

Abstract
A subset of proteins containing NlpC/P60 domains are bacterial peptidoglycan hydrolases that cleave noncanonical peptide linkages and contribute to cell wall remodeling as well as cell separation during late stages of division. Some of these proteins have been shown to cleave peptidoglycan in Mycobacterium tuberculosis and play a role in Mycobacterium marinum virulence of zebra fish; however, there are still significant knowledge gaps concerning the molecular function of these proteins in Mycobacterium avium subspecies paratuberculosis (MAP). The MAP genome sequence encodes five NlpC/P60 domain‐containing proteins. We describe atomic resolution crystal structures of two such MAP proteins, MAP_1272c and MAP_1204. These crystal structures, combined with functional assays to measure peptidoglycan cleavage activity, led to the observation that MAP_1272c does not have a functional catalytic core for peptidoglycan hydrolysis. Furthermore, the structure and sequence of MAP_1272c demonstrate that the catalytic residues normally required for hydrolysis are absent, and the protein does not bind peptidoglycan as efficiently as MAP_1204. While the NlpC/P60 catalytic triad is present in MAP_1204, changing the catalytic cysteine‐155 residue to a serine significantly diminished catalytic activity, but did not affect binding to peptidoglycan. Collectively, these findings suggest a broader functional repertoire for NlpC/P60 domain‐containing proteins than simply hydrolases.
Keywords: Mycobacterium, Johne's disease, crystal structure, peptidoglycan, proteins, antigens, paratuberculosis
Short abstract
Introduction
Mycobacterium avium subspecies paratuberculosis (MAP) causes a chronic intestinal disease of ruminants including cattle, sheep, and goats. This disease, termed Johne's disease, has a worldwide distribution and >90% prevalence in U.S. dairy cattle herds,1 which results in an economic toll of more than 200 million dollars annually.2 The bacterium is transmitted from cow‐to‐calf via the fecal–oral route and through contaminated milk. Thus, a multipronged research approach that encompasses epidemiology, pathogenesis, diagnostics, vaccine development, and ideal herd management strategies is warranted to reduce the impact of Johne's disease on farms.3, 4, 5
This economically significant veterinary pathogen is among the Mycobacteria which all contain a unique cell wall structure that includes mycolic acids covering the lipoarabinogalactan layer with a foundational lattice of muropeptide cross‐linked peptidoglycan.6 Implicit with this sturdy cell wall are hydrolytic mechanisms required for cell division, peptidoglycan remodeling, nutrient uptake, as well as other biological processes. Thus the peptidoglycan layer in particular must be constantly modified during growth and at least partially disassembled to separate daughter cells during the final stages of cell division. This is accomplished by three classes of enzymes: glycosidases, which cleave bonds between the repeating disaccharides; amidases, which hydrolyze the bond between the peptide moiety and the N‐acetylmuramic acid; and finally, the NlpC/P60 endopeptidases, which cleave the interchain bonds of the muropeptide. The high lipid content and heavy cross‐linking of these layers in the cell wall is thought to enable mycobacteria to resist antibiotics, UV exposure, heat treatment, and other environmental insults.7, 8, 9, 10
Proteins containing the NlpC/P60 superfamily domain (pfam00877) are important in bacterial physiology11 and virulence.12, 13 A Mycobacterium tuberculosis gene, Rv2190c, encodes an NlpC/P60 protein that is required for virulence in the mouse model of TB and was shown to be immunogenic in the context of infection.12 The NlpC/P60 domain typically has hydrolytic activity and enzymes containing this domain include acyltransferases, amidases, and endopeptidases.11 NlpC/P60 proteins have also been localized at the septa of dividing bacteria and mutations are phenotypically manifested by septation defects,14, 15 suggesting a role in separation of daughter cells during cell division or general maintenance of the cell wall.16 Two genes in Mycobacterium marinum, a fish pathogen, which encode NlpC/P60 domain‐containing proteins are required for infection in zebra fish and cord formation seen in virulent mycobacteria,13 thus adding to the diverse functions associated with proteins containing this domain.
The MAP genome sequence encodes a total of five NlpC/P60 domain‐containing proteins, which have been designated MAP_0036, MAP_1203 (ortholog of RipA in M. tuberculosis), MAP_1204 (RipB in M. tuberculosis), MAP_1272c (RipD in M. tuberculosis), and MAP_1928c (Rv2190c in M. tuberculosis) in the bovine strain K‐10.17 This is the same number of NlpC/P60 proteins annotated in M. tuberculosis 12 and Corynebacterium glutamicum genomes.15 MAP_1203 shows increased expression, nearly 25‐fold, when MAP is exposed to milk,18 a hyperosmolar environment that triggers a more invasive form of the bacterium. Although both MAP_1204 and MAP_1272c proteins are immunogenic, MAP_1272c in particular has been observed as a strong antigen in multiple studies.19, 20, 21 Our group has also evaluated MAP_1204 and MAP_1272c as part of a subunit vaccine cocktail against Johne's disease in mice.21 Those results showed a significant decrease in MAP colonization of the vaccinated mouse intestine. Two monoclonal antibodies were also developed against MAP_1272c and their binding sites and biochemical properties have been determined.19 We have now extended those studies by examining the structure and catalytic properties of these proteins in MAP. Here we demonstrate that while both proteins display a conserved substrate‐binding groove, one NlpC/P60 domain‐containing protein hydrolyzes peptidoglycan while a second one is nonhydrolytic.
