

Abstract
Increasing evidence suggests that an overactive endocannabinoid system (ECS) may contribute to the development of diabetes by promoting energy intake and storage, impairing both glucose and lipid metabolism, by exerting pro‐apoptotic effects in pancreatic beta cells and by facilitating inflammation in pancreatic islets. Furthermore, hyperglycaemia associated with diabetes has also been implicated in triggering perturbations of the ECS amplifying the pathological processes mentioned above, eventually culminating in a vicious circle. Compelling evidence from preclinical studies indicates that the ECS also influences diabetes‐induced oxidative stress, inflammation, fibrosis and subsequent tissue injury in target organs for diabetic complications. In this review, we provide an update on the contribution of the ECS to the pathogenesis of diabetes and diabetic microvascular (retinopathy, nephropathy and neuropathy) and cardiovascular complications. The therapeutic potential of targeting the ECS is also discussed.
Linked Articles
This article is part of a themed section on Endocannabinoids. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v173.7/issuetoc
Abbreviations
- 2‐AG
2‐arachidonoylglycerol
- AEA
anandamide
- CB1/2 receptor
cannabinoid receptor 1/2
- DN
diabetic nephropathy
- DNR
diabetic neuropathy
- ECS
endocannabinoid system
- ROS/RNS
reactive oxygen/nitrogen species
- T1DM
type 1 diabetes mellitus
- T2DM
type 2 diabetes mellitus
- ZDF rat
Zucker diabetic fatty rat
Tables of Links
| TARGETS | |
|---|---|
| GPCRs a | Transporters c |
| AT1 receptor | Ca2+‐ATPase |
| CB1 receptor | Enzymes d |
| CB2 receptor | Adenylate cyclase (AC) |
| CCR2 | Diacylglycerol lipase (DGL) |
| Catalytic receptors b | Fatty acid amide hydrolase (FAAH) |
| Insulin receptor | Monoacylglycerol lipase (MGL) |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,c,dAlexander et al., 2013a, 2013b, 2013c, 2013d).
Introduction
The major psychoactive component of Cannabis sativa, Δ9‐tetrahydrocannabinol (THC), was identified 50 years ago. Since then, much effort has been directed to identifying the endogenous compounds whose biological actions are mimicked by THC and to clarify their role in various physiological and pathological processes. The endogenous cannabinoid system (ECS) comprises the endocannabinoids (ECs), the enzymes that regulate their production and degradation, and the receptors through which they signal. Anandamide (AEA) and 2‐arachidonoylglycerol (2‐AG), the most studied ECs, are bioactive lipid mediators produced from cell membrane phospholipids. ECs are synthesized ‘on demand’, AEA predominantly via hydrolysis of N‐arachidonoyl phosphatidylethanolamine by a phospholipase D and 2‐AG from diacylglycerol by diacylglycerol lipase (DGL), although parallel biosynthetic pathways also exist. Once synthesized, AEA or 2‐AG are immediately released to target their receptors and then rapidly degraded by fatty acid amide hydrolase or monoacylglycerol lipase (MGL) respectively. The effects of ECs are mediated primarily by the Gi/o‐coupled cannabinoid receptor 1 or 2 (CB1 receptor /CB2 receptor), with the possible involvement of additional receptors, such as GPR‐55. AEA signals predominantly via CB1 receptors, while 2‐AG is a full agonist at both CB1 and CB2 receptors. Receptor activation results in a variety of biochemical responses, including inhibition of voltage‐gated Ca2+ channels and adenylate cyclase activity, leading to lower cAMP levels, as well as activation of K+ channels, phospholipases and MAPK pathways, the latter via G protein‐independent mechanisms (Howlett et al., 2010; Horváth et al., 2012).
CB1 receptors are expressed at very high levels in the CNS, whereas CB2 receptors are predominantly found in immune, inflammatory and haematopoietic cells (Pacher and Mechoulam, 2013). However, these receptors are present in several other cell types and the ECS has been implicated in a growing number of pathophysiological processes. Thus, pharmacological modulation of the ECS emerges as a promising therapeutic strategy in a variety of pathological conditions, including neurodegenerative, cardiovascular, gastrointestinal, liver and renal diseases (Pacher et al., 2006; Pacher and Kunos, 2013). Here, we provide a brief overview of emerging evidence suggesting an important role of the ECS in the pathogenesis of type 2 diabetes mellitus (T2DM) and its chronic complications. The therapeutic potential of targeting the ECS in diabetes and diabetic complications will also be discussed.
Diabetes and diabetic complications
Diabetes mellitus affects 387 million people worldwide and this number is expected to rise to 592 million by 2035 (International Diabetes Federation, 2014). The diabetes pandemic has been attributed to the growing prevalence of obesity, a major risk factor for T2DM. It has been estimated that almost 80% of T2DM cases could be prevented by adequate control of body weight. Diabetes is the seventh leading cause of death in the United States and both macrovascular and microvascular complications are the major cause of morbidity and mortality in diabetic patients. People with diabetes are two to six times more likely to develop macrovascular complications. Nearly half of all diabetic patients develop diabetic retinopathy and diabetes is the leading cause of blindness in adults, being responsible for 10 000 new cases of blindness every year in the United States alone. Diabetic nephropathy (DN) affects ∼30% of patients with diabetes and diabetes is known to account for over 50% of all patients receiving renal transplants in the United States. About 60% of non‐traumatic lower‐limb amputations among people aged 20 years or older occur in people with diabetes and diabetic neuropathy (DNR) is a major underlying cause (International Diabetes Federation, 2014).
Intervention studies have convincingly demonstrated that hyperglycaemia is a major pathogenic factor for diabetic complications (Varga et al., 2015). The underlying mechanisms are not fully understood; however, formation of advanced glycation end products, activation of the polyol, hexosamine and PKC pathways have been implicated. Oxidative stress through formation of both reactive oxygen and nitrogen (ROS/RNS) species is a common upstream event in the activation of these deleterious metabolic/signalling pathways (Varga et al., 2015). Furthermore, inflammatory processes orchestrated by infiltrating monocytes/macrophages also contribute to target organ damage (Forbes and Cooper, 2013).
The role of the ECS in the pathogenesis of T2DM
Both insulin resistance in peripheral tissues and a relative deficiency in insulin secretion by islet beta cells are key components in the development of T2DM. Studies performed in the last two decades have highlighted the central role of the ECS in the development of obesity and its deleterious effects on both glucose and lipid metabolism that can contribute to the development of insulin resistance and T2DM (Figure 1). The well‐established role of the ECS in metabolism has been recently reviewed (Silvestri and Di Marzo, 2013) and will only be briefly summarized. Recent emerging data suggest that the ECS also contributes to beta cell loss in T2DM by modulating inflammatory and cell death processes. These novel findings which may open an entirely new avenue to target the ECS in T2DM will be highlighted and discussed.
Figure 1.
