

Abstract
Protein structures are stabilized by a variety of noncovalent interactions (NCIs), including the hydrophobic effect, hydrogen bonds, electrostatic forces and van der Waals’ interactions. Our knowledge of the contributions of NCIs, and the interplay between them remains incomplete. This has implications for computational modeling of NCIs, and our ability to understand and predict protein structure, stability, and function. One consideration is the satisfaction of the full potential for NCIs made by backbone atoms. Most commonly, backbone‐carbonyl oxygen atoms located within α‐helices and β‐sheets are depicted as making a single hydrogen bond. However, there are two lone pairs of electrons to be satisfied for each of these atoms. To explore this, we used operational geometric definitions to generate an inventory of NCIs for backbone‐carbonyl oxygen atoms from a set of high‐resolution protein structures and associated molecular‐dynamics simulations in water. We included more‐recently appreciated, but weaker NCIs in our analysis, such as n→π* interactions, Cα‐H bonds and methyl‐H bonds. The data demonstrate balanced, dynamic systems for all proteins, with most backbone‐carbonyl oxygen atoms being satisfied by two NCIs most of the time. Combinations of NCIs made may correlate with secondary structure type, though in subtly different ways from traditional models of α‐ and β‐structure. In addition, we find examples of under‐ and over‐satisfied carbonyl‐oxygen atoms, and we identify both sequence‐dependent and sequence‐independent secondary‐structural motifs in which these reside. Our analysis provides a more‐detailed understanding of these contributors to protein structure and stability, which will be of use in protein modeling, engineering and design.
Keywords: protein folding, protein structure, protein stability, bioinformatics, hydrogen bonding, noncovalent interactions, n→π* interactions
Introduction
Almost 80 years after Pauling and Mirsky predicted the importance of the hydrogen bond in protein structure formation,1 the forces governing the folding of a protein's amino‐acid sequence into its three‐dimensional structure are still not fully understood.2 Protein structures are stabilized by a variety of noncovalent interactions (NCI) including the hydrophobic effect, van der Waals’ interactions, electrostatic forces, and hydrogen bonds.3, 4
To complicate matters further, NCIs are context dependent. For example, hydrogen bonds vary in strength depending on the identities and relative geometries of the donor and acceptor groups, and also the local environment.2 In addition, weaker donor groups such as Cα‐H and methyl‐H are also possible contributors to protein stability.5, 6, 7, 8, 9 More specifically, other hydrogen‐bond‐like, NCIs have been implicated, including the n→π* interaction10, 11, 12 and methyl‐π interactions.13, 14 These particular interactions are much weaker than canonical hydrogen bonds: the latter are typically worth 3–10 kcal/mol15, 16; whereas, n→π* interactions are estimated at 0.7–1.2 kcal/mol,10, 16 and methyl‐π interactions at 0.9–1.5 kcal/mol.17, 18 These share common features with hydrogen bonds; notably, the overlap of van der Waals’ radii and orbital overlap, which result in structure stabilization through electron delocalization. Recently, we demonstrated an interplay between hydrogen bonds and n→π* interactions,16 in particular with asparagine and aspartic acid residues, which form both hydrogen bonds and n→π* interactions via their side chain carbonyl groups.
Thus, the contributions of and interplay between the various possible NCIs in proteins are complicated, and not straightforward to dissect. However, one thing is clear: for a folded protein to be stable, NCIs must combine to outweigh the contributions to the free energy made up by the entropy lost upon folding and any enthalpically favorable interactions made between the unfolded state and water. In respect of the latter, the degree to which any NCI is made or satisfied relative to the unfolded state is important.
Most commonly, backbone hydrogen bonding in proteins has been depicted quite straightforwardly: NH groups “donate protons” to proximal carbonyl‐oxygen “acceptor” atoms [Fig. 1(A)]; alternatively, this can be viewed as the oxygen atom donating electron density from a lone pair of electrons into the antibonding orbital, σ*, of the N—H bond. Moreover, in each of the two common structures in proteins—the α‐helix and the β‐sheet—each backbone‐carbonyl oxygen atom makes a single such C=O⋯H—N hydrogen bond.19 However, these depictions are at odds with the standard model from physical organic chemistry, in which the carbonyl oxygen atom is sp 2 hybridized, and therefore, presents two lone pairs, either or both of which could participate in hydrogen bonds or other NCIs. Thus, by invoking only one hydrogen bond, and utilizing only one of these lone pairs, the backbone carbonyl atoms of a folded protein could be considered as already unsatisfied as compared with fully solvent‐accessible atoms in the unfolded state. In turn, these lost hydrogen bonds could be considered as adding to the free‐energy debt of the folded state. In support of this, model studies of unfolded alanine peptides reveal an enthalpy deficit for helix formation, which is not provided for by hydrogen bonds,20 and which cannot be fully accounted for by modeling interactions of the peptide with water.
Figure 1.
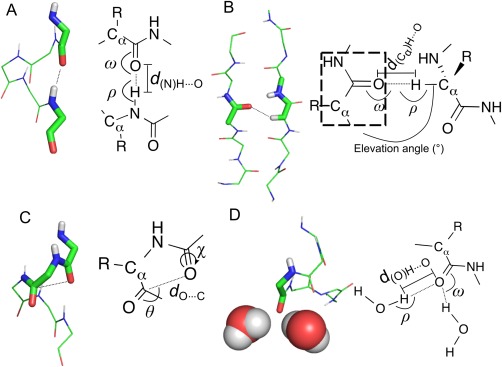
Backbone‐carbonyl‐oxygen non‐covalent interaction (NCIC=O) considered in this analysis. (A) “Standard” hydrogen bonds, as exemplified by NHi→C=Oi−4 hydrogen bonds found in an α‐helix (NHbb, dNH⋯O ≤ 2.44 Å25; ω ≥ 90°; ρ ≥ 90°). Other donor groups include (i) side‐chain NH, e.g., from lysine or arginine, (NHsc, parameters as for NHbb); and (ii), side‐chain hydroxyl groups (OHsc, d(O)H⋯O ≤ 2.31 Å25; ω ≥ 90°; ρ ≥ 90°). (B) Hydrogen bonds with a Cα—H group donor (CαH, d(Cα)H⋯O ≤ 2.68 Å25; ω ≥ 90°; ρ ≥ 90°, elevation angle < 50°), or alternatively donated by other methyl or ethyl groups from protein sidechains9 (CHX, parameters as for CαH). (C) n→π* interactions, shown with a main‐chain carbonyl group acceptor (dC⋯O ≤ 3.22 Å; 95° ≥ θ ≥ 125°; Cα⋯C⋯O⋯H dihedral χ ≥ 120°57); but these can also have a side‐chain acceptor, e.g., asparagine or glutamine, (n→π* sc, parameters as for n→π*). (D) Hydrogen bonds made with water (HOH, d(O)H⋯O ≤ 2.31 Å25; ω ≥ 90°; ρ ≥ 90°).