Results
Characteristics of MAP_1204 and MAP_1272c
Based on BLAST analysis of the NCBI sequence database combined with experimental data,19 we suggest that the MAP_1272c protein is 279 amino acids in length (29.2 kDa) and not 316 as annotated in the K‐10 genome sequence.17 MAP_1204 encodes a 244 amino acid protein with a calculated molecular weight of 25.4 kDa. Both proteins contain an N‐terminal signal sequence predicted by the SignalP 4.1 server.22 MAP_1204 cleavage is predicted between Ala‐33 and Asp‐34, whereas MAP_1272c cleavage is predicted between Ala‐29 and Asp‐30 of the revised annotation. While MAP_1204 contains a canonical NlpC/P60 domain near its C‐terminal end (amino acids 128–243) similar to most mycobacterial NlpC/P60 proteins, the domain is more centrally located in MAP_1272c (Fig. 1). Specifically, it is present at 52–166 amino acids in MAP_1272c, followed by a glutamine‐rich region (169–273) containing the repeat sequence (QQAPLQ)6 .
Figure 1.
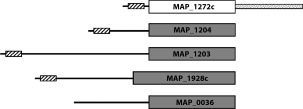
Schematic of the five NlpC/P60 domain‐containing proteins present in the MAP genome. Locations of the NlpC/P60 domain within each protein are shown drawn to scale based upon their preproteins; enzymatically active domains are colored grey while inactive domains are white. The locations of signal peptide regions are designated with a striped box. The dotted region at the C‐terminus of MAP_1272c represents a polyglutamine repeat of unknown structure and function.
NlpC/P60 domain structures in MAP_1272c and MAP_1204
A truncated form of MAP_1272c, consisting of residues 27–168, was overexpressed, purified, and crystallized. These crystals belong to the primitive monoclinic space group P21 and diffracted X‐rays to 1.75 Å Bragg‐limiting resolution. The truncated MAP_1272c structure (Protein Data Bank entry 3GT2) was determined ab initio by single‐wavelength anomalous dispersion (SAD) phasing using diffraction data collected from crystals of protein labeled in vivo with selenium‐substituted methionine. This structure was refined to R/Rfree values of 18.3 and 20.1%, respectively (Table 1). Similarly, a truncated form of MAP_1204, consisting of residues 109–244, was overexpressed, purified, and crystallized. These crystals belong to the primitive cubic space group P213 and diffracted X‐rays to 2.4 Å Bragg‐limiting resolution. The truncated MAP_1204 structure (Protein Data bank Entry 3I86) was determined by molecular replacement using the refined truncated MAP_1272c structure as a search model. This structure was subsequently refined to R/Rfree values of 22.0 and 28.0%, respectively (Table 2).
Table 1.
Data Collection Statistics, Determination, and Refinement for MAP_1272c
| Parameters | Data collection and structure solution |
|---|---|
| Beamline | APS 22‐ID |
| Wavelength (Å) | 0.97242 |
| Space group | P21 |
| Unit cell dimensions (Å; °) | a = 34.72, b = 53.21, c = 38.33; β = 102.95 |
| Molecule/ASU | 1 |
| Resolution limits (Å) | 22.68–1.75 (1.81–1.75)a |
| Completeness (%) | 96.1 (78.1) |
| Total reflections | 47,147 |
| Unique reflections | 13,278 |
| R merge (%)b | 7.2 (21.6) |
| ≪I>/<σ≫ | 9.45 (3.31) |
| Redundancy | 3.6 |
| Heavy atom sites (Se) | 7 |
| Figure of Merit <m> | 0.64 |
| Refinement statistics | |
| R cryst/R free (%)c | 18.3/20.1 |
| RMS deviations from ideality | |
| Bond length (Å) | 0.006 |
| Bond angle (°) | 0.971 |
| Dihedral angle (°) | 15.002 |
| Ramachandran core (disallowed) (%) | 93.3 (1.9) |
| Average B factor (Å2) | 23.63 |
| RMSD of B factor (Å2) | 6.37 |
| Proteins atoms modeled | 993 |
| Ordered solvent molecules | 100 |
Numbers in parentheses correspond to the highest resolution shell.
R merge = ΣhΣi|I i(h) − <I(h)>|/ΣhΣi I i(h), where I i(h) is the i th measurement of reflection h and <I(h)> is the weighted mean of all measurements of h.
