

Abstract
The Gfo/Idh/MocA protein family contains a number of different proteins, which almost exclusively consist of NAD(P)‐dependent oxidoreductases that have a diverse set of substrates, typically pyranoses. In this study, to clarify common structural features that would contribute to their function, the available crystal structures of the members of this family have been analyzed. Despite a very low sequence identity, the central features of the three‐dimensional structures of the proteins are surprisingly similar. The members of the protein family have a two‐domain structure consisting of a N‐terminal nucleotide‐binding domain and a C‐terminal α/β‐domain. The C‐terminal domain contributes to the substrate binding and catalysis, and contains a βα‐motif with a central α‐helix carrying common essential amino acid residues. The β‐sheet of the α/β‐domain contributes to the oligomerization in most of the proteins in the family.
Keywords: NAD(P), oxidoreductase, Gfo/Idh/MocA, pyranose
Abbreviations
- AAOR
aldose‐aldose oxidoreductase
- AFR
1,5‐anhydro‐d‐fructose reductase
- A‐zyme
α‐N‐acetylgalactosidase
- BVR
biliverdin reductase
- DD
dihydrodiol dehydrogenase
- G6PD
glucose‐6‐phosphate dehydrogenase
- Gal80p
transcriptional inhibitor Gal80p
- Gfo
GFOR, glucose‐fructose oxido‐reductase
- GFOR
glucose‐fructose oxidoreductase
- IDH
inositol dehydrogenase
- KijD10
C‐3″‐ketoreductase
- MocA
rhizopine catabolism protein MocA
- PDB
protein data bank
- RMSD
root‐mean‐square deviation
- WlbA
uridine‐5′‐phosphate‐2,3‐diacetamido‐2,3‐dideoxy‐d‐mannuronic acid dehydrogenase.
Introduction
Recently, there have been a number of publications describing crystal structures of NAD(P)‐dependent oxidoreductases (or dehydrogenases) belonging to the Gfo/Idh/MocA protein family. The abbreviations Gfo, Idh, and MocA refer to glucose‐fructose oxidoreductase, inositol 2‐dehydrogenase, and the rhizopine catabolism protein MocA, which could catalyze dehydrogenase reaction involved in rhizopine catabolism. In 1994, the first protein structure of the Gfo/Idh/MocA family was described when Rowland et al. solved the structure of glucose‐6‐phosphate dehydrogenase (G6PD).1 The earliest mention of Gfo/Idh/MocA family we discovered dates back to 2001 in a publication describing the sequencing of the whole genome of a virulent isolate of Streptococcus pneumonia.2 A typical protein structure in this family consists of two main domains: a N‐terminal dinucleotide‐binding domain containing a typical Rossmann fold3 and a C‐terminal α/β‐domain participating in substrate binding and oligomerization.
This structural family contains enzymes that catalyze various different chemical reactions such as oxidation and reduction of carbohydrates,4, 5, 6, 7, 8, 9 oxidation of trans‐dihydrodiols,10 reduction of biliverdin,11 and hydrolyzation of glycosidic bonds.12 All the enzymes in this family utilize NAD(P) as a hydride donor or acceptor. Moreover, structurally, the transcriptional repressor Gal80p is a member of the family, although it does not have enzymatic activity.13 It is quite intriguing that even though the sequence identity between most of the proteins in the Gfo/Idh/MocA family seems very low (under 20%) and the extent of different functions is large, the overall three‐dimensional folds are similar. We analyzed the structural and functional features of the proteins in the Gfo/Idh/MocA family to get an insight about the central features of this two‐domain protein scaffold, which is utilized in the catalysis of a diverse set of different substrates.