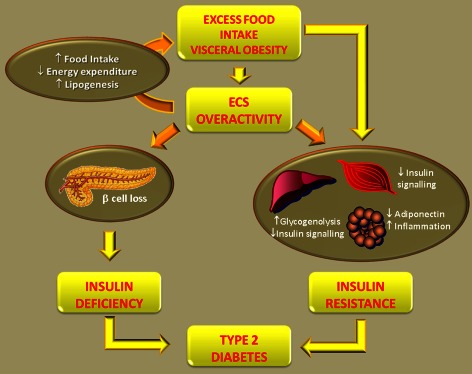
Role of the ECS in the development of T2DM. Excess food intake and obesity enhance the ECS tone. A hyperactive ECS further contributes to visceral fat accumulation and obesity by reducing energy expenditure and by enhancing both food intake and lipogenesis. Therefore, the ECS is involved in the development of obesity‐dependent insulin resistance. Moreover, an overactive ECS has direct deleterious effects on insulin sensitivity independent of weight gain in peripheral organ of metabolism (liver, adipose tissue, skeletal muscle). Finally, the ECS indirectly contribute to beta cell failure through activation of the Nlrp3‐ASC inflammasome in infiltrating macrophages, resulting in beta cell apoptosis. Both insulin resistance and relative insulin deficiency lead to the development of T2DM.
ECS in obesity and insulin resistance
In the CNS, activation of CB1 receptors enhances food intake by modulating the activity of hypothalamic neurons and, subsequently, the release of orexigenic and anorexigenic neuropeptides. Furthermore, CB1 receptor signalling affects reward and reinforcement circuits in the mesolimbic system, leading to a preference for highly palatable food. The CB1 receptor is also present in peripheral organs important in the control of metabolism and activates anabolic pathways, favouring energy storage. In white adipocytes, CB1 receptor activation increases de novo fatty acid synthesis, enhances triglyceride accumulation and reduces lipolysis, whereas in brown adipose tissue, the CB1 receptor counteracts the uncoupling of respiration from ATP production. Furthermore, the CB1 receptor increases hepatic lipogenesis and drives defective oxidative metabolism through impaired mitochondrial oxidative phosphorylation in skeletal muscle (Kunos and Tam, 2011; Silvestri and Di Marzo, 2013; Boon et al., 2014). The ECS has thus been proposed to be ‘part of a thrifty phenotype selected to cope with food shortage and make the best out of periods of plenty’ (Di Marzo, 2012).
In abdominal obesity, the ECS is generally up‐regulated in both central and peripheral tissues, as indicated by high EC levels and/or CB1 receptor overexpression. The exact underlying mechanisms are unclear; however, ECs are lipid mediators and their biosynthesis can be directly influenced by dietary fat intake. This hyperactive ECS can contribute to further fat accumulation by enhancing food intake as well as by favouring lipogenesis and reducing energy expenditure in peripheral organs (Blüher et al., 2006; Tedesco et al., 2010; Kunos and Tam, 2011; Silvestri et al., 2011; Silvestri and Di Marzo, 2013). Consistently, both pharmacological and genetic CB1 receptor blockade reduces body weight in animal models of obesity (Kunos and Tam, 2011). The effect of CB1 receptor inhibition on food intake is transient and weight loss occurs predominantly through blockade of peripheral CB1 receptors. However, recent data suggest that the central ECS also controls peripheral energy metabolism (O'Hare et al., 2011). As visceral adiposity is a major determinant of insulin resistance, it is not surprising that ECS overactivity favours the development of obesity‐associated metabolic abnormalities.
Emerging data suggest that a dysregulated ECS has also direct deleterious effects on insulin sensitivity and glucose metabolism independently of weight gain. In adipose tissue, activation of the ECS enhances glucose uptake to increase energy storage in the form of de novo synthesized lipids, down‐regulates adiponectin thereby affecting insulin sensitivity at distant organs and may favour local inflammation (Murumalla et al., 2011; Ge et al., 2013). In skeletal muscle, the CB1 receptor interferes with glucose uptake by inhibiting signalling pathways activated by insulin, including those required for plasma membrane translocation of glucose transporters. In the liver, activation of hepatic CB1 receptors can reduce systemic insulin sensitivity independently from body weight. Indeed, mice that express CB1 receptors exclusively on hepatocytes remain lean when fed a high‐fat diet, but they develop hepatic and systemic insulin resistance, whereas mice with hepatocyte‐specific CB1 receptor deletion become obese, but remain insulin‐sensitive (Liu et al., 2012). Several mechanisms may underlie these findings: hepatic CB1 receptor activation reduces insulin clearance by reducing the hepatic expression of the insulin‐degrading enzyme and inhibits insulin signalling through IRS1 and Akt2, resulting in increased hepatic glucose production due primarily to increased glycogenolysis (Liu et al., 2012). Furthermore, CB1 receptor activation induces endoplasmic reticulum stress resulting in elevated hepatic levels of long‐chain ceramides that in turn inhibit insulin signalling (Cinar et al., 2014). Collectively, these data provide strong evidence that a deranged ECS due to conditions leading to obesity, such as a high‐fat diet, may then contribute to further fat accumulation and insulin resistance through excess CB1 receptor activity and thus set the stage for the development of T2DM.
There is relatively little knowledge on the role of CB2 receptors in the control of metabolic processes; however, recent studies suggest CB2 receptors may affect inflammatory aspects of both obesity and T2DM. Surprisingly, CB2 receptor agonists potentiated obesity‐associated inflammation, insulin resistance and hepatic steatosis and CB2 receptor deficiency improved insulin sensitivity (Deveaux et al., 2009; Agudo et al., 2010). Furthermore, CB2 receptor overexpression in the brain induces hyperglycaemia and a lean phenotype in adult mice (Romero‐Zerbo et al., 2012). However, these studies need additional confirmation with improved CB2 selective ligands (particularly given the potent anti‐inflammatory role of CB2 agonists reported in numerous pathological disease models (Pacher and Mechoulam, 2011).
ECS and pancreatic beta cells
Data on the expression of ECS components in pancreatic islet cells are contradictory and vary among species; however, most studies agree that beta cells express both ECs and CB1 receptors and that CB1 receptor activation enhances insulin release (Horváth et al., 2012; Malenczyk et al., 2013). Recent studies have explored the possibility that the ECS may favour the development of T2DM by inducing beta cell apoptosis. Zucker diabetic fatty rats (ZDF), homozygous for non‐functional leptin receptors (ZDF/Gmi fa/fa)), are a valuable animal model to address this issue because they develop spontaneous diabetes with progression similar to human T2DM. Indeed, young ZDF rats are insulin‐resistant and normoglycaemic, while older ZDF become hyperglycaemic because of progressive beta cell failure. In this model, ibipinabant, a global CB1 receptor antagonist, attenuates beta cell loss independently of its effects on body weight (Rohrbach et al., 2012). Furthermore, a peripherally restricted CB1 receptor antagonist JD5037 delays the progression of T2DM and beta cell function loss, confirming that EC acting through peripheral CB1 receptors can contribute to beta cell failure (Jourdan et al., 2013).
In beta cells, insulin itself positively regulates beta cell survival and resistance to apoptosis in an autocrine manner and recent in vitro studies suggest that the CB1 receptor forms a heteromeric complex with the insulin receptor and thus inhibits insulin signalling by blocking insulin receptor kinase activity. This causes reduced phosphorylation of the pro‐apoptotic Bad, thereby causing beta cell death (Kim et al., 2012). Although these in vitro findings suggest that EC may induce beta cell death by acting directly on beta cells, a recent study has convincingly shown that beta cell failure in adult ZDF rats is not associated with CB1 receptor signalling in beta cells, but rather in pro‐inflammatory macrophages infiltrating pancreatic islets. Specifically, CB1 receptor activation in macrophages induced activation of the Nlrp3‐ASC inflammasome, resulting in the proteolytic activation and release of IL‐1β and IL‐18, which act as paracrine signals to induce beta cell apoptosis (Jourdan et al., 2013). The dominant role of macrophages in progressive beta cell death does not, however, exclude the possibility that high glucose acting on beta cells may trigger the inflammatory process by inducing IL‐1β and MCP‐1 (CCL2) release and thus macrophage infiltration. Data on CB2 receptor expression in beta cells are controversial; however, given the key role of the CB2 receptor in inhibiting inflammatory processes, it would be of interest to explore the potential protective role of signalling through this receptor in inflammatory cell‐mediated beta cell death.