R = Σh|F obs(h) − F calc(h)|/Σh|F obs|. R cryst and R free were calculated from the working and test reflection sets, respectively. The test set constituted 10% of the total reflections not used in refinement.
Table 2.
Data Collection Statistics, Determination, and Refinement for MAP_1204
| Parameters | Data collection and structure solution |
|---|---|
| Beamline | APS 22‐BM |
| Wavelength (Å) | 1.000 |
| Space group | P21 3 |
| Unit cell dimensions (Å) | a = 97.11 |
| Resolution limits (Å) | 43.43–2.40 (2.64–2.40)a |
| Completeness (%) | 99.6 (100) |
| Total reflections | 12,254 |
| Unique reflections | 1,213 |
| R merge (%)b | 16.9 (85.6) |
| ≪I>/<σ≫ | 14.1 (2.2) |
| Redundancy | 13.0 |
| Refinement statistics | |
| R cryst/R free (%)c | 22.0/28.0 |
| RMS deviations from ideality | |
| Bond length (Å) | 0.008 |
| Bond angle (°) | 1.21 |
| Dihedral angle (°) | 16.995 |
| Ramachandran core (disallowed) (%) | 91.1 (1.9) |
| Average B factor (Å2) | 30.21 |
| RMSD of B factor (Å2) | 7.72 |
| Proteins atoms modeled | 2042 |
| Ordered solvent molecules | 45 |
Numbers in parentheses correspond to the highest resolution shell.
R merge = Σh Σi|I i(h) − <I(h)>|/Σh Σi I i(h), where I i(h) is the i th measurement of reflection h and <I(h)> is the weighted mean of all measurements of h.
R = Σh|F obs(h) − F calc(h)|/Σh ΣF obs|. R cryst and R free were calculated from the working and test reflection sets, respectively. The test set constituted 5% of the total reflections not used in refinement.
The structures of the two NlpC/P60 domains are characterized by a helix–loop–helix in their N‐terminal regions, while their C‐terminal regions contain a β hairpin motif consisting of six antiparallel strands [Fig. 2(A)]. As might be expected for two proteins that share 52% sequence identity with one another, the structures of MAP_1204 and MAP_1272c share a high level of structural homology when superimposed (Supporting Information, Fig. S1); 135 of 136 residues align within 5.0 Å distance with a root‐mean‐square deviation (RMSD) of 0.69 Å. Consistent with these minor differences in tertiary structure, both MAP_1272c and MAP_1204 share a striking feature when viewed as molecular surfaces. Specifically, an elongated groove is formed at the interface between the helix–loop–helix and β hairpin regions [Fig. 2(B)]. This grove very likely represents a substrate access channel since residues that comprise the canonical catalytic triad of NlpC/P60 domains lie at the center of the groove (as is discussed below). Nevertheless, comparison of the electrostatic surface potentials within this groove shows notable differences between MAP_1272c and MAP_1204 [Fig. 2(C)]. Whereas the MAP_1204 channel is largely uncharged, the corresponding surface from MAP_1272c appears highly acidic. Given the proximity of this groove to the NlpC/P60 active site, these electrostatic differences may reflect both the substrate/ligand preferences and distinct physiological roles of MAP_1204 and MAP_1272c, though the specific function of MAP_1272c remains unclear.
Figure 2.
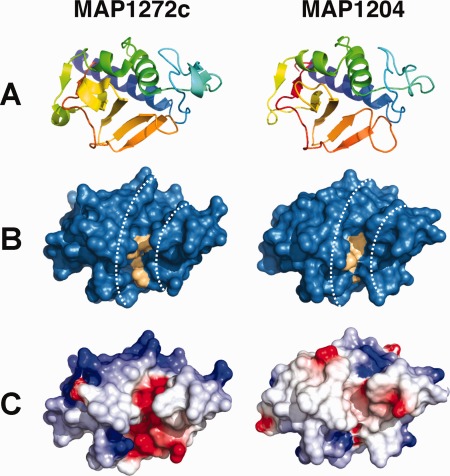
Three‐dimensional structures of NlpC/P60 domains from MAP_1272c and MAP_1204. (A) Refined 1.75 Å resolution structure of MAP_1272c and 2.4 Å resolution structure of MAP_1204. The proteins are rendered as cartoons with the N‐terminus in red and the C‐terminus in blue. (B) Molecular surface of MAP_1272c and MAP_1204, where residues of the catalytic triad are colored light orange. Note that the dashed white lines represent the boundaries of putative substrate/ligand‐binding channels in each protein. (C) Distribution of electrostatic potential at the surface of each protein contoured at ±5e/kT. The orientation of all structures has been kept constant in each panel for clarity.