The satisfaction of backbone hydrogen‐bonding potential in proteins has been studied.21, 22 In their hydrogen‐bonding hypothesis, Fleming and Rose argue that all potential backbone hydrogen‐bond donors and acceptors are satisfied a significant fraction of the time, either via intramolecular hydrogen bonds or hydrogen bonds to water.23 The basis of the hypothesis is that unsatisfied hydrogen‐bonding potential is highly unfavorable energetically and therefore rare. Indeed, revisiting foregoing studies, which suggest that up to 10% of this potential remains unmet in folded proteins,21 Fleming and Rose show that unsatisfied donors and/or acceptors can be satisfied with small adjustments to the X‐ray crystal structures.23, 24 However, Fleming and Rose consider carbonyl groups that make just one hydrogen bond to be satisfied, that is traditional hydrogen‐bonded patterns. By extension of their arguments, it stands to reason that if both lone pairs could be utilized in hydrogen bonding or other NCIs then the consequences for protein stability would be considerable and favorable.
Herein, we re‐examine the satisfaction of hydrogen‐bonding potential in light of (a) the identification of other and significant NCIs, and (b) the revision of hydrogen‐bonding criteria based on electron‐density topology.25, 26, 27 We explore the question of NCI saturation from the perspective of both lone pairs of electrons of the carbonyl‐oxygen atoms. For example, in an α‐helix, the carbonyl group of residue i usually accepts a hydrogen bond from the NH group of the i + 4th residue (the traditional depiction), and additionally makes an n→π* interaction with the carbonyl group of the i + 1th residue,28 thereby satisfying both lone pairs. Another means of satisfying both lone pairs in helices comes from bifurcated hydrogen bonds, in which a carbonyl group accepts hydrogen bonds from amides at the i–3th and i–4th positions.29, 30 For a set of ultra‐high‐resolution protein X‐ray crystal structures, we identify and categorize NCIs made by the carbonyl‐oxygen groups (hereafter referred to as NCIC=O). We find that generally, both lone pairs of electrons are satisfied by two NCIC=O, and that combinations of different NCIC=O correlate with different secondary structure types. In addition, we use molecular‐dynamics (MD) simulations to explore the dynamics of such NCIs, including examples with under‐ and over‐satisfied carbonyl groups. Although not common, where found the latter are sustained over the course of MD simulations, suggesting that they are pertinent and not structural anomalies. In this way, we identify three structurally conserved NCIC=O motifs that are found in helices. Overall, the system is very much dynamic. Undersatisfied groups are balanced by oversatisfied groups, and the whole system tends towards being slightly oversatisfied.
We believe that this study provides a different and more‐nuanced view of NCIs within protein secondary structures, which is currently not widely considered. It will be of use in the refinement of modeling forcefields for proteins, and to help assess and validate protein models in structure determination, and in protein engineering and design.
Results
Data generation
A set of 31 nonredundant, ultra‐high resolution (≤1 Å) structures in which the hydrogen atoms are assigned was obtained from the Protein Data Bank (PDB).31 Multi‐chain assemblies were discounted in order to avoid the complication of interchain interactions, which may or may not be due to crystal artefacts. An inventory of NCIC=O made by each residue was generated using operational definitions for four types of NCI (Fig. 1): traditional hydrogen bonds, CH‐based hydrogen bonds, n→π* interactions, and hydrogen bonds made to water.
Not all of the selected protein structures had complete solvent shells. Therefore, each was simulated for 100 ns using a standard molecular dynamics protocol (see Methods for full details). NCIC=O were identified at 1 ns intervals using the same operational definition as for the static structures.
Backbone carbonyl groups are generally fully satisfied
Our hypothesis was as follows: given that each carbonyl oxygen atom has two lone pairs of electrons, each of these might be expected to make a NCI. Thus, to be fully satisfied, every backbone‐carbonyl oxygen atom should make two NCIs, one for each lone pair. To begin testing this, we examined the number of NCIC=O made by the carbonyl oxygen atom from the original static protein structures (Fig. 2, black bars). We found that approximately half of carbonyl groups (53%) were satisfied by two NCIC=O, and the remainder were under‐ or over‐satisfied.
Figure 2.
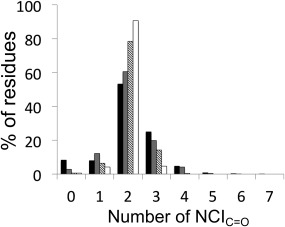
The percentages of NCIC=Os per residue made across all residues in proteins. These were measured in three ways: across all residues in the initial, unsolvated high‐resolution crystal structures (black bars); across all residues and snapshots from the last 81ns of a molecular‐dynamics simulation (gray bars); from the distribution of modal averages of all residues across the same set of molecular‐dynamics simulation snapshots (diagonal bars); across all residues and snapshots for those residues that spend at least half of their molecular‐dynamics simulation at their modal average number of NCIC=O (white bars).