Intervention studies in humans and future perspectives
Clinical trials in obese and T2DM patients have proven the efficacy of the global CB1 receptor inverse agonist rimonabant in reducing body weight and waist circumference and in ameliorating both lipid and glucose control. Based on these promising data, rimonabant was licensed in over 50 countries worldwide for the treatment of obesity. However, the drug was subsequently withdrawn from the market because of an increased risk of psychiatric adverse events, such as anxiety, depression and suicidal ideation, and the therapeutic development of this class of compounds was discontinued (Christensen et al., 2007).
More recently, peripherally restricted CB1 receptor antagonists that poorly cross the blood–brain barrier and are thus devoid of centrally mediated psychiatric side effects have been developed to assess if peripheral CB1 receptor inhibition preserves the metabolic benefit of global CB1 receptor blockade. A proof of principle study by Tam et al. (2010) demonstrated that treatment of diet‐induced obese mice with the peripherally restricted neutral CB1 receptor antagonist AM6545 improved glucose tolerance, insulin sensitivity, plasma lipid profile and also reversed fatty liver, although it was less effective than rimonabant in reducing body weight and it did not affect caloric intake. Subsequent studies have shown that a highly potent, selective and brain impermeable CB1 receptor inverse agonist, JD5037, is even more effective in improving metabolic parameters in rodent models of obesity/diabetes and has hypophagic effects by reversing leptin resistance (Tam et al., 2012), abolishes obesity‐induced hepatic insulin resistance (Cinar et al., 2014) and preserves beta cell function in ZDF rats (Jourdan et al., 2013). These results raise hope that CB1 receptor blockade may still be a viable option to combat dysmetabolism and JD5037 is currently undergoing toxicology screening and may move to clinical testing in the near future.
Diabetic nephropathy
DN is a leading cause of end‐stage renal failure and significantly enhances the cardiovascular risk of diabetic patients. The complication is characterized by both increased glomerular permeability to proteins and a relentless decline in renal functions. Structural changes comprise podocyte abnormalities, including nephrin loss, mesangial expansion and tubulointerstitial fibrosis. It is well established that oxidative stress, inflammation and fibrogenesis play a pivotal role in the development and progression of DN (Forbes and Cooper, 2013). Given the pro‐oxidative, pro‐inflammatory and profibrotic effects of CB1 receptor signalling and the opposing effects of signalling through CB2 receptors, there is growing interest on the potential role of the ECS in the pathogenesis of DN.
The ECS (ECs, their main metabolic enzymes and receptors CB1 CB2) is present within the normal kidney. In healthy animals, the CB1 receptor is expressed by endothelial cells of the renal arteries and weakly by podocytes and tubular epithelial cells, while CB2 receptors are strongly expressed by podocytes. This pattern of expression changes profoundly in diabetes. The CB1 receptor is overexpressed by podocytes in animal models in both T1DM and T2DM (Barutta et al., 2010; Tam et al., 2012; Jourdan et al., 2014). In contrast, there is a deficiency of 2‐AG, the main CB2 receptor ligand, in the renal cortex from mice with early STZ‐induced diabetes and podocyte CB2 receptor expression is markedly down‐regulated in human biopsies from patients with advanced DN (Barutta et al., 2011). Taken together, these data indicate that in diabetic kidneys the protective CB2 receptor signalling is impaired, while the detrimental CB1 receptor signalling is enhanced favouring deleterious consequences. It is likely that both hyperglycaemia and hypertension are important determinants of these alterations as in cultured podocytes exposure to high glucose was shown to increase CB1 receptor expression (Nam et al., 2012), while mechanical stress, mimicking glomerular capillary hypertension, down‐regulates CB2 receptors (Barutta et al., 2014). Moreover, proteinuria may lower constitutive tubular CB2 receptor expression in advanced DN as exposure of tubular epithelial cells to albumin down‐regulates CB2 receptor expression (Jenkin et al., 2013).
Intervention studies in animal models of DN have uncovered a potentially important role of the ECS in the pathogenesis of DN. The first evidence was provided in murine models of the metabolic syndrome. Treatment with rimonabant prevented proteinuria, ameliorated renal function and reduced the glomerular damage in obese ZDF rats and improved both albumin‐creatinine ratio and glomerulosclerosis in JCR : LA‐cp rats (a strain which is a close model of the human syndrome characterized by obesity, hyperlipidaemia, insulin resistance and a high risk for cardiovascular disease) (Janiak et al., 2007; Russell et al., 2010). More recently, a study performed in db/db mice, a model of T2DM, has shown that rimonabant markedly decreases urinary albumin excretion and mesangial expansion and suppresses synthesis of profibrotic and proinflammatory cytokines (Nam et al., 2012). However, CB1 receptor blockade also significantly improved insulin resistance and lipid profile in these animals, and the observed renoprotection may be due, at least in part, to improvement of metabolic abnormalities. Convincing proof for the direct role of CB1 receptors in the development of DN arose from a study performed in STZ‐induced diabetes, a model of T1DM, in which protective metabolic effects of CB1 receptor blockade cannot confound outcomes. In this model, treatment with the selective CB1 receptor reverse agonist AM251 significantly reduced albuminuria and prevented down‐regulation of nephrin and podocin, suggesting that enhanced podocyte CB1 receptor signalling may contribute to the development of albuminuria by lowering the expression of podocyte proteins crucial to maintaining glomerular permselectivity (Barutta et al., 2010). There was no effect of CB1 receptor blockade on markers of renal fibrosis and it is unclear whether the differential effect on fibrogenesis observed in animal models of T1DM versus T2DM reflects true differences in underlying mechanisms or whether it is animal strain‐related. In vitro, CB1 receptor activation is profibrotic, as it mediates the effects of high glucose both in inducing podocyte collagen overexpression (Nam et al., 2012) and promoting mesangial cell apoptosis (Lim et al., 2011); however, it is still controversial if CB1 receptor are present in mesangial cells in vivo (Barutta et al., 2014). A recent study using ZDF rats (Jourdan et al., 2014) provided additional mechanistic insight into the role of CB1 receptors in the pathogenesis of DN. This study demonstrated that peripheral CB1 receptor blockade was not only effective in preventing the characteristic hallmarks/symptoms of DN (albuminuria, reduced glomerular filtration, activation of renin‐angiotensin system, oxidative/nitrative stress, podocyte loss and increased CB1 receptor expression in glomeruli), but could also reverse these changes after they developed. This study also provided evidence that the enhanced CB1 receptor signalling in diabetic kidneys promotes up‐regulation of the local angiotensin II receptor‐NADPH oxidase signalling promoting ROS generation in podocytes and cell death (Jourdan et al., 2014). However, this study has not explored the effect of peripheral CB1 receptor inhibition on BP in obese hypertensive ZDF rats (Jourdan et al., 2014). In the light of recent results demonstrating that acute and chronic systemic CB1 receptor blockade improved BP regulation and metabolic profile in an angiotensin II‐dependent hypertensive(mRen2)27 rat model (Schaich et al., 2014), one can speculate that CB1 receptor inhibition in ZDF rats could ameliorate hypertension, which could in turn contribute, at least in part, to its beneficial effects in DN.
