

Abstract
Pseudomonas putida SQ1 was isolated for its ability to utilize the plant sugar sulfoquinovose (6-deoxy-6-sulfoglucose) for growth, in order to define its SQ-degradation pathway and the enzymes and genes involved. Here we describe the features of the organism, together with its draft genome sequence and annotation. The draft genome comprises 5,328,888 bp and is predicted to encode 5,824 protein-coding genes; the overall G + C content is 61.58 %. The genome annotation is being used for identification of proteins that might be involved in SQ degradation by peptide fingerprinting-mass spectrometry.
Keywords: Pseudomonas putida SQ1, aerobic, Gram-negative, Pseudomonadaceae, plant sulfolipid, organosulfonate, sulfoquinovose biodegradation
Introduction
Pseudomonas putida strain SQ1 belongs to the family of Pseudomonadaceae in the class of Gammaproteobacteria. The genus Pseudomonas was first described by Migula (in the year 1894 [1]) and the species Pseudomonas putida by Trevisan (in 1889 [2]). P. putida strain KT2440 was the first strain whose genome had been sequenced (in 2002 [3]), and it is the most well-studied P. putida strain thus far [4]. Currently, there are more than 30 genome sequences of P. putida strains available (e.g., 12 complete and 24 draft genomes in NCBI; January 2015), including the complete genome sequence of type strain NBRC 14164T [5]. P. putida species are highly abundant in water, soil and in the rhizosphere [6, 7], can be plant-beneficial [8], and are extensively studied for their capabilities to degrade a broad range of substrates, especially aromatic compounds [9–12].
P. putida strain SQ1 was isolated for its ability to utilize the sulfonated plant sugar sulfoquinovose (6-deoxy-6-sulfoglucose) as a sole source of carbon and energy for growth, and was enriched from a sample of littoral sediment of pre-Alpine Lake Constance, Germany [13]. SQ is the polar headgroup of the plant sulfolipid sulfoquinovosyl diacylglycerol, which is present in the photosynthetic membranes of all higher plants, mosses, ferns and algae and most photosynthetic bacteria [14]. SQ is one of the most abundant organosulfur compounds in the biosphere, following glutathione, cysteine, and methionine, and the global production of SQ is estimated at 10 gigatons (1010 tons) per year [15]. Hence, the complete degradation of SQ concomitant with a recycling of the bound sulfur in form of inorganic sulfate is an important process of the carbon and sulfur cycle, e.g. in soils.
Until today only one bacterial degradation pathway for SQ has been identified, ‘sulfoglycolysis’ in Escherichia coli K-12 [16]. In this pathway, SQ is catabolized in analogy to glucose-6-phosphate via an adapted Embden-Meyerhof-Parnas (glycolysis) pathway, involving four newly identified enzymes and genes, and four newly identified metabolites. The pathway yields dihydroxyacetone phosphate (DHAP), which drives energy metabolism and growth of E. coli, and sulfolactaldehyde, which is reduced to dihydroxypropanesulfonate and excreted [16]. For Pseudomonas species, it is well-known that these bacteria lack the key enzyme for glycolysis, phosphofructokinase, but that the alternative Entner-Doudoroff pathway is operative, i.e., an oxidative entry into glucose-6-phosphate catabolism via a dehydrogenase enzyme. We detected a SQ-dehydrogenase activity in crude extract of SQ-grown P. putida SQ1 cells, and we therefore suspect that a ‘Sulfo-Entner-Doudoroff’-type of pathway might be operative in P. putida SQ1 for catabolism of SQ, but not sulfoglycolysis.
A draft genome sequence of strain SQ1 has been established and annotated in the IMG pipeline, and the annotation has been transferred to a proteomics (Mascot) database for peptide fingerprinting-mass spectrometry: in our present (unpublished) work, the database is used to identify enzymes and genes that are specifically induced during growth with SQ, e.g. in comparison to cells grown with glucose, by two-dimensional protein gel electrophoresis. Here, we present a summary classification and a set of features for Pseudomonas putida strain SQ1, together with the description of the shotgun genomic sequencing and annotation.