Considering NlpC/P60 domains more broadly, extensive homology between MAP_1272c and MAP_1204 is also found in M. tuberculosis RipD (Rv1566c), which is the most closely related structure in the PDB (4JXB; 87% identity to MAP_1272c).23 However, when the MAP structures are superimposed onto other more distantly related NlpC/P60 domains, such as those found in Bacillus cereus BCE_2878 (3H41; 36% identity)24 and Desulfovibrio vulgaris DVU_0896 (3M1U; 20% identity; unpublished), more structural diversity becomes evident (Fig. 3). While this diversity expectedly becomes more significant as identity decreases, it is important to note that most of the divergence is seen in regions removed from the NlpC/P60 catalytic core. Much similar to the changes in electrostatic potential mentioned above, these structural differences at the active site periphery have probably accompanied alterations in substrate/ligand specificity and function during the evolution of NlpC/P60 domain‐containing proteins.
Figure 3.
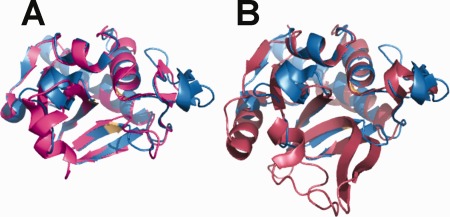
Superposition of NlpC/P60 domains from MAP_1272c with non‐mycobacterial NlpC/P60 domain‐containing proteins. (A) Shown is superposition of MAP_1272c (blue) and B. cereus BCE_2878 (magenta) and (B) superposition of NlpC/P60 domains from MAP_1272c (blue) and D. vulgaris DVU_0896 (rust). Note that residues of the MAP_1272c catalytic triad are colored light orange for the purposes of orientation. The orientation of all structures is identical to that shown in Figure 2.
Divergent catalytic triads in MAP_1204 and MAP_1272c
A catalytic triad that consists of a cysteine, histidine, and a polar residue enables the hydrolytic activity that is conserved within the NlpC/P60 superfamily.11 In this regard, all but one of the MAP NlpC/P60 proteins contain the signature motif “FDCSGL,” where the cysteine residue is part of the catalytic triad. The lone exception to this is MAP_1272c, where the Cys residue at position 82 is replaced by Ala [Fig. 4(A)]. Amino acid sequence alignment of other mycobacterial homologs of MAP_1272c shows the same alanine residue is conserved (Supporting Information, Fig. S2). However, alignment with all MAP NlpC/P60 proteins, except MAP_1272c, to corresponding mycobacterial homologs shows the cysteine residue is preserved (Supporting Information, Figs. S3–S6).
Figure 4.
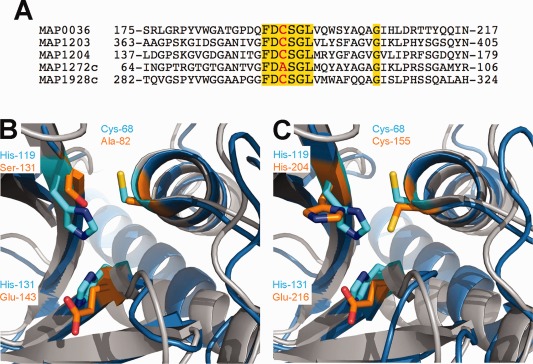
Comparison of the catalytic triads of MAP NlpC/P60 proteins, and E. coli Spr. (A) Shown is a partial alignment of the five MAP proteins surrounding the cysteine catalytic residue. Conserved amino acids among all 5 proteins are highlighted and the catalytic residue is shown in red. (B) Three‐dimensional overlay of MAP_1272c (blue) with E. coli Spr (grey). Residues comprising the catalytic triads have been rendered as ball‐and‐stick and colored distinctly for MAP_1272c (orange) and Spr (cyan). (C) Overlay of MAP_1204 (blue) with E. coli Spr (grey). Residues comprising the catalytic triads have been rendered as ball‐and‐stick and colored distinctly for MAP_1204 (orange) and Spr (cyan). The identity and position of each residue of interest is inset.
Similar to MAP_1272c and MAP_1204, the putative cell wall anchored hydrolase, Spr, from Escherichia coli also belongs to the NlpC/P60 superfamily.25 The catalytic triad for Spr includes Cys‐68, His‐119, and His‐131.25 Superimposition of the Spr and MAP_1272c structures reveals that the amino acid side chains in the corresponding locations of MAP_1272c are Ala‐82, Ser‐131, and Glu‐143 [Fig. 4(B)]. Although it is interesting to note that these three side chains do not adopt the same relative orientations in MAP_1272c as is found in Spr, the lack of a substrate or substrate analog in either crystal precludes a more thorough consideration of the functional consequences of such differences. Nevertheless, there is no proton in the alanine side chain that is sufficiently acidic as to be capable of generating a nucleophile that facilitates substrate hydrolysis. Intriguingly, this limitation is not seen for MAP_1204, where side chain orientations of catalytic triad residues Cys‐155, His‐204, and Glu‐216 are congruent with one another [Fig. 4(C)].