Recent studies have highlighted an important protective role for CB2 receptors in DN. In STZ‐induced diabetes, activation of CB2 receptors by the selective CB2 agonist AM1241 reduced albuminuria, glomerular monocyte accrual and nephrin down‐regulation (Barutta et al., 2011). Conversely, knocking down CB2 receptors worsened slit diaphragm protein down‐regulation, proteinuria, overexpression of extracellular matrix components, mesangial matrix expansion, monocyte infiltration and renal function loss in diabetic mice (Barutta et al., 2014). CB2 receptor activation reduced MCP‐1 signalling, whereas CB2 receptor deficiency markedly increased the expression of the MCP‐1 receptor CCR2 in the renal cortex, as well as in both cultured podocytes and monocytes (Montecucco et al., 2008; Barutta et al., 2011; 2014). By lowering CCR2 expression in monocytes, CB2 receptor agonists may reduce the recruitment of inflammatory cells that can contribute to renal injury through the release of ROS, toxic products and cytokines. On the other hand, CB2 receptor‐induced CCR2 down‐regulation on podocytes may prevent the direct deleterious effects of MCP‐1 on this cell type, including nephrin down‐regulation (Tarabra et al., 2009; Giunti et al., 2010). Of interest, recent experiments employing adoptive transfer of bone marrow have clarified that the worsening of DN in CB2 receptor‐deficient mice is mainly due to CB2 receptor loss on podocytes rather than on monocytes (Barutta et al., 2014).
Studies performed in experimental cisplatin‐induced nephropathy have shown that both CB1 receptor blockade and CB2 receptor activation reduce tissue injury, cell death and interrelated inflammation and oxidative/nitrosative stress (Mukhopadhyay et al., 2010a, 2010b). This suggests that CB1 and CB2 receptors also have opposing effects on tubular epithelial cells that may be of relevance in the pathogenesis of diabetes‐induced tubulointerstitial injury. In keeping with this notion, palmitic acid that promotes tubulointerstitial damage in T2DM, induces CB1 receptor expression in cultured proximal tubular epithelial cells and CB1 receptor mediates palmitic acid‐induced endoplasmic reticulum stress and apoptosis. Furthermore, AEA causes proximal tubular epithelial cell hypertrophy and this effect is reduced by CB1 receptor antagonists and enhanced by CB2 receptor antagonists. Although, tubular hypertrophy initially leads to increased capacity of the proximal tubules to reabsorb albumin, an increase in albumin re‐absorption can activate fibrotic cytokines, contributing to tubulointerstitial injury (Jenkin et al., 2012).
Collectively, these data suggest a beneficial effect of both CB1 receptor blockade and CB2 receptor activation in DN (Figure 2) . This is of significant therapeutic relevance as 20% of patients with incipient DN still progress to overt nephropathy despite optimal treatment, and there is an increasing need for novel therapeutic strategies. Further studies are required to establish the therapeutic potential of peripheral restricted CB1 receptor antagonists or CB2 receptor agonists in DN and to find out whether the addition of these compounds to current DN treatment protocols results in extra benefit.
Figure 2.
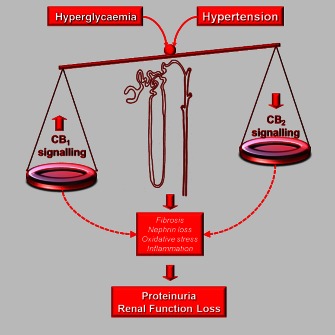
Opposing effects of CB1 receptor and CB2 receptor in DN. The CB1 receptor has deleterious pro‐oxidative and pro‐inflammatory effects, while opposing protective effects are induced by CB2 receptor activation. In diabetes, hyperglycaemia and hypertension alter the balance between CB1 receptor and CB2 receptor signalling as CB1 receptor expression is enhanced, while CB2 receptor is down‐regulated. This imbalance favours oxidative stress, inflammatory and profibrotic processes and contributes to the development of proteinuria by enhancing nephrin loss and of renal function loss by exacerbating fibrogenesis in both the mesangium and tubulointerstitium.
Diabetic neuropathy
DNR affects as many as 60% of patients with long‐standing diabetes. Distal symmetrical polyneuropathy (DSP), the most common type of DNR, is due to axon degeneration secondary to both metabolic abnormalities and injury of endoneural microvessels. Almost a third of patients with DSP describe burning, electric or stabbing pain (allodynia/hyperalgesia) (Peltier et al., 2014) and there is considerable interest in the possibility of exploiting the antinociceptive properties of the ECS for therapeutic gain.
Treatment with CB1 receptor agonists has antinociceptive effects in STZ‐induced diabetes (Horváth et al., 2012; Vera et al., 2012). Peripherally restricted CB1 receptor agonists, devoid of central side effects, are likely to be equally effective as analgesia is predominantly due to activation of CB1 receptors on peripheral nociceptors (Agarwal et al., 2007). However, as discussed earlier, CB1 receptor activation contributes to the development of T2DM and its complications in addition to deleterious cardiovascular effects, which is a major obstacle to their therapeutic use (Pacher and Kunos, 2013). CB2 receptor agonists also exert antinociceptive effects in diabetic mice, which appear to be predominantly related to inhibition of microglia‐driven inflammation (Vincenzi et al., 2013). In contrast to CB1 receptor agonists, CB2 receptor agonists do not have unwanted central side effects and appear to be protective in most of the diabetic complications. However, CB2 receptor agonism has been reported to have deleterious effects on metabolism (Deveaux et al., 2009; Agudo et al., 2010), which is still a matter of debate and requires further clarification. Furthermore, positive results in animals do not imply efficacy in humans, as some mixed CB1/CB2 receptor agonists have so far performed poorly in patients, despite efficacy in rodents (in part because of the metabolic and cardiovascular adverse effects attributable to CB1 receptor stimulation). A clinical trial performed in 30 patients with painful DNR randomized to either Sativex, containing both THC and cannabidiol, or placebo, has failed to show any benefit of Sativex (Selvarajah et al., 2010), although depression was a major confounding factor during the study.
Besides the potential importance of the ECS as a therapeutic target in painful DNR, there is also evidence for its potential role in the pathogenesis of DNR, although the data are often conflicting. Expression of CB1 receptors was found to be reduced in dorsal root ganglia of diabetic rats and CB1 receptor activation attenuated neural damage and normalized neurite outgrowth in cells exposed to a high glucose milieu (Zhang et al., 2009). On the other hand, in vivo studies suggest that inhibition rather than activation of CB1 receptors may be beneficial. In STZ‐induced diabetes, treatment with rimonabant partially prevented loss of intraepidermal nerve fibre density and increased current perception threshold. These effects were paralleled by reduced skin capillary loss, increased blood flow and diminished tissue TNF‐α levels, suggesting that the observed effects may be related to the anti‐inflammatory and vasoprotective properties of rimonabant (Liu et al., 2010). Furthermore, in diabetic mice, rimonabant attenuated mechanical allodynia, reduced oxidative stress in peripheral nerves, inhibited TNF‐α overexpression in the spinal cord and moderated nerve growth factor deficiency, suggesting that CB1 receptor blockade interferes with mechanisms leading to nerve injury and favours nerve regeneration. Accordingly, the histological analysis of sciatic nerves showed a marked degeneration of myelinated fibres in diabetic mice that were reduced by rimonabant treatment (Comelli et al., 2010).