Organism Information
Classification and features
P. putida SQ1 is a rod-shaped (Fig. 1), motile, Gram-negative bacterium that grows aerobically in complex medium (e.g. LB-medium), or prototrophically in mineral-salts medium with a single carbon source (e.g., succinate, glucose, SQ). Strain SQ1 grows overnight on LB-agar plates and forms beige-whitish, smooth colonies (Table 1). Pseudomonas putida SQ1 has been deposited in the Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures under reference number DSM 100120.
Fig. 1.
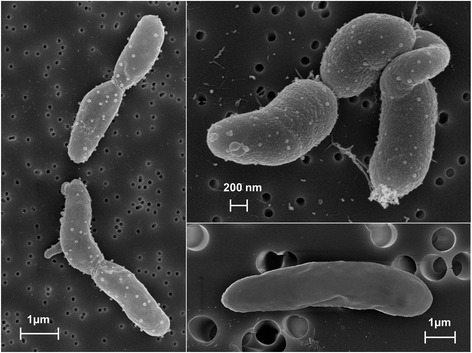
Scanning electron micrographs of Pseudomonas putida SQ1. Cells derived from a liquid culture (LB medium)
Table 1.
Classification and general features of Pseudomonas putida SQ1 [32]
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Current classification | Domain Bacteria | TAS [33] | |
| Phylum Proteobacteria | TAS [34] | ||
| Class Gammaproteobacteria | TAS [34, 35] | ||
| Order Pseudomonadales | TAS [36, 37] | ||
| Family Pseudomonadaceae | TAS [38, 39] | ||
| Genus Pseudomonas | TAS [1, 38–40] | ||
| Species putida | TAS [1, 2] | ||
| Strain SQ1 | TAS [13] | ||
| Gram stain | Negative | TAS [13] | |
| Cell shape | Rod-shaped | TAS [13] | |
| Motility | Motile | TAS [13] | |
| Sporulation | Non-sporulating | TAS [13] | |
| Temperature range | Mesophile | TAS [13] | |
| Optimum temperature | 30 °C | TAS [13] | |
| pH range; Optimum | Not tested; 7.2 | TAS [13] | |
| Carbon source | Succinate, glucose, sulfoquinovose | IDA,TAS [13] | |
| Energy source | Chemoorganotroph | IDA,TAS [13] | |
| MIGS-6 | Habitat | Aerobic habitat | TAS [13] |
| MIGS-22 | Oxygen requirement | Aerobic | TAS [13] |
| MIGS-15 | Biotic relationship | Free-living | NAS |
| MIGS-14 | Pathogenicity | Potentially pathogenic, Risk group 2 (classification according to German TRBA) | |
| MIGS-4 | Geographic location | Isolated from littoral sediment of Lake Constance, Germany | TAS [13] |
| MIGS-5 | Collection date | 2011 | TAS [13] |
| MIGS-4.1 MIGS-4.2 | Latitude | 47°41'44.77"N | |
| Longitude | 9°11'34.76"E | ||
| MIGS-4.4 | Altitude | 399 m |
aEvidence codes – IDA: Inferred from Direct Assay; TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [32]
Based on its 16S rRNA gene sequence, strain SQ1 is a member of the genus and species Pseudomonas putida, which is placed in the family Pseudomonadaceae within the order Pseudomonadales of Gammaproteobacteria, as illustrated by a phylogenetic tree shown in Fig. 2. Currently, 1,732 genome sequences of member of the order Pseudomonadales of Gammaproteobacteria, and 707 genome sequences within the family Pseudomonadaceae have been established (IMG JGI, January 2015).
Fig. 2.