It is possible that these structures are not all properly solvated, and that a more‐complete picture might be obtained by fully solvating the protein structures ahead of the analysis. In addition, proteins are dynamic systems, and static poses may not reveal the full picture. Therefore, each structure was subjected to MD simulation to enable identification of NCIC=Os over a period of time. A disadvantage of using MD forcefields, however, is that necessarily they approximate NCIs. Such parameterization may itself introduce bias into the simulations and how they are interpreted. Hydrogen bonding of carbonyl‐oxygen atoms to water molecules is a case in point: in most forcefields, these are dealt with implicitly rather than explicitly through the application of Coulomb's Law on atomic point charges and the steric bulk of the interacting atoms alone; nonetheless, these tend to result in two hydrogen bonds on average, consistent with each lone pair of the carbonyl oxygen making the hydrogen bonds. Whilst capable of capturing some of the known geometric preferences of hydrogen bonds, these approximations may bias the data away from some of the NCI that might be captured in single high‐resolution structures: however, we could not do the analysis without properly solvated structures, and these could only be reliably obtained by looking at ensembles of MD snapshots. Therefore, we collected data from 81 ns of MD simulation for each structure, taking one snapshot at nanosecond intervals, and then examined the number of NCIC=O made by each carbonyl oxygen at each time‐point in four ways: First, a frequency distribution of the number of NCIC=O made by each carbonyl group in each nanosecond snapshot of the MD simulation showed that 60% of carbonyl groups participated in 2 NCIC=O during the course of their simulation (Fig. 2, gray and Table 1). A smaller, but still significant proportion (∼30%) participated in 1 or 3 NCIC=O, and this was close to a normal distribution with a mean NCIC=O of 2, as compared with the static snapshot picture. Second, we looked at the modal average of NCIC=O for each carbonyl group along the length of the simulation (Fig. 2, diagonal lines), which showed that ∼80% of backbone carbonyl groups were fully satisfied, i.e., making 2 NCIC=O, but with a much smaller contribution from those groups participating in 1 or 3 NCIC=O (6% and 14%, respectively). Finally, when we considered the distribution of numbers of NCIC=O of only those residues that spent half or more of their time through the MD simulations in their modal average state (Fig. 2, white), we found that just over 90% of residues made 2 NCIC=O, and that the over‐ and undersatisfied residues balanced out at ∼5% each. For comparison, previous work on forcefield development32 has shown that carbonyl‐oxygen atoms in model amides simulated in water have 2 water‐molecule neighbors (equivalent to 2 NCIC=O) approximately two‐thirds of the time, with an even distribution between 1 and 3 for the remainder of the time.
Table 1.
Summary of NCIC=O inventory
| (A) Mean number of residues (n = 81 MD snapshots) with NCIC=O = x. Modal average in parentheses. | ||||||||
|---|---|---|---|---|---|---|---|---|
| Secondary structure | x = 0 | 1 | 2 | 3 | 4 | 5 | 6 | Total residues |
| α‐helix | 45 (7) | 213 (63) | 998 (1390) | 584 (580) | 163 (17) | 17 (0) | 0 (0) | 2121 |
| β‐strand | 31 (9) | 226 (163) | 732 (965) | 204 (86) | 23 (0) | 1 (0) | 0 (0) | 1246 |
| 310/π‐helix | 9 (2) | 28 (12) | 177 (229) | 55 (33) | 11 (2) | 1 (0) | 0 (0) | 312 |
| Turn | 33 (8) | 95 (43) | 693 (884) | 143 (59) | 22 (1) | 1 (0) | 0 (0) | 901 |
| Bend | 16 (6) | 35 (24) | 260 (293) | 36 (7) | 4 (0) | 0 (0) | 0 (0) | 312 |
| None | 22 (3) | 65 (41) | 435 (507) | 60 (8) | 5 (0) | 0 (0) | 0 (0) | 550 |
| (B) Total number of each NCIC=O identified (over 81 MD snapshots taken at 1 ns intervals) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Secondary structure | NHbb | NHsc | n→π* | Cα—H | O—H | CHx | HOH | Total NCI |
| α‐helix | 123568 | 8148 | 88839 | 1493 | 8315 | 62514 | 88088 | 380605 |
| β‐strand | 66133 | 4026 | 9772 | 35391 | 3366 | 24373 | 51239 | 194300 |
| 310/π‐helix | 9363 | 2549 | 8034 | 760 | 968 | 5378 | 21267 | 48319 |
| Turn | 17076 | 9228 | 18349 | 3648 | 2757 | 116866 | 94363 | 162287 |
| Bend | 2177 | 3897 | 4128 | 1587 | 983 | 5801 | 36260 | 58433 |
| None | 4724 | 5642 | 7115 | 2964 | 2447 | 9482 | 59711 | 92085 |
| (C) Average number of each NCI type per residue (mean per snapshot per residue) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Secondary structure | NHbb | NHsc | n→π* | Cα—H | O—H | CHx | HOH | Total NCIC=O per residue |
| α‐helix | 0.72 | 0.05 | 0.52 | 0.01 | 0.05 | 0.36 | 0.51 | 2.22 |
| β‐strand | 0.66 | 0.04 | 0.10 | 0.35 | 0.03 | 0.24 | 0.51 | 1.93 |
| 310/π‐helix | 0.37 | 0.10 | 0.32 | 0.03 | 0.04 | 0.21 | 0.84 | 1.91 |
| Turn | 0.23 | 0.13 | 0.25 | 0.05 | 0.04 | 0.23 | 1.29 | 2.22 |
| Bend | 0.09 | 0.15 | 0.16 | 0.06 | 0.04 | 0.23 | 1.43 | 2.17 |
| None | 0.11 | 0.13 | 0.16 | 0.07 | 0.05 | 0.21 | 1.34 | 2.07 |
Types of NCI made correlate with secondary structure
Given the above observation that NCIC=O = 2 for the majority of peptide units, and that this contrasts with traditional models and depictions of regular secondary structures founded on single C=O⋯H—N hydrogen bonds, we asked what types of additional NCIs were being made by the oxygen atoms (Fig. 3).
Figure 3.