Taken together, the studies summarized earlier suggest that CB1 receptor signalling enhances the inflammatory and oxidative processes leading to both neuronal and microvessel damage, in addition to having some neuroprotective and antinociceptive properties. Therefore, CB1 receptor effects may vary substantially in different experimental settings and species, which may underlie the conflicting data. Further research is required to reconcile controversies and to establish whether and what type of modulation of ECS activity is a feasible therapeutic strategy in DNR.
Diabetic cardiomyopathy
Both major cannabinoid receptors as well as EC synthetic and metabolizing enzymes are expressed in the myocardium and vasculature. Based on preclinical studies, under normal physiological conditions, the ECS appears to play only a very limited, if any, role in cardiovascular regulation. However, it emerges as an important player in triggering or promoting disease pathology/progression in cardiovascular disease (Pacher et al., 2006). Similarly to the DN discussed earlier, it appears that activation of CB1 and CB2 receptors has opposing consequences in various major cardiovascular pathologies. ECs acting via CB1 receptors generally promote hypotension, bradycardia and negative inotropy via receptors located on sympathetic and parasympathetic nerve terminals, cardiomyocytes and endothelial cells (Pacher et al., 2006). In addition, ECs through CB1 receptor‐dependent/independent pathways may also promote ROS generation and activation of pro‐apoptotic stress signalling pathways (e.g. p38 and JNK MAPKs) in murine and human cardiomyocytes, endothelial and smooth muscle cells, and promote profibrotic signalling in fibroblasts/myofibroblasts (Mukhopadhyay et al., 2010c; Rajesh et al., 2010; 2012; Tiyerili et al., 2010). Emerging evidence also suggests that EC activation of CB1 receptors promotes pro‐inflammatory signalling in macrophages and enhances recruitment of various inflammatory cells to the site of insult, facilitating cardiovascular inflammation, vascular or myocardial remodelling and tissue injury (Steffens and Pacher, 2015). In agreement with this, ECs and CB1 receptors have been implicated in the pathogenesis of cardiac dysfunction, cell death and inflammation in various forms of shock, heart failure and atherosclerosis (Pacher et al., 2006). In contrast, activation of CB2 receptors in immune cells attenuates chemotaxis, adhesion of inflammatory cells to the activated endothelium and activation of these immune cells. CB2 receptor activation also attenuates endothelial cell activation and pro‐inflammatory response, decreases smooth muscle proliferation and may exert protective effects in cardiomyocytes (Steffens and Pacher, 2012). These effects are responsible for the benefits of CB2 receptor agonists reported in myocardial, cerebral and other models of ischaemic/reperfusion injury (Pacher and Hasko, 2008). However, the role of CB2 receptors in cardiomyocytes requires additional confirmation in light of concerns with the specificity of the commercially available CB2 receptor antibodies (Steffens and Pacher, 2012). ECs may also exert numerous CB1/CB2 receptor‐independent effects (e.g. vasodilation/vasoconstriction, anti‐inflammatory/pro‐inflammatory, etc.) in the cardiovascular or other organ systems via degradation to arachidonic acid metabolites or through putative novel cannabinoid or other (e.g. TRPV1) receptors depending on the context and concentration/dose used (Pacher and Kunos, 2013; Stanley and O'Sullivan, 2014).
Although diabetes is a well‐recognized risk factor for cardiovascular disease and heart failure, the mechanisms of the development and progression of diabetic cardiomyopathy, which involve complex interplay of oxidative/nitrative stress with metabolic, pro‐inflammatory and cell death pathways, are still not completely understood (Varga et al., 2015).
Using a mouse model of type 1 diabetic cardiomyopathy, Rajesh et al. (2012) investigated the role of EC‐CB1 receptor signalling in myocardial dysfunction, inflammation, remodelling and cell death. They found increased levels of AEA and increased CB1 receptor expression in diabetic hearts, accompanied by enhanced accumulation of advanced glycation end products (AGEs), oxidative/nitrative stress, inflammation, cell death and fibrosis. This also paralleled with enhanced angiotensin II type 1(AT1) receptors, p47(phox) NADPH oxidase signalling, β‐myosin heavy chain isozyme switch, decreased expression of sarcoplasmic/endoplasmic reticulum Ca2+‐ATPase and both diastolic and systolic cardiac dysfunction (Rajesh et al., 2012). These pathological processes were markedly attenuated by CB1 receptor blockade with globally acting CB1 receptor antagonists or by genetic deletion of CB1 receptors. These effects were glucose‐independent, as inhibition of CB1 receptors had no effect on the elevated blood glucose levels following destruction of pancreatic beta cells by multiple injections of streptozotocin, yet CB1 receptor blockade not only prevented but also reversed the pathological remodelling and diabetic cardiac dysfunction in this type I diabetes model. In db/db mice, chronic CB1 receptor inhibition attenuated myocardial fibrosis and remodelling, similar to its earlier described beneficial effects in DN (Nam et al., 2012). CB1 receptor inhibition also improved cardiac function and remodelling after experimental myocardial infarction and metabolic syndrome by mechanisms similar to those described earlier (Slavic et al., 2013). Furthermore, acute and chronic systemic CB1 receptor blockade improved BP regulation and metabolic profile in an angiotensin II‐dependent hypertensive(mRen2)27 rat model (Schaich et al., 2014).
Supporting the pathological function of an overactive ECS in cardiometabolic diseases, increased plasma levels of AEA and 2‐AG were strongly correlated with adverse coronary circulatory events or impaired coronary endothelial function in human obese subjects (Quercioli et al., 2011; Pacher and Kunos, 2013). These studies even suggested that plasma EC levels be considered as biomarkers of cardiovascular risk in obese populations.
Collectively, the earlier studies strongly suggest that activation of CB1 receptors by ECs contributes to the pathogenesis of diabetic cardiovascular dysfunction by facilitating AT1 receptor expression/AT1 receptor‐NADPH oxidase‐ROS signalling, MAPK activation, AGE accumulation, oxidative/nitrative stress, inflammation and fibrosis (Figure 3) .
Figure 3.
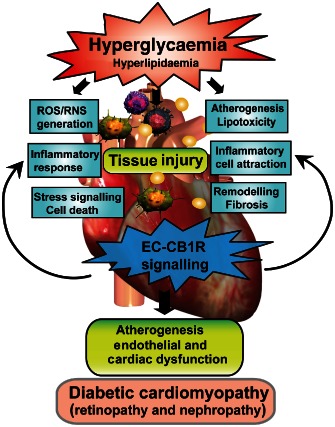
Role of the EC‐CB1 receptor signalling in diabetic cardiovascular complications. Hyperglycaemia and hyperlipidaemia associated with diabetes promotes increased ROS/RNS generation in endothelium, vascular smooth muscle and cardiomyocytes, induces stress signalling, profibrotic changes and cell death in the myocardial cells, as well as leads to activation and recruitment of inflammatory cells with consequent pro‐inflammatory response. Hyperglycaemia also directly or indirectly leads to enhanced EC‐CB1 receptor signalling, which in turn amplifies these pathological processes facilitating tissue injury, cardiovascular dysfunction and eventually development of diabetic cardiovascular complications such as cardiomyopathy, nephropathy, retinopathy and enhanced atherosclerosis.