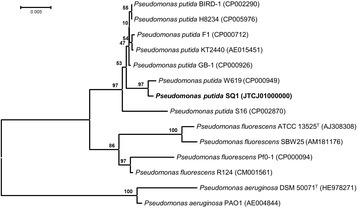
Phylogenetic tree based on the 16S rRNA gene sequence of P. putida SQ1, and sequences of other strains of the species P. putida, P. aeruginosa and P. fluorescens. The sequences were aligned with the CLUSTAL W program and the tree was built with the neighbor-joining algorithm integrated in the MEGA 6.0 program [31]. The phylogenetic tree was tested with 1000 bootstrap replicates; bootstrap values are shown at each node. The scale bar represents a 0.005 % nucleotide sequence divergence
Genome sequencing information
Genome project history
The DNA sample was submitted to GATC Biotech (Konstanz, Germany) in December 2012 where the whole-genome shotgun sequencing phase was completed in April 2013; the whole-genome shotgun sequencing was performed by GATC using the Illumina HiSeq2000 platform and a 100-bp paired-end library. After the mapping and de-novo assembly of the unmapped reads, which was done at the Genomics Center of the University of Konstanz, the draft genome sequence was uploaded into the IMG Pipeline for annotation and presented for public access on December 2014. The draft genome annotation is available at IMG under the IMG submission ID 14279, and was also deposited in Genbank under the accession number JTCJ00000000. Table 2 presents the project information and its association with MIGS version 2.0 compliance [17].
Table 2.
Project information
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | Permanent draft |
| MIGS-28 | Libraries used | 100-bp paired-end library |
| MIGS-29 | Sequencing platforms | Illumina HiSeq2000 |
| MIGS-31.2 | Fold coverage | >10x |
| MIGS-30 | Assemblers | Velvet v1.2.10 |
| MIGS-32 | Gene calling method | Prodigal |
| Genbank ID | JTCJ00000000 | |
| Genbank Date of Release | December 16, 2014 | |
| GOLD ID | Gi0045313 | |
| BIOPROJECT | PRJNA266268 | |
| MIGS 13 | Source Material Identifier | DSM 100120 |
| Project relevance | Study of unknown degradation pathway |
Growth conditions and genomic DNA preparation
Genomic DNA was extracted from an overnight culture of P. putida SQ1 grown at 30 °C in LB medium (500-ml scale), using JGI`s Bacterial Genomic DNA isolation protocol (CTAB protocol 2012).
Genome sequencing and assembly
The whole-genome shotgun sequencing was performed under contract by GATC Biotech (Konstanz, Germany) using the Illumina HiSeq2000 platform and a 100-bp paired-end library, which resulted in 23,816,201 sequenced reads (1.85 × 109 total bases). The trimming, mapping, as well as the de novo assembly of the unmapped raw reads, was performed at the Genomics Center of the University of Konstanz, Germany. First, the remaining adapters were removed and reads were trimmed by quality in CLC Genomics Workbench v6.5 (CLC bio, Aarhus, Denmark). In the next step, Bowtie v2.2.3 [18] was used to align the filtered reads against the genome of the closest relative, P. putida strain W619, to which 21,943,994 reads matched. These mapped reads were assembled with a reference-guided approach using the Columbus module implemented in Velvet v1.2.10 [19]. Velvet was then used to de novo assemble also all unmatched 1,872,207 reads (8.5 % of total reads). The whole process resulted in a total number of 1,634 contigs larger than 200 bp; the largest contig is 37,533 bp. The size of the draft genome is 5.3 Mb with 4,750,611 DNA coding bases, which is a normal size compared to other known P. putida genomes (range 3.0 to 7.1 Mb). The average G + C content is 61.58 %. At this time, no additional work is planned for this genome sequencing project (labeled as Permanent Draft).
Genome annotation
Genes were identified and auto-annotated in the DOE-IMG pipeline [20]. Genes were identified using Prodigal [21] and the predicted CDGs were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant database, UniProt [22], TIGRFam [23], Pfam [24], KEGG [25], COG [26], and InterPro [27] databases. The tRNAscan-SE tool [28] was used to identify tRNA sequences, whereas ribosomal RNA sequences were identified by searches against models of the ribosomal RNA genes built from SILVA [29]. The RNA components of the protein secretion complex and the RNaseP were identified by searching the genome of the corresponding Rfam profiles using INFERNAL.
Genome properties
The draft genome assembly of P. putida SQ1 consists of 1,634 contigs with an overall G + C content of 61.58 %. For these contigs, 5,925 complete genes or partial genes at ends of contigs have been predicted, 5,824 (98.30 %) of which for protein-coding genes. 4,624 (78.04 %) of these were assigned to a putative function with the remaining annotated as hypothetical proteins. The draft genome annotation predicted also 101 (1.70 %) sequences of RNA coding genes. The properties and the statistics of the draft genome annotation are summarized in Table 3 and the distribution of genes into COGs functional categories is presented in Table 4.