Members of the Gfo/Idh/MocA Protein Family
We found 89 coordinate entries for structures belonging to the Gfo/Idh/MocA family using the literature, structure, and sequence searches within the worldwide Protein Data Bank (PDB)14 as well as DALI‐server.15 The searches were performed using the sequences of aldose‐aldose oxidoreductase (AAOR) and glucose‐fructose oxidoreductase (GFOR), and also the protein family name as search items. The number of studied proteins was reduced by excluding similar enzymes from different sources and structures without a defined function (no publication available). The final analysis contained 11 different proteins: 1) G6PD from Leuconostoc mesenteroides,1 2) GFOR from Zymomonas mobilis,5, 16, 17 3) biliverdin reductase (BVR) from Rattus norwegicus,11, 18 4) 1,5‐anhydro‐D‐fructose reductase (AFR) from Sinorhizobium meliloti,6 5) α‐N‐acetylgalactosidase (A‐zyme) from Elizabethkingia meningosepticum,12 6) transcriptional inhibitor Gal80p from Kluyveromyces lactis,13 7) dihydrodiol dehydrogenase (DD) from Macaca fascicularis,10, 19 8) uridine‐5′‐phosphate‐2,3‐diacetamido‐2,3‐dideoxy‐D‐mannuronic acid dehydrogenase (WlbA) from Thermus thermophiles,7 9) myo‐inositol dehydrogenase (IDH) from Bacillus subtilis,8, 20 10) C‐3″‐ketoreductase (KijD10) from Actinomadura kijaniata,9 and 11) the novel AAOR from Caulobacter crescentus.4 The structures are listed in Table 1 with their PDB IDs, EC numbers (concluded from the catalyzed reaction or published in the article related to the structure), catalytic activity, number of amino acid residues, quaternary structures, utilized cofactors, and the calculated root‐mean square deviations (RMSD) superimposing the secondary structures to Cc AAOR. The sequence identities between the proteins were generally low, under 20%, in spite of many common structural features, such as a NAD(P) binding site. The higher sequence identities existed between AAOR and GFOR (49%), AAOR and AFR (26%), and AFR and Kijd10 (24%).
Table 1.
Crystal Structures Used in the Comparison
| Protein | Organism | Abbreviation | PDB ID | EC | Catalytic activity | Amino acids | Quaternary structure | Cofactor | RMSD Å1 | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| Aldose‐aldose oxidoreductase | C. crescentus | AAOR | 5A02, 5A03, 5A04, 5A05, 5A06 | 1.1.99.‐ | Oxidoreductase | 339 | dimer1 | NADP | 0 | 4 |
| α‐N‐Acetylgalactosidase | E. meningosepticum | A‐zyme | 2IXA, 2IXB | 3.2.1.49 | Hydrolase | 444 | dimer1 | NAD | 2.6 | 12 |
| 1,5‐Anhydro‐D‐fructose reductase | S. meliloti | AFR | 4KOA | 1.1.1.263 | Reductase | 332 | dimer1 | NADP | 1.9 | 6 |
| 1.1.1.292 | ||||||||||
| Biliverdin reductase | R. norwegicus | BVR | 1GCU, 1LC0, 1LC3 | 1.3.1.24 | Reductase | 295 | monomer | NAD | 2.4 | 11 |
| C‐3″‐ketoreductase | A. kijaniata | KijD10 | 3RBV, 3RC1, 3RC2, 3RC7, 3RC9, 3RCB | 1.1.1.‐ | Reductase | 350 | tetramer1 | NADP | 2.4 | 9 |
| Dihydrodiol dehydrogenase | M. fascicularis | DD | 2O48, 2O4U, 2POQ, 3OHS | 1.3.1.20 | Dehydrogenase | 334 | dimer1 | NADP | 2.5 | 10 |
| Glucose‐fructose oxidoreductase | Z. mobilis | GFOR | 1OFG, 1EVJ, 1H6A, 1H6B, 1H6C, 1H6D, 1RYD, 1RYE | 1.1.99.28 | Oxidoreductase | 381 | tetramer1 | NADP | 0.9 | 5, 16, 17 |
| Glucose‐6‐phosphate dehydrogenase | L. mesenteroides | G6PD | 1DPG | 1.1.1.49 | Dehydrogenase | 485 | dimer2 | NADP | 3.1 | 1 |
| myo‐inositol dehydrogenase | B. subtilis | myo‐IDH | 3MZ0, 3NT2, 3NT4, 3NT5, 3NTO, 3NTQ, 3NTR, 4L8V, 4L9R | 1.1.1.18 | dehydrogenase | 344 | tetramer1 | NAD/NADP | 2.3 | 8, 20 |
| 1.1.369 | ||||||||||
| Transcriptional repressor Gal80p | K. lactis | Gal80p | 2NVW | – | – | 479 | dimer1 | – | 2.4 | 13 |
| Uridine‐5′‐phosphate‐2,3‐diacetamido‐2,3‐dideoxy‐D‐mannuronic acid dehydrogenase | T. thermophilus | WlbA | 3OA0, 3O9Z | 1.1.1.374 (?) | Dehydrogenase | 318 | tetramer2 | NAD | 2.2 | 7 |
1) RMSD to the refererence protein AAOR.