Diabetic retinopathy
The previously mentioned mechanisms are also critical in the development of other microvascular complications of diabetes (e.g. DN (discussed in the earlier parts) and retinopathy (El‐Remessy et al., 2011; Horváth et al., 2012), as indicated by the beneficial effects of CB1 receptor inhibition or genetic deletion (El‐Remessy et al., 2011). CB1 receptor inhibition limits the vascular inflammation and cell death in a mouse model of diabetic retinopathy and in a human retinal cell line exposed to high glucose (El‐Remessy et al., 2011) and attenuates hyperglycaemia‐induced apoptosis in retinal pigment epithelial cells (Horváth et al., 2012). The role of CB2 receptors in diabetic retinopathy is still unexplored.
Thus, inhibition of peripheral CB1 receptors with a new generation of peripherally restricted antagonists/inverse agonists may represent a promising strategy in the treatment of diabetic cardiovascular complications, including retinopathy and nephropathy.
Cannabidiol for diabetes and diabetic complications
Numerous experimental studies have also demonstrated beneficial effects of cannabidiol, which does not interact with classical cannabinoid receptors in vivo, in primary diabetes and various diabetic complications, including retinopathy, cardiomyopathy and neuropathy (reviewed in Horváth et al., 2012), the detailed discussion of which is beyond the scope of this paper. In these studies, the beneficial effects of cannabidiol were largely attributed to its antioxidant, anti‐inflammatory and tissue protective effects (Horvath and Pacher, 2012). A recent study also demonstrated that cannabidiol improved mitochondrial function and biogenesis in a myocardial injury model (Hao et al., 2015), which could also contribute to its beneficial properties observed in diabetes and diabetic complications. In light of these preclinical data and recent orphan drug approval of cannabidiol by the FDA for the treatment of refractory childhood epilepsy and glioblastoma, there is a strong rationale to explore its therapeutic potential in human diabetes and diabetic complications.
Conclusion and perspectives
CB1 receptor blockade is beneficial in animal models of obesity and metabolic syndrome, and these findings have been confirmed in humans. Furthermore, recent preclinical studies suggest that ‘peripherally restricted’ CB1 receptor antagonists may represent a novel therapeutic strategy to minimize or avoid neuropsychiatric liability while retaining metabolic efficacy in obesity, insulin resistance and beta cell loss. These new compounds deserve further development and clinical testing as they might have a significant clinical impact. Alternative strategies to counteract EC overactivity would be to develop drugs that lower EC levels through modulating their biosynthesis and/or degradation, or to develop dietary interventions that would lower the abundance of EC precursors. Future studies will clarify if these new approaches are feasible.
Cannabinoid‐based therapies may also protect against diabetic complications. The opposing effects of CB1 and CB2 receptors on inflammation, oxidative stress and fibrogenesis probably explain the beneficial effects of CB1 receptor blockade and CB2 receptor activation in the setting of diabetic complications. Although data on the functional consequences of CB1 receptor gene polymorphism are still lacking, an association between a common CB1 receptor polymorphism and the presence of both nephropathy and retinopathy has been recently reported in T2DM patients (Buraczynska et al., 2014). Thus, second‐generation CB1 receptor antagonists may have promise in the treatment of diabetic complications.
Regarding the therapeutic potential of CB2 receptor agonists, it is important to emphasize that their effect on worsening insulin resistance, if confirmed by other studies using more specific ligands, may hamper their use in the treatment of T2DM complications. It is important to note that the CB2 receptor agonists used in studies so far may not have been entirely specific, particularly at high doses, and may have induced unwanted, CB1 receptor‐mediated effects (Pacher and Mechoulam, 2011). Therefore, it is very important to develop more selective CB2 receptor agonists.
In conclusion, modulation of the ECS in diabetes and diabetic complications with peripherally restricted synthetic CB1 receptor antagonists and/or CB2 receptor agonists holds therapeutic promise. Furthermore, marijuana‐derived constituents, such as cannabidiol, may also have therapeutic potential in diabetes and diabetic complications (Horvath and Pacher, 2012).
Author contributions
All authors contributed to writing and editing the manuscript.
Conflict of interest
Authors declare no conflict of interest.
Acknowledgements
Research in the authors' laboratory was supported by European Federation for the Study of Diabetes, Società Italiana di Diabetologia, Piedmont Region and Intramural Research Program of NIAAA. We apologize to all the investigators whose important works have not been cited due to space restrictions. P. P. dedicates this review to a friend/collaborator Itai Bab.
Gruden, G. , Barutta, F. , Kunos, G. , and Pacher, P. (2016) Role of the endocannabinoid system in diabetes and diabetic complications. Br J Pharmacol, 173: 1116–1127. doi: 10.1111/bph.13226.
References
- Agarwal N, Pacher P, Tegeder I, Amaya F, Constantin CE, Brenner GJ et al (2007). Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nat Neurosci 10: 870–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agudo J, Martin M, Roca C, Molas M, Bura AS, Zimmer A et al (2010). Deficiency of CB2 cannabinoid receptor in mice improves insulin sensitivity but increases food intake and obesity with age. Diabetologia 53: 2629–2640. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013a). The Concise Guide to PHARMACOLOGY 2013/14: G protein‐coupled receptors. Br J Pharmacol 170: 1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013b). The Concise Guide to PHARMACOLOGY 2013/14: catalytic receptors. Br J Pharmacol 170: 1676–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013c). The Concise Guide to PHARMACOLOGY 2013/14: transporters. Br J Pharmacol 170: 1706–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013d). The Concise Guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barutta F, Corbelli A, Mastrocola R, Gambino R, Di Marzo V, Pinach S et al (2010). Cannabinoid receptor 1 blockade ameliorates albuminuria in experimental diabetic nephropathy. Diabetes 59: 1046–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barutta F, Piscitelli F, Pinach S, Bruno G, Gambino R, Rastaldi MP et al (2011). Protective role of cannabinoid receptor type 2 in a mouse model of diabetic nephropathy. Diabetes 60: 2386–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barutta F, Grimaldi S, Franco I, Bellini S, Gambino R, Pinach S et al (2014). Deficiency of cannabinoid receptor of type 2 worsens renal functional and structural abnormalities in streptozotocin‐induced diabetic mice. Kidney Int 86: 979–990. [DOI] [PubMed] [Google Scholar]
- Blüher M, Engeli S, Klöting N, Berndt J, Fasshauer M, Bátkai S et al (2006). Dysregulation of the peripheral and adipose tissue endocannabinoid system in human abdominal obesity. Diabetes 55: 3053–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon MR, Kooijman S, van Dam AD, Pelgrom LR, Berbée JF, Visseren CA et al (2014). Peripheral cannabinoid 1 receptor blockade activates brown adipose tissue and diminishes dyslipidemia and obesity. FASEB J 28: 5361–5375. [DOI] [PubMed] [Google Scholar]