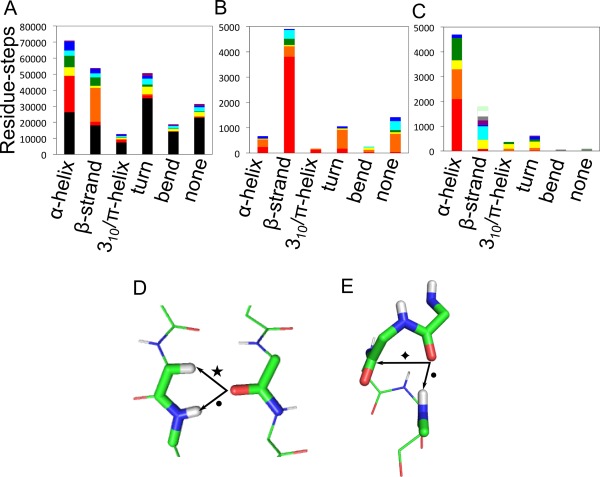
Distributions of types of NCIC=O made in different secondary structure. (A) Where 2 × NCIC=O are made per residue; (B) 1 × NCIC=O; and (C) 3 × NCIC=O. For clarity, only those combinations of NCI representing at least 2% of all residues are shown in (A), which accounts for 89% of residues overall. Key for panel (A): black bars, 2 × HOH; red, 1 × n→π* plus 1 × NHbb; orange, 1 × CαH plus 1 × NHbb; yellow, 1 × n→π* plus 1 × HOH; green, 1 × NHbb plus 1 × CHX; turquoise, 1 × HOH plus 1 × CHX; dark blue, 1 × NHbb plus 1 × HOH; purple, 1 × NHsc plus 1 × HOH. (B and C) Residues were included in the plots for panels (B) and (C) if their modal average number of NCIC=O was 1 or 3, and spent at least 50% of the duration of MD‐simulation in these categories. Key for panel (B): red bars, 1 × NHbb; orange, 1 × NHsc; yellow, 1 × n→π*; green, 1 × CαH; turquoise, 1 × OHsc, blue, 1 × CHX. Key for panel (C): red bars, 1 v NHbb, 1 × n→π*, 1 × CHX; orange, 2 × NHbb plus 1 × n→π*; yellow, 1 × NHbb, 1 × n→π*, 1 × HOH; green, 1 × n→π*, 1 × OHsc, 1 × NHbb; turquoise, 1 × CαH, 1 × HOH, 1 × NHbb; blue, 1 × n→π*, 1 × CαH, 1 × NHbb; purple, 1 × NHbb, 1 × NHsc, 1 × n→π*; gray, 1 × NHbb, 1 × CαH, 1 × CHX; white, 2 × NHbb, 1 × CHX; mint green, 1 × NHbb, 1 × NHsc, 1 × CαH. (D, E) The most‐common NCIC=O combinations identified in the two most‐prevalent secondary structure types. (D) β‐Strand residues with a backbone NH hydrogen bond (NHbb, •) plus a Cα–H hydrogen bond (Cα–H, ★), (PDB 1G66, residues A6, A84‐A85). (E) α‐Helical residues residues with a NHbb (•) plus an n→π* interaction ( ), (PDB 1G66, residues A26‐A30). Secondary structures were assigned by Promotif,42 which uses a modified version of the Kabsch and Sander DSSP algorithm.58 Categories “E” and “B” were combined into a single β‐structure category.
), (PDB 1G66, residues A26‐A30). Secondary structures were assigned by Promotif,42 which uses a modified version of the Kabsch and Sander DSSP algorithm.58 Categories “E” and “B” were combined into a single β‐structure category.
First, we found that just under half (49%) of all residues in all secondary structure types that made 2 × NCIC=O were fully satisfied by hydrogen bonds to water [Fig. 3(A)]. As might be expected, this proportion was greatest for the nonstructured, bend and turn regions (70%, 72%, and 66% respectively), which are more‐exposed to solvent, and lowest for regular α‐helical and β‐strand conformations (34% and 32%, respectively).
Turning to conformations not wholly satisfied by hydrogen bonds to water, we found that nearly half (44%) of the residues in α‐helical conformations that made 2 × NCIC=O did so with one traditional NHi→C=Oi‐4 hydrogen bond, plus one C=Oi→C=Oi + 1 n→π* interaction [Fig. 3(A)]. Approximately equal, but smaller proportions of α‐helical residues, either made one backbone NH hydrogen bond, plus either one hydrogen bond to water (10%), or one CHX (where X = 1, 2, or 3) hydrogen bond (14%), or made one n→π* interaction plus one hydrogen bond to water (10%). The preponderance and potential importance of n→π* interactions in the α‐helix has been noted.13 However, how these arise is worth reiterating. The NHi→C=Oi−4 hydrogen bonds in α‐helices are unusual: typically, hydrogen‐bond energies are maximized when the angle between the donor and C=O bond axis is ≈120°33; however, in the α‐helix this angle approaches ≈180°, i.e., the hydrogen bond is aligned with the C=O bond vector. This results in demixing of carbonyl lone pairs from sp 2‐like orbitals away from the “rabbit ears” model and into s‐type orbital along the C=O bond vector and an orthogonal p‐type orbital. The first lone pair participates in the NHi→C=Oi‐4 hydrogen bond, or n→σ* interaction, leaving the second lone pair available to make an n→π* interaction with the adjacent carbonyl group.16
Our analysis also revealed that half of carbonyl groups found in β‐structure not satisfied by hydrogen bonds to water were satisfied by one backbone NH hydrogen bond (NHbb), plus one Cα‐H hydrogen bond (CαH), (55%), Figure 3(A). Both of these bridge strands [Fig. 3(D)], in what are termed i → j interactions. Although the role of CαH interactions has been identified in several studies,5, 34, 35 the consensus is that they are weak, and of lower importance than hydrogen bonds with traditional donors, i.e., protons attached to electronegative nitrogen and oxygen atoms. However, our data, which show that CαH interactions are made by most residues in β‐sheets, suggests that they are common and made significant proportion of the time. Thus, they could also be important contributors to protein stability. Moreover, they help account for the full satisfaction of the carbonyl‐oxygen lone pairs of electrons.
Under‐satisfied residues participating in 1 NCIC=O
As argued by Rose and colleagues,36 backbone polar groups that are under‐satisfied in their hydrogen‐bonding potential almost certainly disfavor protein folding by reducing protein stability. Our hypothesis and consideration of both lone pairs on carbonyl oxygen atoms potentially increases the number of such unsatisfied groups. We investigated these by considering residues with a modal average number of just 1 NCIC=O, with the additional requirement that the residue had to maintain this number in at least half the snapshots taken from the MD simulations. This was done to ensure that we were considering sustained interactions, and not ephemeral arrangements that may have arisen as the simulations fluctuated.
Figure 3(B) shows that the largest contribution of residues of this type, approximately half, are in β‐structure and make a single NHbb. Indeed, across all secondary structure types, a single hydrogen bond made to an NH group (red and orange bars), either backbone or side chain, accounts for 79% of all residues in this NCIC=O = 1 category.