Topological Conservation
Monomers from every structure were compared by superimposing them according to their secondary structure elements in PyMOL.21 All the structures had two main domains: the N‐terminal domain containing the classical nucleotide‐binding Rossmann fold and the C‐terminal α/β‐domain that participates in substrate binding and frequently also in oligomerization. The different monomers have been presented in Figure 1. Rossmann fold is already extensively characterized.3, 22 The C‐terminal domain consisted of a two‐layer α/β‐sandwich, in which the β‐sheet was predominantly antiparallel, having six to nine β‐strands. Both faces of the β‐sheet were usually hydrophobic. One face participated in the homodimer formation by packing against a similar face of a β‐sheet from the second polypeptide chain around a two‐fold symmetry axis. The other face of the β‐sheet packed against α‐helices of the α/β‐sandwich. These helices were in the N‐ and C‐terminal regions of the α/β‐domain and in the polypeptide chain after the first β‐strand of the β‐sheet of the α/β‐domain. The α‐helices were mainly located between the β‐sheet and the N‐terminal dinucleotide‐binding domain. The orientation of the two domains against each other between the proteins belonging to the family was very similar even though the sequence identities are very low.
Figure 1.
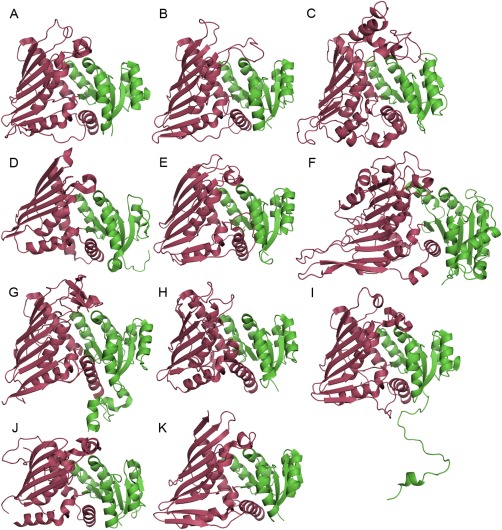
The cartoon representation of monomers of the proteins used in the comparison: (A) AAOR, (B) AFR, (C) A‐zyme, (D) BVR, (E) DD, (F) G6DP, (G) Gal80p, (H) IDH, (I) GFOR, (J) WlbA, (K) KijD10. The N‐terminal nucleotide‐binding domains are in green and the C‐terminal domains in red.
The chosen structures were also compared by inspecting the topology of the proteins. Figure 2 shows the topology diagrams of AAOR and G6PD, which were the most distantly related family members according to the RMS deviations. From the diagrams we can see that the nucleotide‐binding domains are almost identical. The structural differences occur mainly at the N‐terminal α/β‐domain, which is considerably larger in G6PD than in AAOR. Nevertheless, the β‐sheet of the α/β‐domains could be divided into three sections similarly. There were two β‐hairpin‐like motifs consisting of three to four adjacent mainly antiparallel β‐strands at the both ends of the β‐sheet. The first strand of the β‐sheet was located between them. This first strand was parallel to the first motif and antiparallel to the second motif. This first β‐strand was topologically important, because the catalytic residues were located in the conserved helix after this β‐strand. Together these secondary structure elements formed the βα‐motif, a characteristic feature of Gfo/Idh/MocA protein family (Figs. 2 and 3).
Figure 2.
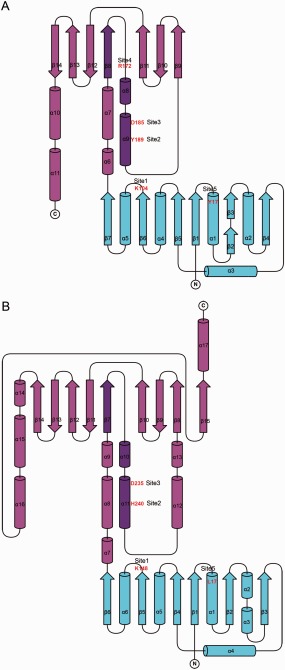
Topology diagrams of two Gfo/Idh/MocA family members (A) AAOR and (B) G6PD. The cofactor‐binding domains are presented in cyan and the C‐terminal α/β‐domains in purple. The central βα‐motifs have been highlighted with a darker color, and the amino acid residues at the key sites in the active sites have been labeled in red. The topology diagrams were drawn with TopDraw27 from the CCP4 Program Suite.28
Figure 3.