Table 3.
Nucleotide and gene count levels of the genome of P. putida SQ1
| Attribute | Genome (total) | |
|---|---|---|
| Value | % of totala | |
| Genome size (bp) | 5,328,888 | 100.00 |
| DNA coding | 4,750,611 | 89.15 |
| DNA G + C (bp) | 3,281,384 | 61.58 |
| DNA scaffolds | 1,634 | 100.00 |
| Total genes | 5,925 | 100.00 |
| Protein coding genes | 5,824 | 98.30 |
| RNA genes | 101 | 1.70 |
| rRNA operon count | 9 | 0.15 |
| Genes with function prediction | 4,624 | 78.04 |
| Genes in paralog clusters | 4,497 | 75.90 |
| Genes assigned to COGs | 3,249 | 54.84 |
| Genes with Pfam domains | 4,781 | 80.69 |
| Genes with signal peptides | 535 | 9.03 |
| Genes with transmembrane helices | 1,270 | 21.43 |
| CRISPR count | 1 | |
a) The total is based on either the size of the genome in base pairs or the total number of protein coding genes predicted in the annotated draft genome
Table 4.
Number of genes associated with general COG functional categories in P. putida SQ1
| Code | Value | % agea | Description |
|---|---|---|---|
| J | 167 | 4.60 | Translation, ribosomal structure and biogenesis |
| A | 1 | 0.03 | RNA processing and modification |
| K | 323 | 8.90 | Transcription |
| L | 107 | 2.95 | Replication, recombinant and repair |
| B | 1 | 0.03 | Chromatin structure and dynamics |
| D | 28 | 0.77 | Cell cycle control, Cell division, chromosome partitioning |
| V | 40 | 1.10 | Defense mechanisms |
| T | 204 | 5.62 | Signal transduction mechanisms |
| M | 179 | 4.93 | Cell wall/membrane/envelope biogenesis |
| N | 99 | 2.73 | Cell motility |
| U | 107 | 2.95 | Intracellular trafficking, secretion, and vesicular transport |
| O | 143 | 3.94 | Posttranslational modification, protein turnover, chaperones |
| C | 221 | 6.09 | Energy production and conversion |
| G | 183 | 5.04 | Carbohydrate transport and metabolism |
| E | 369 | 10.17 | Amino acid transport and metabolism |
| F | 87 | 2.40 | Nucleotide transport and metabolism |
| H | 158 | 4.35 | Coenzyme transport and metabolism |
| I | 147 | 4.05 | Lipid transport and metabolism |
| P | 216 | 5.95 | Inorganic ion transport and metabolism |
| Q | 91 | 2.51 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 423 | 11.66 | General function prediction only |
| S | 335 | 9.23 | Function unknown |
| - | 2,676 | 45.16 | Not in COGs |
a) The total is based on the total number of protein coding genes in the annotated genome
Currently, there are 50 genome sequencing projects for Pseudomonas putida strains registered in the JGI Genomes Online Database (GOLD), and 32 P. putida genome sequences (finished or permanent draft) are accessible within the IMG database (January 2015) for direct comparison; their genome sizes range between 3.0 Mb (P. putida MR3) and 7.1 Mb (P. putida S12), and their overall G + C content ranges between 60.81 % (P. putida MR3) and 63.14 % (P. putida CSV86). The genome sequence of P. putida W619 was chosen as reference genome for the mapping, as this genome showed the highest overall nucleotide sequence identity (91.9 %) of all genomes of P. putida strains that had been available at the time of sequencing. For comparison, the genome of the most well-studied P. putida strain, strain KT2440, shows 49.3 % overall nucleotide sequence identity to that of strain SQ1.
The genome of strain SQ1 (5.3 Mb) is smaller compared to these of strains W619 (5.8 Mb) and KT2440 (6.2 Mb). The IMG abundance profiles for these three P. putida genomes indicated a lower abundance of transposases (COG3436 and COG3547) in strains SQ1 (2 total) and W619 (2 total) in comparison to KT2440 (21 total), as well as a lower abundance of ABC-type periplasmic, transmembrane or permease component genes (COG0834, COG0765, COG0715, COG0683, COG1132, COG0747 and COG4177) in strains SQ1 (46 total) and W619 (47 total) in comparison to KT2440 (68 total).
