L, Papaioannou AI, Roussou A, Triantafillidou C, Baou K, Malagari K, Argentos S, Kotanidou A, et al. Survival in Idiopathic pulmonary fibrosis acute exacerbations: the non-steroid approach. BMC Pulm Med. 2015;15:162. doi: 10.1186/s12890-015-0146-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pesci A, Ricchiuti E, Ruggiero R, De Micheli A. Bronchoalveolar lavage in idiopathic pulmonary fibrosis: what does it tell us? Respir Med. 2010;104(Suppl 1):S70–73. doi: 10.1016/j.rmed.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 23.Tabuena RP, Nagai S, Tsutsumi T, Handa T, Minoru T, Mikuniya T, Shigematsu M, Hamada K, Izumi T, Mishima M. Cell profiles of bronchoalveolar lavage fluid as prognosticators of idiopathic pulmonary fibrosis/usual interstitial pneumonia among Japanese patients. Respiration. 2005;72:490–498. doi: 10.1159/000087673. [DOI] [PubMed] [Google Scholar]
- 24.Kinder BW, Brown KK, Schwarz MI, Ix JH, Kervitsky A, King TE., Jr Baseline BAL neutrophilia predicts early mortality in idiopathic pulmonary fibrosis. Chest. 2008;133:226–232. doi: 10.1378/chest.07-1948. [DOI] [PubMed] [Google Scholar]
- 25.Kishaba T, Shimaoka Y, Fukuyama H, Nagano H, Nei Y, Yamashiro S, Tamaki H. Clinical characteristics of idiopathic pulmonary fibrosis patients with gender, age, and physiology staging at Okinawa Chubu Hospital. J Thorac Dis. 2015;7:843–849. doi: 10.3978/j.issn.2072-1439.2015.04.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hyldgaard C. A cohort study of Danish patients with interstitial lung diseases: burden, severity, treatment and survival. Dan Med J. 2015;62:B5069. [PubMed] [Google Scholar]
- 27.Ley B, Collard HR, King TE., Jr Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183:431–440. doi: 10.1164/rccm.201006-0894CI. [DOI] [PubMed] [Google Scholar]
- 28.Greene KE, King TE, Jr, Kuroki Y, Bucher-Bartelson B, Hunninghake GW, Newman LS, Nagae H, Mason RJ. Serum surfactant proteins-A and -D as biomarkers in idiopathic pulmonary fibrosis. Eur Respir J. 2002;19:439–446. doi: 10.1183/09031936.02.00081102. [DOI] [PubMed] [Google Scholar]
- 29.Takahashi H, Fujishima T, Koba H, Murakami S, Kurokawa K, Shibuya Y, Shiratori M, Kuroki Y, Abe S. Serum surfactant proteins A and D as prognostic factors in idiopathic pulmonary fibrosis and their relationship to disease extent. Am J Respir Crit Care Med. 2000;162:1109–1114. doi: 10.1164/ajrccm.162.3.9910080. [DOI] [PubMed] [Google Scholar]
- 30.Barlo NP, van Moorsel CH, Ruven HJ, Zanen P, van den Bosch JM, Grutters JC. Surfactant protein-D predicts survival in patients with idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. 2009;26:155–161. [PubMed] [Google Scholar]
- 31.Selman M, King TE, Pardo A, American Thoracic S, European Respiratory S, American College of Chest P Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134:136–151. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- 32.Chung MP, Monick MM, Hamzeh NY, Butler NS, Powers LS, Hunninghake GW. Role of repeated lung injury and genetic background in bleomycin-induced fibrosis. Am J Respir Cell Mol Biol. 2003;29:375–380. doi: 10.1165/rcmb.2003-0029OC. [DOI] [PubMed] [Google Scholar]
- 33.Azuma A, Nukiwa T, Tsuboi E, Suga M, Abe S, Nakata K, Taguchi Y, Nagai S, Itoh H, Ohi M, et al. Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2005;171:1040–1047. doi: 10.1164/rccm.200404-571OC. [DOI] [PubMed] [Google Scholar]
- 34.Taniguchi H, Ebina M, Kondoh Y, Ogura T, Azuma A, Suga M, Taguchi Y, Takahashi H, Nakata K, Sato A, et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2010;35:821–829. doi: 10.1183/09031936.00005209. [DOI] [PubMed] [Google Scholar]
- 35.Collard HR, Yow E, Richeldi L, Anstrom KJ, Glazer C, investigators IP Suspected acute exacerbation of idiopathic pulmonary fibrosis as an outcome measure in clinical trials. Respir Res. 2013;14:73. doi: 10.1186/1465-9921-14-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ryerson CJ, Cottin V, Brown KK, Collard HR. Acute exacerbation of idiopathic pulmonary fibrosis: shifting the paradigm. Eur Respir J. 2015;46:512–520. doi: 10.1183/13993003.00419-2015. [DOI] [PubMed] [Google Scholar]
- 37.Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, Cottin V, Flaherty KR, Hansell DM, Inoue Y, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–2082. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]