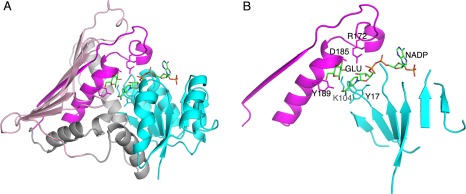
The two‐domain structure of AAOR. (A) The N‐terminal NADP binding domain is in cyan, and the C‐terminal α/β‐domain in grey and pink (the α‐helical part in grey and the β‐sheet part in pink). The first β‐strand and following polypeptide chain containing the short and the long central α‐helices are highlighted in magenta. (B) The simplification of the major structural elements of the Gfo/Idh/MocA protein fold (as in AAOR). NADP is shown as a stick model. The side chains for the residues in the five key sites are shown as stick models. Also a glucose ligand (GLU, a substrate for AAOR) is shown.
The current CATH‐database23 includes half of the proteins used in this study (GFOR, WlbA, BVR, myo‐IDH, Gal80p, and G6PD). The N‐terminal domain was recognized as a Rossmann fold. The C‐terminal domain was included in a large dihydrodipicolinate reductase domain superfamily including rather variable structures. The six Gfo/Idh/MocA family proteins did not form a single structural cluster. Meso‐diaminopimelate dehydrogenase from Symbiobacterium thermophilum 24 was included in the dihydrodipicolinate reductase domain superfamily and could be considered as a member of the Gfo/Idh/MocA protein family, but it had a six‐stranded completely antiparallel β‐sheet without the βα‐motif described before. Furthermore, aspartate dehydrogenase from Thermotoga maritima 25 could be considered a candidate for the Gfo/Idh/MocA family, but it did not have the central α‐helix after the first β‐strand in the α/β‐domain, and the β‐sheet was smaller and only five‐stranded.
Nucleotide Binding
All of the enzymes in the Gfo/Idh/MocA family catalyzed reactions using a nucleotide cofactor. In many of the enzymes, the dissociation of the cofactor was possible, because there were several structures solved without the bound cofactor, but, for example, in AAOR, the cofactor was tightly bound and regenerated in a reaction cycle without any observed cofactor dissociation.26 In every structure where the NAD(P) was bound to the enzyme, it was bound in the expected binding‐site at the C‐terminal edge of the Rossmann fold in a crevice formed between the loops from the β‐strands in which there was a change in the direction of the strand order. The cofactor was a prerequisite for the substrate binding and was the reason all the complex structures available in PDB also included the bound cofactor.
Eight crystal structures included the bound cofactor, and these are presented in Figure 4. Three enzymes, which had NADP as a cofactor (AAOR, GFOR, and KijD10), had a very similar cofactor binding position and conformation in which the adenine group was in the syn‐conformation (only the NADP in AAOR is presented in Fig. 4). Having a bound NAD cofactor, Myo‐IDH, A‐zyme and WlbA had the adenine group of the cofactor in the anti‐conformation (only the NAD in myo‐IDH is presented in Fig. 4). The conformation of NADP in AFR was different. It has a syn‐conformation for the nicotinamide ring, and also the orientation of the adenine group was twisted. Furthermore, the conformation of the NAD bound in BVR was unique forming a kind of curved molecule, also having the nicotinamide ring in syn‐conformation.
Figure 4.
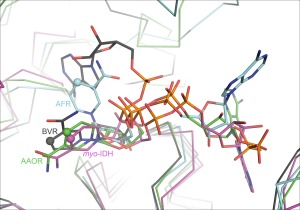
The variation of the bound NAD(P) cofactor in the crystal structures of Gfo/Idh/MocA proteins. Different colors have been used for different cofactor conformations. NADP in AAOR is shown as green stick model, NADP in AFR in cyan, NAD in myo‐IDH, in purple, and NAD in RVB in grey. The C4 in the nicotinamide ring in every cofactor has been highlighted in spherical atom.