It is interesting to speculate whether these residues do make other, as yet unforeseen NCIC=O. We found that the C=O groups in this category regularly made sub‐van der Waals’ contacts with the backbone amide proton of the same residue, and/or with the Cα proton of the adjacent residue (Fig. S2, Supporting Information). Neither of these potential NCIs were formally considered in our analysis as they have not been previously documented or recognized as stabilizing; although, weakly stabilizing NCIs between C=O and NH groups have been observed in small‐molecule systems.37, 38
Additionally, in the nonstructured regions and in turns, we see a larger preponderance of hydrogen bonds donated by a side‐chain NH group. This highlights the importance of side chain—main chain interactions in these regions and has been noted by others (e.g., Refs. 39, 40, 41).
Over‐satisfied residues participating in 3 NCIC=O
Under‐satisfied NCIC=Os are one thing, but residues that make more than two NCIC=O are curious given that there are just two lone pairs of electrons per carbonyl group. Intrigued by the significant proportion (14%) of these over‐satisfied C=O groups, we investigated them by considering only residues where the modal average NCIC=O was 3 in the MD simulation, and again, stipulating that the residue had to be in this state for at least 50% of the snapshots taken from the simulations [Fig. 3(C)]. This identified 11,843 snapshots from 263 individual residues. Three significant groups emerged, all involving α‐helical residues. The largest group formed one NHbb plus one n→π* interaction and one CHX hydrogen bond [63 unique examples from 2391 snapshots, Fig. 4(A)]. These were found at all positions across α‐helices and showed no preference for termini.
Figure 4.
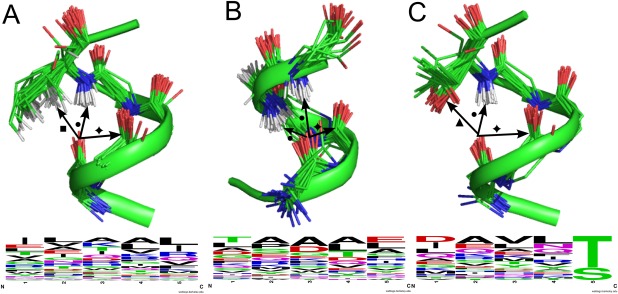
Local structures with over‐satisfied backbone‐carbonyl‐oxygen atoms, i.e., with 3 × NCIC=O. (A) α‐helical motifs with 1 × NHbb (•), 1 × n→π* interaction ( ) and one 1 × CHX (▪). The residue providing the CHx has been truncated for clarity. (B) Motifs at the α‐helical Ntermini with 2 × NHbb (•) plus 1 × n→π* interaction (
) and one 1 × CHX (▪). The residue providing the CHx has been truncated for clarity. (B) Motifs at the α‐helical Ntermini with 2 × NHbb (•) plus 1 × n→π* interaction ( ). (C) α‐helical C‐termini with 1 × OHsc, (▲), 1 × NHbb (•) and 1 × n→π* interaction (
). (C) α‐helical C‐termini with 1 × OHsc, (▲), 1 × NHbb (•) and 1 × n→π* interaction ( ), and associated WebLogos59 indicating the amino‐acid frequencies from sequences in our dataset that display this motif. Structural images prepared with PyMOL (http://www.pymol.org). PDB codes and residue identifiers for each example can be found in the Supporting Information.
), and associated WebLogos59 indicating the amino‐acid frequencies from sequences in our dataset that display this motif. Structural images prepared with PyMOL (http://www.pymol.org). PDB codes and residue identifiers for each example can be found in the Supporting Information.
The second largest group of over‐satisfied residues formed two NHbb, plus an additional n→π* interaction [42 unique examples from 1435 snapshots, Fig. 4(B)]. These were found in α‐helical structures, with two‐thirds coming from the “little h” category defined by Promotif,42 i.e., the first or last turn of an α‐helix. The majority were found at the N‐termini of α‐helices, where they may have a sequence‐independent role in helix‐capping [Fig. 4(B)]; that is, different from other identified capping motifs, which involve side chain—main chain contacts. The overwhelming majority of these formed bifurcated hydrogen bonds, with donors coming from the i–3rd and i–4th residue. Over all residue‐steps, these accounted for 19.4% of all hydrogen bonds to main‐chain amide groups. Interestingly, when these interactions did fluctuate down to two NCIC=O in the MD simulations, it was usually one of the NHbb that was lost, and not the n→π* interaction, which perhaps runs contrary to expectations given that the latter is considered the weaker of the two interactions.16
A third type of three‐NCIC=O cluster was found in the C‐terminal turns of α‐helices [31 unique examples from 1039 snapshots, Fig. 4(C)]. This comprised one NHbb, a hydrogen bond donated by a side‐chain hydroxyl group (OHsc), and an n→π* interaction. Both the OHsc and the NHbb were donated either by serine or threonine residues. This helix‐capping motif has been identified by Richardson & Richardson, who note both hydrogen bonds, but not the additional n→π* interaction.43
A small subset of residues (13 unique examples from 567 snapshots) that form 3 × NCIC=O was found in the general β‐strand/extended secondary structure class. These have one NHbb, one CαH and an additional hydrogen bond to water. Although these are not well conserved structurally, owing to the different underlying structures found in parallel and antiparallel β‐sheets, similarities can be identified within these groups: they occur in exposed β‐strands where a backbone carbonyl group is exposed and makes a close contact with a water molecule in addition to the NHbb and CαH interactions.
Prevalence of “weaker” interactions
It is interesting to note the prevalence of weaker interactions found in this study and how they compare with other foregoing studies. We found n→π* interactions in 31% of residue‐steps, which agrees with the average of 34% found previously.12 Ten percentage of the Cα‐H groups made hydrogen bonds to C=O groups, which is in line with the proportion identified by Derewenda et al.5 Turning to CHX bonds (donated by side‐chain CH3, CH2, or CH groups), we find that 10% of all such available groups in the dataset formed these weak hydrogen bonds, a much reduced proportion compared with the 36% found by Yesselman et al.9 However, this discrepancy can probably be explained in that our analysis only considers hydrogen bonds accepted by main‐chain C=O groups and not other hydrogen bond acceptors.