MAP_1272c, but not MAP_1204, lacks hydrolytic activity against Bacillus subtilis peptidoglycan
Given the apparent absence of a catalytic site in MAP_1272c, we hypothesized that this mycobacterial NlpC/P60 protein may not have hydrolytic activity akin to that predicted for the E. coli Spr protein.25 To test this hypothesis, a suitable assay was designed to assess bacterial survival in the presence of exogenous quantities of peptidoglycan remodeling enzymes. The concept of this assay is straightforward in that the integrity of the bacterial cell should be compromised in the presence of NlpC/P60 hydrolase activity, and that cell wall integrity is essential for cell viability. Because the long generation time of MAP makes its use impractical for survival assays, we used Bacillus subtilis as a model Gram‐positive system for the studies presented here. The B. subtilis peptidoglycan substrate was also chosen since its muropeptide sequence (l‐Ala, d‐Glu, meso‐diaminopimelic acid, d‐Ala) is identical with that present in mycobacterial peptidoglycan.26
To establish whether MAP proteins were capable of acting upon B. subtilis peptidoglycan layers, we first assessed B. subtilis survival in the presence of MAP_0318. MAP_0318 is 85% identical to M. tuberculosis Rv3717, which is an N‐acetylmuramyl‐l‐alanine amidase known to play a role in cell wall formation and remodeling.27 Indeed, incubation with exogenously added MAP_0318 significantly diminished B. subtilis survival compared to the irrelevant control protein, IpaD, from Shigella flexneri [Fig. 5(A)]. A significant decrease in B. subtilis viability was also observed in the presence of MAP_1204 [Fig. 5(A)], suggesting that this NlpC/P60 protein is capable of hydrolyzing substrates found in B. subtilis peptidoglycan. To test whether this activity required an intact catalytic triad, we overexpressed and purified a mutant form of MAP_1204 where the active site Cys‐155 was replaced with serine (MAP_1204‐C155S). As expected, B. subtilis cells exposed to this mutant did not display a significant decrease in cell viability [Fig. 5(A)], even though this site‐directed mutation exhibited nearly identical structural features to its wild‐type counterpart as judged by circular dichroism spectropolarimetry (Supporting Information, Fig. S7). Similarly, B. subtilis exposed to exogenously added MAP_1272c showed little or no decrease in viability [Fig. 5(A)]; in fact, this result was largely indistinguishable from that obtained for the MAP_1204 active site mutant.
Figure 5.
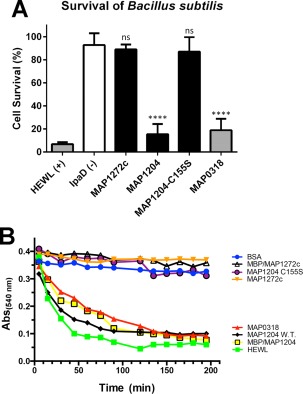
Measurement of hydrolytic activity using B. subtilis and peptidoglycan substrates. (A) Effect of MAP NlpC/P60 domains on viability of B. subtilis. Actively growing B. subtilis cells were incubated with 32 μM of each NlpC/P60 domain protein, known peptidoglycan modifying enzymes (e.g., hen egg white lysozyme (HEWL, 4 μM), or a negative control protein (i.e., S. flexneri IpaD) and plated to assess bacterial viability as described in materials and methods. Total colony forming units were counted, and the results were compared to a mock treatment (culture medium alone) to assess the percent loss of viability. The mean values of each treatment (±SD) were calculated and compared to that of the negative control (IpaD) by an unpaired t‐test to assess statistical significance. ****p ≤ 0.0001; ns, not significant. (B) Hydrolytic activity of MAP_1204 and MAP_1272c on B. subtilis peptidoglycan measured at 540 nm over time. BSA and HEWL are positive and negative controls, respectively.
This study was expanded using B. subtilis peptidoglycan fragments obtained commercially to rule out any potential nonspecific effects on B. subtilis viability. This purified peptidoglycan was incubated with each protein and degradation was measured for optical clearance at 540 nm over time. Both the nontagged and MBP‐tagged MAP_1204 proteins hydrolyzed peptidoglycan as did MAP_0318 [Fig. 5(B)]. Conversely, MAP_1272c and MAP_1204 C155S did not degrade peptidoglycan.
Collectively, these data demonstrate that MAP_1204 is an active peptidoglycan hydrolase and indicate that mutation of the proposed catalytic Cys‐155 residue greatly diminishes the enzyme's activity. Furthermore, the functional differences in the MAP_1204 and MAP_1204 C155S proteins are likely due to loss of specific side chains rather than altered protein structure (Supporting Information, Fig. S7). These results are also consistent with the notion that MAP_1272c lacks the hydrolytic activity characteristic of most other members of the NlpC/P60 family. Similarity searches suggest that this catalytic cysteine is missing only in mycobacteria. Thus, we propose that MAP_1272c is a noncatalytic member of the NlpC/p60 family found only in Mycobacterium spp.