Catalyzed Reactions
The enzymes in the Gfo/Idh/MocA family catalyzed various reactions. The cofactor NAD(P)(H) had naturally an essential role either in removing a hydride ion (H‐) from the substrate, which is oxidized (dehydrogenase reaction), or in adding a hydride ion to the substrate, which is subsequently reduced (reductase reaction). The sites in the substrates for the removal or addition of hydrides are shown in Figure 5. The pyranose‐type of carbohydrate is the most frequently utilized substrate. AAOR, GFOR, and GP6D act on the C1 carbon of the substrates. Both AAOR and GFOR are able to oxidize D‐glucose to D‐glucono‐1,5‐lactone but they are also able to reduce open‐chain forms of carbohydrates to sugar alcohols. Especially AAOR catalyzes also the reactions of a panel of different sugars.4, 5 GP6D is more specific, it catalyses oxidation (dehydrogenation) of D‐glucose‐6‐phosphate to 6‐phospho‐D‐glucono‐1,5‐lactone.1 AFR reduces C2 carbon of 1,5‐anhydro‐D‐fructose, the central intermediate of the anhydrofructose pathway.6 The substrate of myo‐IDH is myo‐inositol, but also substituted myo‐inositol derivatives can act as substrates. The enzyme catalyzes the oxidation of C2 carbon, which has an axial hydroxyl group.20 WlbA and KijD10 remove the hydride from the C3 carbon of the substrate. WlbA oxidates UDP‐N‐acetyl‐D‐glucosaminuric acid, an intermediate for the synthesis of bacterial O‐antigens.7 KijD10 catalyzes the reduction of dTDP‐3,‐4‐diketo‐2,6‐dideoxy‐D‐glucose, an intermediate of the biosynthesis of L‐digotoxose.9 A‐zyme catalyzes the hydrolytic removal of the terminal α‐1,3‐linked N‐acetylgalactosamine (GalNac) which determines the blood group A. Although A‐zyme is a glycosidase, the suggested reaction mechanism includes the removal from and the addition of the hydride to the C3 carbon during the catalytic cycle.12 DD catalyzes the oxidation of trans‐dihydrodiols of aromatic hydrocarbons to their corresponding catechols.10 Although the substrate is not a carbohydrate, the size of the substrates of DD is similar to the pyranose rings. Only the substrate for BVR is clearly different to the substrates of the other enzymes belonging to the Gfo/Idh/MocA family. BVR catalyzes the reduction of the γ‐methene bridge of biliverdin.11
Figure 5.
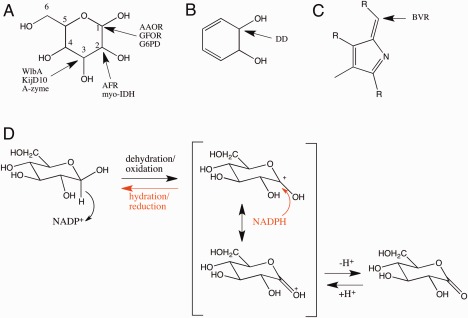
The positions of the substrate carbons in which a hydride is abstracted (oxidation) or added (reduction) in different type of enzymes. (A) pyranose ring, (B) trans‐dihydrodiol, (C) biliverdin. (D) The representative reaction mechanism for D‐glucose oxidation and D‐glucono‐1,5‐lactone reduction. NADP+ or NADPH is a key catalytic part of the enzyme. Depending on the enzyme amino acid residues may participate in the deprotonation/protonation step.
The Active Sites and the Catalytic Amino Acid Residues
The active site residues, especially catalytic residues, could be deduced by using sequence comparison and crystal structures in which the enzyme crystal has been soaked or co‐crystallized with the appropriate ligand. Unfortunately, the available complex structures were limited for the Gfo/Idh/MocA family proteins. Therefore, the identification of the key residues in the active site involved in the substrate binding and catalysis was restricted. However, our analysis indicated the positions for five residues in the active site (sites 1‐5 in Fig. 2) that were frequently located near the substrate binding area (Table 2). The first two residue sites were putative catalytic residues, which could either donate or accept a proton from the substrate. The third site was typically occupied by a negatively charged residue (Asp or Glu), and the fourth site was, in general, a positively charged residue (Lys or Arg). The residues occupying similar positions in the polypeptide chain, but which are probably non‐functional or too distant from the active site in the structure, are in parenthesis. In the table, the fifth residue site was packed against the reactive nicotinamide ring. This residue varied considerably having aliphatic, aromatic or even positively charged side chain.