- Buraczynska M, Wacinski P, Zukowski P, Dragan M, Ksiazek A (2014). Common polymorphism in the cannabinoid type 1 receptor gene (CNR1) is associated with microvascular complications in type 2 diabetes. J Diabetes Complications 28: 35–39. [DOI] [PubMed] [Google Scholar]
- Christensen R, Kristensen PK, Bartels EM, Bliddal H, Astrup A (2007). Efficacy and safety of the weight‐loss drug rimonabant: a meta‐analysis of randomised trials. Lancet 370: 1706–1713. [DOI] [PubMed] [Google Scholar]
- Cinar R, Godlewski G, Liu J, Tam J, Jourdan T, Mukhopadhyay B et al (2014). Hepatic cannabinoid‐1 receptors mediate diet‐induced insulin resistance by increasing de novo synthesis of long‐chain ceramides. Hepatology 59: 143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comelli F, Bettoni I, Colombo A, Fumagalli P, Giagnoni G, Costa B (2010). Rimonabant, a cannabinoid CB1 receptor antagonist, attenuates mechanical allodynia and counteracts oxidative stress and nerve growth factor deficit in diabetic mice. Eur J Pharmacol 637: 62–69. [DOI] [PubMed] [Google Scholar]
- Deveaux V, Cadoudal T, Ichigotani Y, Teixeira‐Clerc F, Louvet A, Manin S et al (2009). Cannabinoid CB2 receptor potentiates obesity‐associated inflammation, insulin resistance and hepatic steatosis. PLoS ONE 4: e5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V (2012). De‐liver‐ance’ from CB(1): a way to counteract insulin resistance? Gastroenterology 142: 1063–1066. [DOI] [PubMed] [Google Scholar]
- El‐Remessy AB, Rajesh M, Mukhopadhyay P, Horvath B, Patel V, Al‐Gayyar MM et al (2011). Cannabinoid 1 receptor activation contributes to vascular inflammation and cell death in a mouse model of diabetic retinopathy and a human retinal cell line. Diabetologia 54: 1567–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes JM, Cooper ME (2013). Mechanisms of diabetic complications. Physiol Rev 93: 137–188. [DOI] [PubMed] [Google Scholar]
- Ge Q, Maury E, Rycken L, Gérard J, Noël L, Detry R et al (2013). Endocannabinoids regulate adipokine production and the immune balance of omental adipose tissue in human obesity. Int J Obes (Lond) 37: 874–880. [DOI] [PubMed] [Google Scholar]
- Giunti S, Barutta F, Cavallo Perin P, Gruden G (2010). Targeting the MCP‐1/CCR2 system in diabetic kidney disease. Curr Vasc Pharmacol 8: 849–860. [DOI] [PubMed] [Google Scholar]
- Hao E, Mukhopadhyay P, Cao Z, Erdelyi K, Holocav E (2015). Cannabidiol protects against doxorubicin‐induced cardiomyopathy by modulating mitochondrial function and biogenesis. Mol Med 21: 38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horváth B, Mukhopadhyay P, Haskó G, Pacher P (2012). The endocannabinoid system and plant‐derived cannabinoids in diabetes and diabetic complications. Am J Pathol 180: 432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett AC, Blume LC, Dalton GD (2010). CB(1) cannabinoid receptors and their associated proteins. Curr Med Chem 17: 1382–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Diabetes Federation (2014). Diabetes‐Atlas [Online]. Available at: http://www.idf.org/diabetesatlas (accessed 5/4/2015).
- Janiak P, Poirier B, Bidouard JP, Cadrouvele C, Pierre F, Gouraud L et al (2007). Blockade of cannabinoid CB1 receptor improves renal function, metabolic profile, and increased survival of obese Zucker rats. Kidney Int 72: 1345–1357. [DOI] [PubMed] [Google Scholar]
- Jenkin KA, Verty AN, McAinch AJ, Hryciw DH (2012). Endocannabinoids and the renal proximal tubule: an emerging role in diabetic nephropathy. Int J Biochem Cell Biol 44: 2028–2031. [DOI] [PubMed] [Google Scholar]
- Jenkin KA, McAinch AJ, Briffa JF, Zhang Y, Kelly DJ, Pollock CA et al (2013). Cannabinoid receptor 2 expression in human proximal tubule cells is regulated by albumin independent of ERK1/2 signaling. Cell Physiol Biochem 32: 1309–1319. [DOI] [PubMed] [Google Scholar]
- Jourdan T, Godlewski G, Cinar R, Bertola A, Szanda G, Liu J et al (2013). Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates beta cell loss in type 2 diabetes. Nat Med 19: 1132–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdan T, Szanda G, Rosenberg A, Tam J, Earley B, Godlewski G et al (2014). Overactive cannabinoid 1 receptor in podocytes drives type‐2 diabetic nephropathy. Proc Natl Acad Sci U S A 111: E5420–E5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Lao Q, Shin YK, Carlson OD, Lee EK, Gorospe M et al (2012). Cannabinoids induce pancreatic β‐cell death by directly inhibiting insulin receptor activation. Sci Signal 5: ra23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunos G, Tam J (2011). The case for peripheral CB1 receptor blockade in the treatment of visceral obesity and its cardiometabolic complications. Br J Pharmacol 163: 1423–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JC, Lim SK, Park MJ, Kim GY, Han HJ, Park SH (2011). Cannabinoid receptor 1 mediates high glucose‐induced apoptosis via endoplasmic reticulum stress in primary cultured rat mesangial cells. Am J Physiol Renal Physiol 301: F179–F188. [DOI] [PubMed] [Google Scholar]
- Liu J, Zhou L, Xiong K, Godlewski G, Mukhopadhyay B, Tam J et al (2012). Hepatic cannabinoid receptor‐1 mediates diet‐induced insulin resistance via inhibition of insulin signaling and clearance in mice. Gastroenterology 142: 1218–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu WJ, Jin HY, Park JH, Baek HS, Park TS (2010). Effect of rimonabant, the cannabinoid CB1 receptor antagonist, on peripheral nerve in streptozotocin‐induced diabetic rat. Eur J Pharmacol 637: 70–76. [DOI] [PubMed] [Google Scholar]
- Malenczyk K, Jazurek M, Keimpema E, Silvestri C, Janikiewicz J, Mackie K et al (2013). CB1 cannabinoid receptors couple to focal adhesion kinase to control insulin release. J Biol Chem 288: 32685–32699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecucco F, Burger F, Mach F, Steffens S (2008). CB2 cannabinoid receptor agonist JWH‐015 modulates human monocyte migration through defined intracellular signaling pathways. Am J Physiol Heart Circ Physiol 294: H1145–H1155. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay P, Pan H, Rajesh M, Bátkai S, Patel V, Harvey‐White J et al (2010a). CB1 cannabinoid receptors promote oxidative/nitrosative stress, inflammation and cell death in a murine nephropathy model. Br J Pharmacol 160: 657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Rajesh M, Pan H, Patel V, Mukhopadhyay B, Bátkai S et al (2010b). Cannabinoid‐2 receptor limits inflammation, oxidative/nitrosative stress, and cell death in nephropathy. Free Radic Biol Med 48: 457–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Rajesh M, Batkai S, Patel V, Kashiwaya Y, Liaudet L et al (2010c). CB1 cannabinoid receptors promote oxidative stress and cell death in murine models of doxorubicin‐induced cardiomyopathy and in human cardiomyocytes. Cardiovasc Res 85: 773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murumalla R, Bencharif K, Gence L, Bhattacharya A, Tallet F, Gonthier MP et al (2011). Effect of the cannabinoid receptor‐1 antagonist SR141716A on human adipocyte inflammatory profile and differentiation. J Inflamm (Lond) 8: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam DH, Lee MH, Kim JE, Song HK, Kang YS, Lee JE et al (2012). Blockade of cannabinoid receptor 1 improves insulin resistance, lipid metabolism, and diabetic nephropathy in db/db mice. Endocrinology 153: 1387–1396. [DOI] [PubMed] [Google Scholar]