MAP_1272c does not bind peptidoglycan as efficiently as MAP_1204
Both MAP_1272c and MAP_1204 have what appears to be a substrate‐binding groove on their surface. However, since MAP_1204 hydrolyzes peptidoglycan while MAP_1272c does not, we investigated whether both of these proteins might bind peptidoglycan [Fig. 2(B)]. Purified recombinant proteins were tested for their ability to bind insoluble peptidoglycan fragments from B. subtilis. Consistent with the functional assays in Figure 5, MAP_1204 was able to bind peptidoglycan since this protein accumulated in the pellet with the peptidoglycan [Fig. 6(A)]. Conversely, MAP_1272c was observed primarily in the supernatant suggesting that MAP_1272c has only weak affinity for peptidoglycan. The positive control, lysozyme, did bind peptidoglycan, whereas the negative control, BSA, did not [Fig. 6(A,B)]. The MBP fusions of MAP_1204 and MAP_1272c showed the same peptidoglycan binding characteristics as their untagged counterparts, yet the size of the MBP fusion proteins eliminated comigration with the peptidoglycan fragments [Fig. 6(B)]. Both the wild‐type and C155S mutant of MAP_1204 bound peptidoglycan in a similar manner [Fig. 6(C)], suggesting that a mutation in the catalytic site did not significantly affect binding, only hydrolysis. Thus, it remains unknown as to what substrate is recognized by the binding groove on the surface of MAP_1272c.
Figure 6.
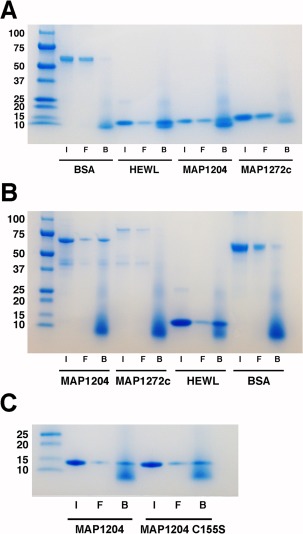
SDS‐PAGE analysis of fractions from the peptidoglycan binding assay. MAP proteins without terminal tags are shown (A) along with positive (HEWL) and negative (BSA) controls. I = input protein, F = free/unbound protein from the supernatant, and B = bound protein from the pellet fraction. (B) MBP–MAP fusion proteins are shown along with positive (HEWL) and negative (BSA) controls. (C) The wild type and mutant (C155S) forms of MAP_1204 bound peptidoglycan in a similar manner. Protein size standards (in kDa) are at the left of each image.
Discussion
Both serendipitous and engineered deletions of NlpC/P60 protein‐encoding genes have been shown by others to reduce pathogenicity and vegetative growth of bacteria.13 As a consequence, members of this family of enzymes are considered attractive targets for next‐generation antibiotic development. A notable validation of NlpC/P60 proteins as antibiotic targets has been done for M. marinum IipA (homolog of MAP_1203), where the gene knockout was constructed and complemented to show a lack of growth and infectivity in macrophages.13 Since MAP is resistant to many antibiotics,28, 29 novel approaches must be explored in treating infections by this organism. Furthermore, since NlpC/P60 proteins are found in all Eubacteria, any compounds that are capable of inhibiting these enzymes have the potential to be developed into new, broad‐spectrum antibiotics. In this regard, the structural information presented here, combined with mutagenesis data consistent with identification of an active site and a slight, but functionally significant variation therein may prove useful in future screening for such inhibitory compounds.
Substitution of the catalytic cysteine of Rv1477 (MAP_1203 homolog) to an alanine demonstrated that Cys383 is an essential catalytic amino acid for peptidoglycan hydrolysis.30 Likewise, a separate study showed that mutation of glutamic acid to alanine in Rv1477 inactivates catalysis by changing the spatial configuration of Cys383.31 However, no one has examined Rv1478 or its counterpart, MAP_1204, using mutational substitutions. Thus, our work extends these findings by demonstrating that the Cys‐to‐Ser substitution in MAP_1204 also renders the NlpC/P60 domain enzymatically inactive but does not affect binding to peptidoglycan.
During the course of preparing this manuscript, it was reported that another mycobacterial protein, RipD/Rv1566c from M. tuberculosis, also displayed a conspicuous lack of hydrolase activity that we observed for MAP_1272c.23 In fact, owing to the high levels of identity between the RipD and MAP_1272c NlpC/P60 domains mentioned above, Both et al.23 used our refined model of MAP_1272c to determine the structure of the RipD core domain. RipD was shown to lack a catalytic cysteine, and the same substitution of a cysteine for alanine within the active site was reported for RipD as for MAP_1272c. Furthermore, RipD contains 8 repeats of QAPVQ at its C‐termini, which is similar to the six QQAPLQ repeats in MAP_1272c. Thus, it appears that RipD is the M. tuberculosis ortholog of MAP_1272c. However, given the implicit biochemical restrictions to forming the NlpC/P60 catalytic triad, and relative permissibility of the third position, which requires only a polar residue,11 it seems likely that noncatalytic NlpC/P60‐containing proteins exist in other bacteria and have yet to be discovered.