Table 2.
Key Sites in the Active Site of Different Gfo/Idh/MocA Proteins
| Protein | Abbreviation | Site1 | Site2 | Site3 | Site4 | Site5 |
|---|---|---|---|---|---|---|
| Catalytic | Catalytic | Nicotinamide bond | ||||
| Aldose‐aldose oxidoreductase | AAOR | K104 | Y189 | D185 | R172 | Y17 |
| α‐N‐Acetylgalactosidase | A‐zyme | (V122) | H228 | Y225 | R213 | R31 |
| 1,5‐anhydro‐D‐fructose reductase | AFR | K94 | H180 | D176 | – | I12 |
| C‐3″‐Ketoreductase | KijD10 | K102 | Y186 | D182 | R170 | I20 |
| Glucose‐fructose oxidoreductase | GFOR | K181 | Y269 | D265 | R252 | Y94 |
| Glucose‐6‐phosphate dehydrogenase | G6PD | K148 | H240 | D235 | – | L17 |
| myo‐inositol dehydrogenase | myo‐IDH | K97 | H176 | (D172) | – | I13 |
| Uridine‐5′‐phosphate‐2,3‐diacetamido‐2,3‐dideoxy‐D‐mannuronic acid dehydrogenase | WlbA | K102 | H188 | N184 | K172 | I13 |
The catalytic residue at the site one, which was typically a lysine, was located at the end of a β‐strand (β6 in the topology diagram of AAOR) in the N‐terminal domain. It made a hydrogen bond with the O2′ of the ribose next to the nicotinamide ring of NAD(P) in many crystal structures. Also the residue at site five was located in the N‐terminal domain. Other sites (2, 3, and 4) were all located in the loop and the central α‐helix following the first β‐strand in the C‐terminal α/β‐domain.
Quaternary Structure
In general, the crystal structures of the proteins, belonging to the Gfo/Idh/MocA family, had a quaternary structure (Fig. 6). Only BVR was found to be monomeric. The most common quaternary structure was a dimeric structure found in AAOR, AFR, A‐zyme, DD, and Gal80p in which the flat face of the β‐sheet of the C‐terminal α/β‐domain was packed against a similar β‐sheet from the second monomer [dimer1, Fig. 6(A)]. Similarly, intermolecular β‐sheet packing could also be found in GFOR, KijD10, and myo‐IDH tetramers but, in addition, these β‐sheets were extended to the second monomer forming together a very extensive β‐sheet [tetramer1, Fig. 6(B)]. GFOR contained also a long N‐terminal structurally irregular polypeptide, which wrapped around the adjacent monomer. In G6PD, the β‐sheet of the α/β−domain also participated in the formation of the monomer‐monomer interface, but the packing of the β‐sheets against each other utilized another face of the β‐sheet [dimer2, Fig. 6(C)]. In fact, the C‐terminal polypeptide included a short α‐helix that packed against the flat β‐sheet face preventing the similar packing against the second monomer as in dimer1. Finally, WlbA tetramer [tetramer2, Fig. 6(D)] represented the arrangement in which the β‐sheet of the α/β domain did not play a role in the formation of the tetramer. In this WlbA structure, the monomer‐monomer interfaces were predominantly located in the N‐terminal nucleotide‐binding domain. These interfaces were not very extensive indicating that WlbA formed a transient tetramer, which could dissociate to a monomer at low protein concentrations.
Figure 6.
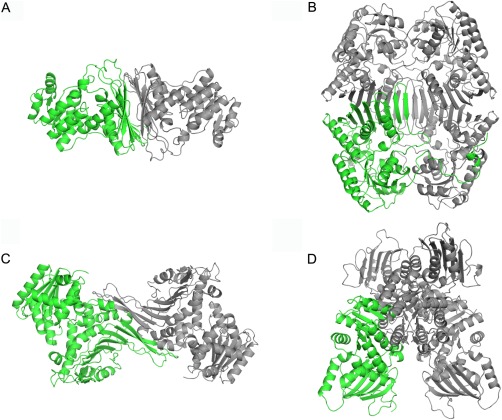
The quaternary structures of Gfo/Idh/MocA family proteins shown as cartoon models: (A) AAOR dimer, (B) GFOR tetramer, (C) G6PD dimer, and (D) WlbA tetramer. One monomer is in green, the other units are in grey.