- O'Hare JD, Zielinski E, Cheng B, Scherer T, Buettner C (2011). Central endocannabinoid signaling regulates hepatic glucose production and systemic lipolysis. Diabetes 60: 1055–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Hasko G (2008). Endocannabinoids and cannabinoid receptors in ischaemia–reperfusion injury and preconditioning. Br J Pharmacol 160: 688–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Kunos G (2013). Modulating the endocannabinoid system in human health and disease‐successes and failures. FEBS J 280: 1918–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Mechoulam R (2011). Is lipid signaling through cannabinoid 2 receptors part of a protective system? Prog Lipid Res 50: 193–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Batkai S, Kunos G (2006). The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev 58: 389–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al; NC‐IUPHAR (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledgebase of drug targets and their ligands. Nucl. Acids Res. 42 (Database Issue): D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltier A, Goutman SA, Callaghan BC (2014). Painful diabetic neuropathy. BMJ 348: g1799. [DOI] [PubMed] [Google Scholar]
- Quercioli A, Pataky Z, Vincenti G, Makoundou V, Di Marzo V, Montecucco F et al (2011). Elevated endocannabinoid plasma levels are associated with coronary circulatory dysfunction in obesity. Eur Heart J 32: 1369–1378. [DOI] [PubMed] [Google Scholar]
- Rajesh M, Mukhopadhyay P, Hasko G, Liaudet L, Mackie K, Pacher P (2010). Cannabinoid‐1 receptor activation induces reactive oxygen species‐dependent and ‐independent mitogen‐activated protein kinase activation and cell death in human coronary artery endothelial cells. Br J Pharmacol 160: 688–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajesh M, Batkai S, Kechrid M, Mukhopadhyay P, Lee WS, Horvath B et al (2012). Cannabinoid 1 receptor promotes cardiac dysfunction, oxidative stress, inflammation, and fibrosis in diabetic cardiomyopathy. Diabetes 61: 716–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrbach K, Thomas MA, Glick S, Fung EN, Wang V, Watson L et al (2012). Ibipinabant attenuates β‐cell loss in male Zucker diabetic fatty rats independently of its effects on body weight. Diabetes Obes Metab 14: 555–564. [DOI] [PubMed] [Google Scholar]
- Romero‐Zerbo SY, Garcia‐Gutierrez MS, Suarez J, Rivera P, Ruz‐Maldonado I, Vida M et al (2012). Overexpression of cannabinoid CB2 receptor in the brain induces hyperglycaemia and a lean phenotype in adult mice. J Neuroendocrinol 24: 1106–1119. [DOI] [PubMed] [Google Scholar]
- Russell JC, Kelly SE, Diane A, Wang Y, Mangat R, Novak S et al (2010). Rimonabant‐mediated changes in intestinal lipid metabolism and improved renal vascular dysfunction in the JCR : LA‐cp rat model of prediabetic metabolic syndrome. Am J Physiol Gastrointest Liver Physiol 299: G507–G516. [DOI] [PubMed] [Google Scholar]
- Schaich CL, Shaltout HA, Brosnihan KB, Howlett AC, Diz DI (2014). Acute and chronic systemic CB1 cannabinoid receptor blockade improves blood pressure regulation and metabolic profile in hypertensive (mRen2)27 rats. Physiol Rep 2(8). pii: e12108. doi: 10.14814/phy2.12108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvarajah D, Gandhi R, Emery CJ, Tesfaye S (2010). Randomized placebo‐controlled double‐blind clinical trial of cannabis‐based medicinal product (Sativex) in painful diabetic neuropathy: depression is a major confounding factor. Diabetes Care 33: 128–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestri C, Di Marzo V (2013). The endocannabinoid system in energy homeostasis and the etiopathology of metabolic disorders. Cell Metab 17: 475–490. [DOI] [PubMed] [Google Scholar]
- Silvestri C, Ligresti A, Di Marzo V (2011). Peripheral effects of the endocannabinoid system in energy homeostasis: adipose tissue, liver and skeletal muscle. Rev Endocr Metab Disord 12: 153–162. [DOI] [PubMed] [Google Scholar]
- Slavic S, Lauer D, Sommerfeld M, Kemnitz UR, Grzesiak A, Trappiel M et al (2013). Cannabinoid receptor 1 inhibition improves cardiac function and remodelling after myocardial infarction and in experimental metabolic syndrome. J Mol Med 91: 811–823. [DOI] [PubMed] [Google Scholar]
- Stanley C, O'Sullivan SE (2014). Vascular targets for cannabinoids: animal and human studies. Br J Pharmacol 171: 1361–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffens S, Pacher P (2012). Targeting cannabinoid receptor CB(2) in cardiovascular disorders: promises and controversies. Br J Pharmacol 167: 313–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffens S, Pacher P (2015). The activated endocannabinoid system in atherosclerosis: driving force or protective mechanism? Curr Drug Targets 16: 334–341. [DOI] [PubMed] [Google Scholar]
- Tam J, Vemuri VK, Liu J, Bátkai S, Mukhopadhyay B, Godlewski G et al (2010). Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J Clin Invest 120: 2953–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam J, Cinar R, Liu J, Godlewski G, Wesley D, Jourdan T et al (2012). Peripheral cannabinoid‐1 receptor inverse agonism reduces obesity by reversing leptin resistance. Cell Metab 16: 167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarabra E, Giunti S, Barutta F, Salvidio G, Burt D, Deferrari G et al (2009). Effect of the monocyte chemoattractant protein‐1/CC chemokine receptor 2 system on nephrin expression in streptozotocin‐treated mice and human cultured podocytes. Diabetes 58: 2109–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedesco L, Valerio A, Dossena M, Cardile A, Ragni M, Pagano C et al (2010). Cannabinoid receptor stimulation impairs mitochondrial biogenesis in mouse white adipose tissue, muscle, and liver: the role of eNOS, p38 MAPK, and AMPK pathways. Diabetes 59: 2826–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiyerili V, Zimmer S, Jung S, Wassmann K, Naehle CP, Lutjohann D et al (2010). CB1 receptor inhibition leads to decreased vascular AT1 receptor expression, inhibition of oxidative stress and improved endothelial function. Basic Res Cardiol 105: 465–477. [DOI] [PubMed] [Google Scholar]
- Varga ZV, Giricz Z, Liaudet L, Hasko G, Ferdinandy P, Pacher P (2015). Interplay of oxidative, nitrosative/nitrative stress, inflammation, cell death and autophagy in diabetic cardiomyopathy. Biochim Biophys Acta 1852: 232–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera G, López‐Miranda V, Herradón E, Martín MI, Abalo R (2012). Characterization of cannabinoid‐induced relief of neuropathic pain in rat models of type 1 and type 2 diabetes. Pharmacol Biochem Behav 102: 335–343. [DOI] [PubMed] [Google Scholar]
- Vincenzi F, Targa M, Corciulo C, Tabrizi MA, Merighi S, Gessi S et al (2013). Antinociceptive effects of the selective CB2 agonist MT178 in inflammatory and chronic rodent pain models. Pain 154: 864–873. [DOI] [PubMed] [Google Scholar]
- Zhang F, Challapalli SC, Smith PJ (2009). Cannabinoid CB(1) receptor activation stimulates neurite outgrowth and inhibits capsaicin‐induced Ca(2) influx in an in vitro model of diabetic neuropathy. Neuropharmacology 57: 88–89. [DOI] [PubMed] [Google Scholar]