Both et al.23 have shown that noncatalytic forms of NlpC/P60 domains have diversified to include peptidoglycan binding in addition to hydrolytic activities. The NlpC/P60 domain of RipD did bind peptidoglycan fragments, but full‐length RipD did not. In our study, neither the full length nor the Nlpc/P60 region of MAP_1272c was able to bind peptidoglycan. Importantly, our crystal structure of MAP_1272c reveals a prominent, negatively charged substrate/ligand‐binding groove on the surface of MAP_1272c. This observation further supports a role for MAP_1272c as a ligand binding protein for a substrate other than peptidoglycan, which tentatively broadens the role of NlpC/P60 domain containing proteins.
Considerations of NlpC/P60 domains aside, both catalytic and noncatalytic members of a protein family have been observed previously. One such example is members of the NTF2‐like protein superfamily.32 Here the catalytic forms include enzymes that function intracellularly in polyketide and natural product biosynthesis, while the extracellular, noncatalytic proteins in this group possess small molecule binding activity.32 Another example can be derived from the chymotrypsin‐like serine proteases found in neutrophils. In this case, neutrophil azurophilic granules contain four enzymatically active serine proteases which include neutrophil elastase, cathepsin G, proteinase‐3, and neutrophil serine protease‐4 along with a single nonactive member, named azurocidin.33, 34, 35 Interestingly, while azurocidin also contains point mutations that render its catalytic triad inactive,36 a body of literature has also reported chemotactic and immunoregulatory activity associated with release of azurocidin during inflammatory events (reviewed in Ref. 35). Thus, noncatalytic members of diverse protein families are not simply restricted to ligand binding roles, but can serve as signaling mediators as well.
When compared to other bacterial pathogens, a lingering issue that confounds many of the overriding scientific advancements in MAP biology and Johne's disease is that there are comparatively few reagent systems available. An extensive recombinant protein repository for MAP provides a vital and powerful tool for proteome‐ and genome‐scale research of this organism.37 Such a resource makes it possible to analyze the activities of proteins in cell growth, maintenance, regulation, survival, and pathogenesis. Additionally, this resource can provide reagents for diagnostics38, 39 and functional proteomic experiments, including both the structural studies described herein as well as defining protein–protein interactions. Finally, tools such as this repository build on genomic information by aiding in the functional annotation of the hundreds of hypothetical proteins. Therefore, this work continues to build on important advances in understanding MAP biology for future countermeasures against Johne's disease.
Materials and Methods
Protein production and purification
Full‐length expression and purification of MAP_1203, MAP_1204, and MAP_1272c has been described elsewhere using the maltose binding protein (MBP) expression system.19, 37 In addition to those MBP fusion proteins, truncated versions of these genes were fused to a polyhistidine tag and Tobacco Etch Virus (TEV) recognition site and subcloned into the E. coli expression vector pT7HMT using methods previously described.40 A series of His‐tagged MAP_1272c truncations have been described in detail elsewhere19; however, this article made extensive use of only a form of MAP_1272c consisting of residues Ala27 to Thr168. Similarly, MAP_1204 was truncated such that only the 136 residues from Val109 to Tyr244 were expressed. The sequence‐confirmed plasmids were transformed into E. coli strain BL21(DE3) for overexpression and purification, which were carried out according to the general protocols outlined previously.40 Upon completing the initial affinity purification, the polyhistidine fusion tag was proteolytically removed by adding recombinant TEV protease as described previously.40 The epitope tag, protease and most contaminating species were removed by IMAC chromatography and the flow through was buffer exchanged into 20 mM sodium formate buffer (pH 3.5). Final purification was achieved with Resource S cation exchange chromatography using a GE Lifesciences Akta fast protein liquid chromatography (FPLC) system using a linear gradient of 0–1M NaCl in 20mM sodium formate pH 3.5.