Discussion
The analysis that we present provides an inventory of non‐covalent interactions (NCIs) for backbone‐carbonyl oxygen atoms in high‐resolution protein structures. Previous analyses have been dismissed as “apples and oranges” comparisons of hydrogen bonds due to the range of strengths that these can have depending on their environment.36, 44, 45 However, as we consider the satisfaction of lone pairs of electrons via several possible NCIs, rather than simply counting “traditional” hydrogen bonds, we suggest that our analysis offers a different perspective on understanding the stabilization of protein structure, and that this helps to explain certain anomalies of previous models. Key points of our hypothesis are that backbone‐carbonyl oxygen atoms can make up to two NCIs, by virtue of their two available lone pairs; and that ideally both of these should be satisfied in the folded state, as presumably they are both involved with hydrogen bonding to solvent in the unfolded state. Thus, if left unsatisfied the stability of the folded state will be sub‐optimal. This is the case in the more‐common models of regular protein secondary structures, which depict just one C=O⋯H—N hydrogen bond per residue.
In support of our hypothesis, we find that the majority of backbone‐carbonyl oxygen atoms do indeed form two NCIs. This is true for static X‐ray crystal structures of proteins. Moreover, these interactions persist during MD simulations. We categorize the various types of additional NCIs as fully as possible, and in the context of known NCIs over and above C=O⋯H—N hydrogen bonds. Table 1 provides a summary of NCIs identified by secondary structure type; a full breakdown per structure is given in the Supporting Information. We find correlations between local backbone structure and the type of NCI made, which we propose further stabilize the secondary and tertiary structures. These observations were largely independent of side‐chain. Specifically, in addition to two bifurcated hydrogen bonds, carbonyl groups in α‐helices tend to make an n→π* interaction; whereas, in β‐structure (parallel or antiparallel) the second lone pair of electrons of the carbonyl group is satisfied through C=O⋯H—C hydrogen bonds.
In addition, we identify and examine examples of residues that appear to be over‐satisfied; that is, where the number of NCIC=O is greater than two. These account for 14% of all residues in our dataset (judged by modal average). These clustered interactions tend to persist during the lifetime of MD simulations, which suggests that they are not structural anomalies. Interestingly, the most‐prevalent clusters are found in helices, and the most‐frequent of those found at helical termini appears to be sequence‐independent, unlike most helix‐capping motifs previously identified.43, 46 A smaller proportion of residues (6%) appear to be under‐satisfied in terms of NCI‐making potential. Proteins systems are clearly dynamic, and therefore we expect a distribution of NCIC=O across all residues, and ideally, it should be balanced. Our analysis points to slight oversatisfaction: it is possible that this is due to errors in the way we have assigned NCIC=O, or that we are not counting other, as yet unidentified, stabilizing interactions. Interestingly, removing some of the fluctuations from the system—by considering only those residues that spend at least half the simulation time with their modal average number of NCIC=O—the systems balance with a 5:90:5 ratio of 1, 2 and 3 NCIC=O made, respectively.
Overall, we can define a density of NCIs made by backbone carbonyl groups (NCIC = O, Table 1). On average across all of the proteins that we analyzed this is 2.12 per residue, which rises to 2.22 per residue for the α‐helical regions, and falls to 1.93 per residue in parallel and antiparallel β‐sheets. For comparison, the average numbers of hydrogen bonds made per residue in our data set (excluding weak C—H hydrogen bonds) are 1.42, 1.33, and 1.24 for these three structural classifications, which is greater than that identified by McDonald and Thornton (mean 1.16 H bonds per backbone C=O).21 Most likely, this discrepancy arises from the use of updated hydrogen‐bonding criteria, and potentially more‐accurate hydrogen‐atom placement in crystal structures and simulations. Such metrics will hopefully help the quest of seeking a quantitative dissection and description of protein stability.
Traditional textbook and literature descriptions of protein folding that cite hydrogen bonds as one of the major stabilizing determinants of protein secondary structures.2, 4, 22 Our analysis is not at odds with this view, but we believe the picture is more detailed and subtle than often portrayed. Recently, others have demonstrated that, for the α‐helix in particular, the classical model of NCIs may not always be appropriate. Kuster et al.29 have demonstrated that a slight crankshaft rotation of backbone torsion angles in protein helices accommodates bifurcated hydrogen bonds, in which one backbone amide makes a hydrogen bond to two carbonyl groups, at the i + 3rd and i + 4th carbonyl groups, without moving the Cα and Cβ atoms from their positions in a classical Pauling α‐helix. These bifurcated hydrogen bonds contribute to the satisfactions of both lone pairs in helices; however, they do not consider other weak NCIs such as the n→π* interaction. Interestingly, they find that 18.5% of helical hydrogen bonds are bifurcated: our data concur with this, but we find that additionally the majority of carbonyl groups making bifurcated hydrogen bonds make an additional n→π* interaction.
Specifically, weaker NCIs such as n→π* interactions and Cα—H hydrogen bonds need to be included to satisfy fully the lone pairs of electrons associated with backbone‐carbonyl oxygen atoms; and the dynamics of the biomolecular systems must be considered. Given the preponderance of these weak interactions, and that they may well be even more readily formed and broken than traditional hydrogen bonds, their roles in protein structure, dynamics, and function may be far reaching. That said, and for the same reasons, gaining thorough experimental, computational and quantitative grasps of these other NCIs will be challenging. Of course, there are concerns and potential caveats in our view and analyses that will need refinement. For example, it is not immediately clear how a model based on satisfying two lone pairs of electrons accommodates carbonyl groups that make 3 NCIs, although there is a dipolar resonance structure for the amide group that places three lone pairs on the carbonyl oxygen. This raises the question of how best we should define and measure NCIs, and, of course, how do we model and assess them computationally and quantitatively. For example, recent work on the Rosetta forcefield has demonstrated that simultaneously modeling the electrostatic and covalent properties of hydrogen bonds improves protein‐structure prediction,47 and work on polarizeable, multipolar forcefields such as AMOEBA48 has challenged the notion of linear hydrogen bonding in α‐helices. Further quantification of the contributions from each NCI and how they cooperate should inform the development of more‐accurate forcefields for molecular modeling and mechanics, and thus afford a deeper understanding of protein structure and stability.
Methods
Inventory generation
The inventory of NCIC=O made by each residue was generated using a python script that measured interatomic distances, angles, and dihedrals, and assigned NCIC=O based on the operational definitions of NCI shown in Figure 1.
MD simulation
To give each protein structure a full solvent shell, each was simulated in a box at least 2 nm larger than the protein in each direction, filled with TIP3P water,32 using the amber99sb‐ildn forcefield49 as implemented in the Gromacs‐4.5.3 suite of MD software.50 Random water molecules were replaced by sodium and chloride ions to give an overall neutrally charged system with an ionic strength of 0.15M. Each simulation was subjected to 2000 steps of energy minimization using steepest descents prior to the MD simulation.