In the draft genome of P. putida SQ1, all genes for the Entner-Doudoroff pathway for glucose/glucose-6-phosphate are represented as part of the two gene clusters (operons) that are highly conserved within P. putida species (e.g., [30]), i.e., predicted genes for glucose-6-phosphate 1-dehydrogenase (IMG locus tag PpSQ1_03570), 6-phosphogluconolactonase (PpSQ1_03569) and 2-keto-3-deoxy-phosphogluconate aldolase (PpSQ1_03568) (gene cluster PP1022-24 in P. putida KT2440, respectively), and glucokinase (PpSQ1_04592), 6-phosphogluconate dehydratase (PpSQ1_02498/04591) and glyceraldehyde-3-phosphate dehydrogenase (gene cluster PP1011-09 in P. putida KT2440, respectively); notably, the prediction of the dehydratase gene is distributed over two contigs of the draft assembly (and therefore has two IMG locus tags), however, the respective contigs are contiguous, as confirmed by PCR with a primer pair spanning over both contigs (this study). Further, all genes for a periplasmic entry into the Entner-Doudoroff pathway (e.g., [30]) were predicted in the draft genome of P. putida SQ1, i.e., for membrane-bound PQQ-dependent glucose dehydrogenases (e.g., PpSQ1_02906) and gluconate dehydrogenase complex (e.g., PpSQ1_00542), and for gluconokinase (PpSQ1_05341), 2-ketogluconate kinase (PpSQ1_05601/ 02858) and 2-ketogluconate 6-phosphate reductase (PpSQ1_02860).
No candidate genes for a sulfoglycolytic pathway for SQ, as found in E. coli K12 [16], were detected in the draft genome sequence of strain SQ1, which supports the notion that a novel, alternative pathway for SQ is operative in strain SQ1 (see Introduction). Neither P. putida strains W619, KT2440 nor F1 grew with SQ when tested ([13] and this study). Further, our preliminary proteomic data (not shown) indicates that enzymes/genes of the ‘classical’ Entner-Doudoroff pathway for glucose/glucose-6-phosphate (see above) are highly induced during growth with glucose, as expected, but not during growth with SQ. We concluded that additional genes in P. putida strain SQ1 are involved in the utilization of SQ, and that these genes might be located on contigs that resulted from the de novo assembly of the un-mapped reads. If appropriate, the proteomic identification of the core enzymes of this novel SQ degradation pathway based on the draft genome sequence established in this study, and their confirmation by biochemical and analytical-chemical methods, will be reported in a future communication.
Conclusions
Here, we present a summary classification and a set of features for Pseudomonas putida strain SQ1, together with the description of the shotgun genomic sequencing and annotation. The draft genome annotation contains no candidate genes for a sulfoglycolytic pathway for SQ, as found in E. coli K12, hence, the pathway operative in P. putida SQ1 represents a second, yet unknown bacterial degradation pathway for SQ. Furthermore, our preliminary proteomic data suggested that the ‘classical’ Entner-Doudoroff enzymes for a utilization of glucose/glucose-6-phophate are not induced during growth with SQ and that, hence, additional enzymes in strain SQ1 are operative during utilization of SQ. Based on the draft genome sequence, these enzymes and genes can now be defined.
Acknowledgements
We wish to thank Jaco Vangronsveld, Hasselt University, Belgium, for sending P. putida strain W619, Joachim Hentschel, University of Konstanz, for SEM operation, and DOE's JGI team for running IMG. A.-K.F wishes to thank Michael Weiss for proofreading and Ralf Schlesiger for help on the phylogenetic tree. The work of A.-K.F. was supported by the Konstanz Research School Chemical Biology (KoRS-CB), the work of P.F. by the University of Konstanz, and the work of D.S. by a DFG grant (SCHL 1936/1) and the University of Konstanz.