Acknowledgment
The linguistic editing of this manuscript was done by Aura Professional English Consulting, Ltd. (www.auraenglish.com)
Statement of Importance: Proteins in the Gfo/Idh/MocA family are typically oxidoreductases of diverse substrates and in this study their structural and functional features were investigated. The Gfo/Idh/MocA family proteins have two domains: an NAD(P) binding domain and a C‐terminal α/β domain. In spite of the low sequence identity, structural features of both domains are similar in all of the proteins. The α/β‐domain has a central β‐sheet that plays a role in the quaternary structure formation, and a βα‐motif that carries the catalytic residues.
References
- 1. Rowland P, Basak AK, Gover S, Levy HR, Adams MJ (1994) The three‐dimensional structure of glucose 6‐phosphate dehydrogenase from Leunostoc mesenteroides refined at 2.0 Å resolution. Structure 2: 1073–1087. [DOI] [PubMed] [Google Scholar]
- 2. Tettelin H, Nelson KE, Paulsen IT, Eisen JA, Read TD, Peterson S, Heidelberg J, DeBoy RT, Haft DH, Dodson RJ, Durkin AS, Gwinn M, Kolonay JF, Nelson WC, Peterson JD, Umayam LA, White O, Salzberg SL, Lewis MR, Radune D, Holtzapple E, Khouri H, Wolf AM, Utterback TR, Hansen CL, McDonald LA, Feldblyum TV, Angiuoli S, Dickinson T, Hickey EK, Holt IE, Loftus BJ, Yang F, Smith HO, Venter JC, Dougherty BA, Morrison DA, Hollingshead SK, Fraser CM (2001) Complete genome sequence of a virulent isolate of Streptococcus pneumoniae . Science 293:498–506. [DOI] [PubMed] [Google Scholar]
- 3. Rao ST, Rossmann MG (1973) Comparison of super‐secondary structures in proteins. J Mol Biol 76:241–256. [DOI] [PubMed] [Google Scholar]
- 4. Taberman H, Andberg M, Koivula A, Hakulinen N, Penttilä M, Rouvinen J, Parkkinen T (2015) Structure and function of Caulobacter crescentus aldose‐aldose oxidoreductase. Biochem J 472:297–307. [DOI] [PubMed] [Google Scholar]
- 5. Kingston RL, Scopes RK, Baker EN (1996) The structure of glucose‐fructose oxidoreductase from Zymomonas mobilis: an osmoprotective periplasmic enzyme containing non‐dissociable NADP. Structure 4:1413–1428. [DOI] [PubMed] [Google Scholar]
- 6. Schu M, Faust A, Stosik B, Kohring GW, Giffhorn F, Scheidig AJ (2013) The structure of substrate‐free 1,5‐anhydro‐D‐fructose reductase from Sinorhizobium meliloti 1021 reveals an open enzyme conformation. Acta Cryst 69:844–849. F [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thoden JB, Holden HM (2010) Structural and functional studies of WlbA: a dehydrogenase involved in the biosynthesis of 2,3‐diacetamido‐2,3‐dideoxy‐d‐mannuronic acid. Biochemistry 49:7939–7948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van Straaten KE, Zheng H, Palmer DR, Sanders DA (2010) Structural investigation of myo‐inositol dehydrogenase from Bacillus subtilis: implications for catalytic mechanism and inositol dehydrogenase subfamily classification. Biochem J 432:237–247. [DOI] [PubMed] [Google Scholar]
- 9. Kubiak RL, Holden HM (2011) Combined structural and functional investigation of a C‐3''‐ketoreductase involved in the biosynthesis of dTDP‐l‐digitoxose. Biochemistry 50:5905–5917. [DOI] [PubMed] [Google Scholar]
- 10. Carbone V, Endo S, Sumii R, Chung RP, Matsunaga T, Hara A, El‐Kabbani O (2008) Structures of dimeric dihydrodiol dehydrogenase apoenzyme and inhibitor complex: probing the subunit interface with site‐directed mutagenesis. Proteins 70:176–187. [DOI] [PubMed] [Google Scholar]
- 11. Kikuchi A, Park SY, Miyatake H, Sun D, Sato M, Yoshida T, Shiro Y (2001) Crystal structure of rat biliverdin reductase. Nature Struct Biol 8:221–225. [DOI] [PubMed] [Google Scholar]