Simulations were performed at 293 K using periodic boundary conditions. Short range electrostatic and van der Waals’ interactions were truncated at 1.4 nm, while long‐range electrostatics were treated with the particle‐mesh Ewald's method,51 and a long‐range dispersion correction was applied. Pressure was controlled by Berendsen's thermostat52 and temperature by the V‐rescale thermostat.53 Simulations were integrated with a leap‐frog algorithm over a 2 fs timestep, constraining bond vibrations with the P‐LINCS method54 and water bonds and angles using the SETTLE method.55 An initial 200 ps simulation was performed in each case with the protein heavy atoms restrained to their initial co‐ordinate positions to relax the system, before a 100 ns period of unrestrained MD. RMSD profiles of MD trajectories were manually inspected for any significant drift from the original structure (Fig. S1, Supporting Information). PDB snapshots were taken from the trajectory at 1 ns intervals from 20–100 ns, to avoid any bias from initial equilibration.
NCIC=O at each time‐point were identified with the same python script used to interrogate the static structures. Results were stored in a relational database for ease of repeated queries (File 1, Supporting Information). The assumption was made that all carbonyl oxygen atoms interacting only with water (i.e., those that were completely exposed) made two hydrogen bonds with water. This avoided any bias resulting from the water model used, as it has been shown recently56 that proteins are under‐solvated in MD simulations.
Supporting information
Supporting Information
Acknowledgements
The thank Robert Newberry and Richard Sessions for helpful discussion and critical reading of the manuscript; and the referees for insightful and constructive comments. DNW holds a Royal Society Wolfson Research Merit Award.
References
- 1. Mirsky A, Pauling L (1936) On the structure of native, denatured, and coagulated proteins. Proc Natl Acad Sci U S A 22:439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pace CN, Scholtz JM, Grimsley GR (2014) Forces stabilizing proteins. FEBS Lett 588:2177–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Anfinsen CB (1973) Principles that govern the folding of protein chains. Science 181:223–230. [DOI] [PubMed] [Google Scholar]
- 4. Dill K (1990) Dominant forces in protein folding. Biochemistry 29:7133–7155. [DOI] [PubMed] [Google Scholar]
- 5. Derewenda ZS, Lee L, Derewenda U (1995) The occurrence of C‐H…O hydrogen bonds in proteins. J Mol Biol 252:248–262. [DOI] [PubMed] [Google Scholar]
- 6. Brandl M, Weiss MS, Jabs A, Sühnel J, Hilgenfeld R (2001) CH…π‐interactions in proteins. J Mol Biol 307:357–377. [DOI] [PubMed] [Google Scholar]
- 7. Steiner T, Koellner G (2001) Hydrogen bonds with π‐acceptors in proteins: frequencies and role in stabilizing local 3D structures. J Mol Biol 305:535–557. [DOI] [PubMed] [Google Scholar]
- 8. Horowitz S, Trievel RC (2012) Carbon‐oxygen hydrogen bonding in biological structure and function. J Biol Chem 287:41576–41582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yesselman JD, Horowitz S, Brooks CL, Trievel RC (2015) Frequent side chain methyl carbon‐oxygen hydrogen bonding in proteins revealed by computational and stereochemical analysis of neutron structures. Proteins Struct Funct Bioinform 83:403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hinderaker MP, Raines RT (2003) An electronic effect on protein structure. Protein Sci 12:1188–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hodges JA, Raines RT (2006) Energetics of an n→π* interaction that impacts protein structure. Org Lett 8:4695–4697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bartlett GJ, Choudhary A, Raines RT, Woolfson DN (2010) n→π* Interactions in proteins. Nat Chem Biol 6:615–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Plevin MJ, Bryce DL, Boisbouvier J (2010) Direct detection of CH/pi interactions in proteins. Nat Chem 2:466–471. [DOI] [PubMed] [Google Scholar]
- 14. Plevin MJ, Hayashi I, Ikura M (2008) Characterization of a conserved ‘threonine clasp’ in CAP‐Gly domains: role of a functionally critical OH/pi interaction in protein recognition. J Am Chem Soc 130:14918–14919. [DOI] [PubMed] [Google Scholar]
- 15. Fersht A (1999) Structure and mechanism in protein science—a guide to enzyme catalysis and protein folding. WH Freeman. [Google Scholar]
- 16. Bartlett GJ, Newberry RW, Vanveller B, Raines RT, Woolfson DN (2013) Interplay of hydrogen bonds and n→π* interactions in proteins. J Am Chem Soc 135:18682–18688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Takagi T, Tanaka A, Sanshiro M, Maezaki H, Tani M, Fujiwara H, Sasaki Y (1987) Computational studies on CH/pi interactions. J Chem Soc Perkin Trans 2 14:1015–1018. [Google Scholar]
- 18. Tsuzuki S, Honda K, Uchimaru T, Mikami M, Tanabe K (2000) The magnitude of the CH/pi interaction between benzene and some model hydrocarbons. J Am Chem Soc 122:3746–3753. [Google Scholar]
- 19. Creighton TE (1993) Proteins structures and molecular properties, 2nd ed. Freeman.