Abbreviations
- SQ
sulfoquinovose
- SQDG
sulfoquinovosyl diacylglycerol
- DHAP
dihydroxyacetone phosphate
- LB
lysogeny broth
- ABC
ATP-binding cassette
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
A.-K.F. prepared the genomic DNA and did the PCR reactions and growth experiments. P.F. did the trimming, mapping, as well as the de novo assembly of the unmapped raw reads. A.-K.F. and D.S. wrote the manuscript.
Contributor Information
Ann-Katrin Felux, Email: ann.katrin.felux@uni-konstanz.de.
Paolo Franchini, Email: paolo.franchini@uni-konstanz.de.
David Schleheck, Email: david.schleheck@uni-konstanz.de.
References
- 1.Migula W. Über ein neues System der Bakterien. Arb Bakteriol Inst Karlsruhe. 1894;1:235–8. [Google Scholar]
- 2.Trevisan V. I generi e le specie delle batteriacee. Zanaboni and Gabuzzi, Milan. 1889;1–35.
- 3.Nelson KE, Weinel C, Paulsen IT, Dodson RJ, Hilbert H. Martins dos Santos VA et al. Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environmental Microbiol. 2002;4(12):799–808. doi: 10.1046/j.1462-2920.2002.00366.x. [DOI] [PubMed] [Google Scholar]
- 4.Nikel PI, Martínez-García E, Lorenzo V. Biotechnological domestication of Pseudomonas using synthetic biology. Nat Rev Microbiol. 2014;12(5):368–79. doi:10.1038/nrmicro3253. [DOI] [PubMed]
- 5.Ohji S, Yamazoe A, Hosoyama A, Tsuchikane K, Ezaki T, Fujita N. The complete genome sequence of Pseudomonas putida NBRC 14164T confirms high intraspecies variation. Genome Announc. 2014;2(1). doi:10.1128/genomeA.00029-14. [DOI] [PMC free article] [PubMed]
- 6.Molina L, Ramos C, Duque E, Ronchel MC, García JM, Wyke L, et al. Survival of Pseudomonas putida KT2440 in soil and in the rhizosphere of plants under greenhouse and environmental conditions. Soil Biol Biochem. 2000;32(3):315–21. doi: 10.1016/S0038-0717(99)00156-X. [DOI] [Google Scholar]
- 7.Dos Santos VA, Heim S, Moore ER, Stratz M, Timmis KN. Insights into the genomic basis of niche specificity of Pseudomonas putida KT2440. Environ Microbiol. 2004;6(12):1264–86. doi: 10.1111/j.1462-2920.2004.00734.x. [DOI] [PubMed] [Google Scholar]
- 8.Chen C, Bauske EM, Musson G, Rodriguez-Kabana R, Kloepper JW. Biological control of Fusarium wilt on cotton by use of endophytic bacteria. Biol Control. 1995;5:83–91. doi: 10.1006/bcon.1995.1009. [DOI] [Google Scholar]
- 9.Jiménez JI, Miñambres B, García JL, Díaz E. Genomic analysis of the aromatic catabolic pathways from Pseudomonas putida KT2440. Environ Microbiol. 2002;4(12):824–41. doi: 10.1046/j.1462-2920.2002.00370.x. [DOI] [PubMed] [Google Scholar]
- 10.Timmis KN. Pseudomonas putida: a cosmopolitan opportunist par excellence. Environ Microbiol. 2002;4(12):779–81. doi: 10.1046/j.1462-2920.2002.00365.x. [DOI] [PubMed] [Google Scholar]
- 11.Gomes NC, Kosheleva IA, Abraham WR, Smalla K. Effects of the inoculant strain Pseudomonas putida KT2442 (pNF142) and of naphthalene contamination on the soil bacterial community. FEMS Microbiol Ecol. 2005;54(1):21–33. doi: 10.1016/j.femsec.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 12.Ward PG, Goff M, Donner M, Kaminsky W, O'Connor KE. A two step chemo-biotechnological conversion of polystyrene to a biodegradable thermoplastic. Environ Sci Technol. 2006;40(7):2433–7. doi: 10.1021/es0517668. [DOI] [PubMed] [Google Scholar]
- 13.Denger K, Huhn T, Hollemeyer K, Schleheck D, Cook AM. Sulfoquinovose degraded by pure cultures of bacteria with release of C3-organosulfonates: complete degradation in two-member communities. FEMS Microbiol Lett. 2012;328(1):39–45. doi: 10.1111/j.1574-6968.2011.02477.x. [DOI] [PubMed] [Google Scholar]
- 14.Benning C. Biosynthesis and function of the sulfolipid sulfoquinovosyl diacylglycerol. Annu Rev Plant Biol. 1998;49:53–75. doi: 10.1146/annurev.arplant.49.1.53. [DOI] [PubMed] [Google Scholar]