- 12. Liu QP, Sulzenbacher G, Yuan H, Bennett EP, Pietz G, Saunders K, Spence J, Nudelman E, Levery SB, White T, Neveu JM, Lane WS, Bourne Y, Olsson ML, Henrissat B, Clausen H (2007) Bacterial glycosidases for the production of universal red blood cells. Nature Biotech 25:454–464. [DOI] [PubMed] [Google Scholar]
- 13. Thoden JB, Sellick CA, Reece RJ, Holden HM (2007) Understanding a transcriptional paradigm at the molecular level. The structure of yeast Gal80p. J Biol Chem 282:1537–1538. [DOI] [PubMed] [Google Scholar]
- 14. Berman HM, Henrick K, Nakamura H (2003) Announcing the worldwide Protein Data Bank. Nature Struct Biol 10:980 [DOI] [PubMed] [Google Scholar]
- 15. Holm L, Rosenström P (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res 38:W545–W549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lott JS, Halbig D, Baker HM, Hardman MJ, Sprengen GA, Baker EN (2000) Crystal structure of a truncated mutant of glucose‐fructose oxidoreductase shows that an N‐terminal arm controls tetramer formation. J Mol Biol 304:575–584. [DOI] [PubMed] [Google Scholar]
- 17. Nurizzo D, Halbig D, Sprenger GA, Baker EN (2001) Crystal structures of the precursor form of glucose‐fructose oxidoreductase from Zymomonas mobilis and its complexes with bound ligands. Biochemistry 40:13857–13867. [DOI] [PubMed] [Google Scholar]
- 18. Whitby FG, Phillips JD, Hill CP, McCoubrey W, Maines MD (2002) Crystal structure of a biliverdin IXα reductase enzyme‐cofactor complex. J Mol Biol 319:1199–1210. [DOI] [PubMed] [Google Scholar]
- 19. Carbone V, Sumii R, Ishikura S, Asada Y, Hara A, El‐Kabbani O (2008) Structure of monkey dimeric dihydrodiol dehydrogenase in complex with isoascorbic acid. Acta Cryst 64:532–542. D [DOI] [PubMed] [Google Scholar]
- 20. Zheng H, Bertwistle D, Sanders DA, Palmer DR (2013) Converting NAD‐specific inositol dehydrogenase to an efficient NADP‐selective catalyst, with a surprising twist. Biochemistry 52:5876–5883. [DOI] [PubMed] [Google Scholar]
- 21. Schrödinger LLC. The PyMOL Molecular Graphics System.
- 22. Day R, Beck DAC, Armen RS, Daggett V (2003) A consensus view of fold space: combining SCOP, CATH, and the Dali Domain Dictionary. Protein Sci 12:2150–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sillitoe O, Lewis TE, Cuff AL, Das S, Ashford P, Dawson NL, Furnham N, Laskowski RA, Lee D, Lees J, Lehtinen S, Studer R, Thornton JM, Orengo CA (2015) CATH: comprehensive structural and functional annotations for genome sequences. Nucleic Acids Res 43:D376–D381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu W, Li Z, Huang CH, Guo RT, Zhao L, Zhang D, Chen X, Wu Q, Zhu D (2014) Structural and mutational studies on the unusual substrate specificity of meso‐diaminopimelate dehydrogenase from Symbiobacterium thermophilum . ChemBioChem 15:217–222. [DOI] [PubMed] [Google Scholar]
- 25. Yang Z, Savchenko A, Yakunin A, Zhang R, Edwards A, Arrowsmith C, Tong L (2003) Aspartate dehydrogenase, a novel enzyme identified from structural and functional studies of TM1643. J Biol Chem 278:8804–8808. [DOI] [PubMed] [Google Scholar]
- 26. Andberg M, Maaheimo H, Kumpula E‐P, Boer H, Toivari M, Penttilä M, Koivula A (2015) Characterization of a unique Caulobacter crescentus aldose‐aldose oxidoreductase having dual activities. Appl Microbiol Biotech doi:10.1007/s00253-015-7011-5. [DOI] [PubMed] [Google Scholar]
- 27. Bond CS (2003) TopDraw: a sketchpad for protein structure topology cartoons. Bioinformatics 19:311–312. [DOI] [PubMed] [Google Scholar]
- 28. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Cryst 67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]