- 20. Avbelj F, Luo P, Baldwin RL (2000) Energetics of the interaction between water and the helical peptide group and its role in determining helix propensities. Proc Natl Acad Sci USA 97:10786–10791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McDonald I, Thornton J (1994) Satisfying hydrogen bonding potential in proteins. J Mol Biol 238:777–793. [DOI] [PubMed] [Google Scholar]
- 22. Rose GD, Fleming PJ, Banavar JR, Maritan A (2006) A backbone‐based theory of protein folding. Proc Natl Acad Sci USA 103:16623–16633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fleming PJ, Rose GD (2005) Do all backbone polar groups in proteins form hydrogen bonds? Protein Sci 14:1911–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Panasik N, Fleming PJ, Rose GD (2005) Hydrogen‐bonded turns in proteins: The case for a recount. Protein Sci 14:2910–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Klein RA (2006) Modified van der Waals atomic radii for hydrogen bonding based on electron density topology. Chem Phys Lett 425:128–133. [Google Scholar]
- 26. Arunan E, Desiraju GR, Klein Ra, Sadlej J, Scheiner S, Alkorta I, Clary DC, Crabtree RH, Dannenberg JJ, Hobza P, Kjaergaard HG, Legon AC, Mennucci B, Nesbitt DJ (2011) Defining the hydrogen bond: an account (IUPAC Technical Report). Pure Appl Chem 83:1619–1636. [Google Scholar]
- 27. Desiraju GR (2011) A bond by any other name. Angew Chem Int Ed 50:52–59. [DOI] [PubMed] [Google Scholar]
- 28. Choudhary A, Raines RT (2011) Signature of n→π* interactions in α‐helices. Protein Sci 20:1077–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kuster DJ, Liu C, Fang Z, Ponder JW, Marshall GR (2015) High‐resolution crystal structures of protein helices reconciled with three‐centered hydrogen bonds and multipole electrostatics. PLoS One 10:e0123146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nemethy G, Phillips D, Leach S, Scheraga H (1967) A second right‐handed helical structure with the parameters of the Pauling‐Corey alpha‐helix. Nature 214:565–565. [DOI] [PubMed] [Google Scholar]
- 31. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jorgensen WL, Swenson CJ (1985) Optimized intermolecular potential functions for amides and peptides. Hydration of amides. J Am Chem Soc 107:1489–1496. [Google Scholar]
- 33. Steiner T (2002) The hydrogen bond in the solid state. Angew Chem Int Ed 41:49–76 [DOI] [PubMed] [Google Scholar]
- 34. Cordier F, Barfield M, Grzesiek S (2003) Direct observation of Cα‐Hα…O=C hydrogen bonds in proteins by interresidue h3JCαC′ scalar couplings. J Am Chem Soc 125:15750–15751. [DOI] [PubMed] [Google Scholar]
- 35. Horowitz S, Dirk LMA, Yesselman JD, Nimtz JS, Adhikari U, Mehl RA, Scheiner S, Houtz RL, Al‐Hashimi HM, Trievel RC (2013) Conservation and functional importance of carbon‐oxygen hydrogen bonding in AdoMet‐dependent methyltransferases. J Am Chem Soc 135:15536–15548. [DOI] [PubMed] [Google Scholar]
- 36. Gong H, Porter LL, Rose GD (2011) Counting peptide‐water hydrogen bonds in unfolded proteins. Protein Sci 20:417–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gould IR, Cornell WD, Hillier IH (1994) A quantum mechanical investigation of the conformational energetics of the alanine and glycine dipeptides in the gas phase and in aqueous solution. J Am Chem Soc 116:9250–9256. [Google Scholar]
- 38. Blanco S, Lesarri A, López JC, Alonso JL (2004) The gas‐phase structure of alanine. J Am Chem Soc 126:11675–11683. [DOI] [PubMed] [Google Scholar]
- 39. Pal TK, Sankararamakrishnan R (2008) Self‐contacts in Asx and Glx residues of high‐resolution protein structures: role of local environment and tertiary interactions. J Mol Graph Model 27:20–33. [DOI] [PubMed] [Google Scholar]
- 40. Vasudev PG, Banerjee M, Ramakrishnan C, Balaram P (2012) Asparagine and glutamine differ in their propensities to form specific side chain‐backbone hydrogen bonded motifs in proteins. Proteins 80:991–1002. [DOI] [PubMed] [Google Scholar]
- 41. Afzal AM, Al‐Shubailly F, Leader DP, Milner‐White EJ (2014) Bridging of anions by hydrogen bonds in nest motifs and its significance for Schellman loops and other larger motifs within proteins. Proteins 82:3023–3031. [DOI] [PubMed] [Google Scholar]
- 42. Hutchinson E, Thornton J (1996) PROMOTIF—a program to identify and analyze structural motifs in proteins. Protein Sci 5:212–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Richardson JS, Richardson DC (1988) Amino acid preferences for specific locations at the ends of alpha helices. Science 240:1648–1652. [DOI] [PubMed] [Google Scholar]
- 44. Fersht AR (1987) The hydrogen bond in molecular recognition. Trends Biochem Sci 12:301–304. [Google Scholar]
- 45. Ben‐Naim A (1991) The role of hydrogen bonds in protein folding and protein association. J Phys Chem 95:1437–1444. [Google Scholar]
- 46. Aurora R, Rose GD (1998) Helix capping. Protein Sci 7:21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. O'Meara MJ, Leaver‐Fay A, Tyka MD, Stein A, Houlihan K, DiMaio F, Bradley P, Kortemme T, Baker D, Snoeyink J, Kuhlman, B (2015) Combined covalent‐electrostatic model of hydrogen bonding improves structure prediction with Rosetta. J Chem Theory Comput 11:609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shi Y, Xia Z, Zhang J, Best R, Wu C, Ponder JW, Ren P (2013) Polarizable atomic multipole‐based AMOEBA force field for proteins. J Chem Theory Comput 9:4046–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lindorff‐Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE (2010) Improved side‐chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct Funct Bioinform 78:1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, Van Der Spoel D, Hess B, Lindahl E (2013) GROMACS 4.5: a high‐throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29:845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG (1995) A smooth particle mesh Ewald method. J Chem Phys 103:8577–8593. [Google Scholar]
- 52. Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR (1984) Molecular dynamics with coupling to an external bath. J Chem Phys 81:3684–3690. [Google Scholar]
- 53. Bussi G, Donadio D, Parrinello M (2007) Canonical sampling through velocity rescaling. J Chem Phys 126:1–7. [DOI] [PubMed] [Google Scholar]
- 54. Hess B (2008) P‐LINCS: a parallel linear constraint solver for molecular simulation. J Chem Theory Comput 4:116–122. [DOI] [PubMed] [Google Scholar]
- 55. Miyamoto S, Kollman PA (1992) SETTLE: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J Comput Chem 13:952–962. [Google Scholar]
- 56. Best RB, Zheng W, Mittal J (2014) Balanced protein−water interactions improve properties of disordered proteins and non‐specific protein association. J Chem Theory Comput 25:5113–5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Newberry RW, Bartlett GJ, Vanveller B, Woolfson DN, Raines RT (2013) Signatures of n→π * interactions in proteins. Protein Sci 23:284–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen‐bonded and geometrical features. Biopolymers 22:2577–2637. [DOI] [PubMed] [Google Scholar]
- 59. Crooks G, Hon G, Chandonia J, Brenner S (2004) WebLogo: a sequence logo generator. Genome Res 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information