- 15.Harwood JL, Nicholls RG. The plant sulpholipid-- a major component of the sulphur cycle. Biochem Soc Trans. 1979;7(2):440–7. doi: 10.1042/bst0070440. [DOI] [PubMed] [Google Scholar]
- 16.Denger K, Weiss M, Felux AK, Schneider A, Mayer C, Spiteller D, et al. Sulphoglycolysis in Escherichia coli K-12 closes a gap in the biogeochemical sulphur cycle. Nature. 2014;507(7490):114–7. doi: 10.1038/nature12947. [DOI] [PubMed] [Google Scholar]
- 17.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26(5):541–7. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18(5):821–9. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mavromatis K, Ivanova NN, Chen IM, Szeto E, Markowitz VM, Kyrpides NC. The DOE-JGI standard operating procedure for the annotations of microbial genomes. Stand Genomic Sci. 2009;1(1):63–7. doi: 10.4056/sigs.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Magrane M, Consortium U. UniProt Knowledgebase: a hub of integrated protein data. The Journal of Biological Databases and Curation. 2011;43:bar009. doi:10.1093/database/bar009. [DOI] [PMC free article] [PubMed]
- 23.Haft DH, Selengut JD, White O. The TIGRFAMs database of protein families. Nucleic Acids Res. 2003;31(1):371–3. doi: 10.1093/nar/gkg128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42(Database issue):D222–30. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40(Database issue):D109–14. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Resesarch. 2000;28(1):33–6. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;37(Database issue):D211–5. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–64. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35(21):7188–96. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Daddaoua A, Krell T, Ramos JL. Regulation of glucose metabolism in Pseudomonas: the phosphorylative branch and entner-doudoroff enzymes are regulated by a repressor containing a sugar isomerase domain. J Biol Chem. 2009;284(32):21360–8. doi: 10.1074/jbc.M109.014555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–9. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA. 1990;87(12):4576–9. doi:10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed]
- 34.Garrity GM, Bell JA, Lilburn T. Phylum XIV. Proteobacteria phyl. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey's Manual of Systematic Bacteriology, Second Edition, Volume 2, Part B. New York: Springer; 2005. p. 1.
- 35.Validation of publication of new names and new combinations previously effectively published outside the IJSEM. List no. 106. Int J Syst Evol Micobiol. 2005;55:2235-8. [DOI] [PubMed]
- 36.Skerman VBD, McGowan V, Sneath PHA. Approved Lists of Bacterial Names. Int J Syst Bacteriol. 1980;30:225–420. doi: 10.1099/00207713-30-1-225. [DOI] [PubMed] [Google Scholar]
- 37.Orla-Jensen S. The main lines of the natural nacterial system. J Bacteriol. 1921;6(3):263–73. doi: 10.1128/jb.6.3.263-273.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garrity GM, Bell JA, Lilburn T. Order IX. Pseudomonadales. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey's Manual of Systematic Bacteriology, Second Edition, Volume 2, Part B. New York: Springer; 2005. p. 323.
- 39.Winslow CE, Broadhurst J, Buchanan RE, Krumwiede C, Rogers LA, Smith GH. The families and genera of the bacteria: preliminary report of the committee of the society of american bacteriologists on characterization and classification of bacterial types. J Bacteriol. 1917;2(5):505–66. doi: 10.1128/jb.2.5.505-566.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Judicial Commission. Opinion 5: conservation of the generic name Pseudomonas Migula 1894 and designation of Pseudomonas aeruginosa (Schroeter) Migula 1900 as type species. Int Bull Bacteriol Nomencl Taxon. 1952;2:121–2.