MAP_1272c from the strain K‐10 genome may have been incorrectly annotated. Its 316 annotated residues start with the sequence VRSQ; however, comparison with other annotations of M. avium complex strains has suggested that this protein is likely only 279 residues long with translation beginning at the Met38 of the annotated K‐10 sequence. Therefore, we adopted the convention used in our previous study that set Met38 as Met1, making the full‐length MAP_1272c protein from 1 to 279 amino acids and the first 25 amino acids comprising the signal sequence.19
Preparation of selenomethionine proteins
Escherichia coli methionine auxotroph strain B834(DE3) was transformed with a plasmid encoding MAP_1272c residues Ala27–Thr168) and then grown in 100 mL Luria Broth (LB) at 37°C to an OD600nm of ∼0.7. Cells were harvested by centrifugation and washed once with minimal media before inoculation into 1 L of M9 minimal salts media supplemented with 5 mg l‐(+)‐selenomethione (SeMet). Cells were grown at 37°C to OD600nm of 0.75 and then cooled to 18°C. IPTG was added to 1 mM along with an additional 5 mg SeMet. After overnight induction at 18°C, the protein was purified identically to the native sample.
Protein crystallization
The truncated MAP_1272c protein was dialyzed extensively against ddH2O and concentrated by centrifugal ultrafiltration to 10 mg/mL as judged by UV absorption spectrophotometry. Initial crystallization screening was carried out by vapor diffusion of hanging drops using commercially available sparse‐matrix kits (Hampton Research, Aliso Viejo, CA). Optimized crystals were produced by the hanging drop vapor diffusion technique. Rod‐shaped crystals grew in drops composed of 1 μL protein and 1 μL of well buffer (0.1M BisTris–HCl, 1.6M ammonium sulfate, 4% (v/v) polyethylene glycol 400), and equilibrated over 500 μL of the same buffer. Crystallization of the selenomethionine‐substituted protein was carried out using an identical protocol. Crystals of the truncated MAP_1204 protein were grown by hanging drop vapor diffusion by mixing 1 μL protein at 10 mg/mL in ddH2O with 1 μL of 1:1 diluted precipitant solution containing 22% (w/v) polyethylene glycol 3,350, 10% (v/v) 2‐propanol, and 0.1M sodium HEPES, pH 7.5. We also obtained crystals of a third NlpC/P60 protein, MAP_1203, but these crystals did not diffract X‐rays in an interpretable manner.
Diffraction data collection, structure solution, and refinement
Individual SeMet‐labeled crystals of truncated MAP_1272c were briefly transferred to a cryoprotection buffer consisting of well buffer with 30% (v/v) ethylene glycol and flash cooled in liquid nitrogen. Three‐hundred and sixty 1° oscillation images were recorded at a high‐energy remote wavelength for the Selenium K‐edge (0.9724 Å; 12750 eV). Individual reflections were indexed, integrated, and merged such that Friedel mates were kept separate using the HKL2000 software package.41 Selenium sites were identified and refined using the AutoSol package prior to automated model building in AutoBuild, as implemented in PHENIX.42 The final model, including SeMet substitutions, was the product of iterative cycles of manual building in COOT43 and refinement using PHENIX. Data collection and refinement statistics are reported in Table 1. With respect to the MAP_1272c polypeptide chain, the final model consists of residues Gly32–Leu166, contiguous. Additional information can be found by consulting the Protein Data Bank (PDB) accession code 3GT2.
Prior to flash cooling in liquid nitrogen, MAP_1204 crystals were briefly transferred to a cryoprotection solution identical to the precipitant but supplemented with 2‐propanol to a final concentration of 18% (v/v). A native MAP_1204 dataset was collected at 12398 eV (1 Å) at APS beamline 22‐BM. Reflections from 360 1° images were processed, indexed, and scaled in HKL2000. The relatively high R merge value associated with this diffraction dataset likely arises from the observational redundancy provided by the high‐symmetry cubic space group of this crystal. Other metrics of data quality, such as ≪I>/<σ≫, are in good agreement with generally accepted criteria for defining the resolution limits of X‐ray diffraction data.44, 45 The refined coordinates of the MAP_1272c truncation were used as a search model to perform molecular replacement in PHENIX.42 The resultant model was iteratively refined and manually modified as described above, with the exception of two cycles of simulated annealing to remove model phase bias. Although two copies of the MAP_1204 polypeptide were found in the asymmetric unit of this crystal form, no noncrystallographic symmetry averaging was used during the process of building and refinement. Final data collection and model refinement statistics are reported in Table 2. The final model accounts for the entire Val109–Tyr244 polypeptide contiguously in both chains. Additional information can be found by consulting the Protein Data Bank (PDB) accession code 3I86.
Circular dichroism (CD) spectropolarimetry
CD spectropolarimetry was used to assess the overall secondary structure content in MAP NlpC/P60 proteins. Purified MAP_1272c, MAP_1204, or MAP_1204 C155S proteins were dissolved in phosphate‐buffered saline (pH 7.4) and were passed through a 0.45 μm centrifugal filter immediately prior to analysis. Samples were diluted to a final concentration such that the OD280nm was equal to 0.5, which corresponds almost exactly to ∼ 0.5 mg/mL for all proteins. CD spectra were measured from 190 to 260 nm in scanning mode using a Jasco J‐815 spectropolarimeter. Five replicate spectra were collected in normal sensitivity mode with a 1 nm bandwidth at a speed of 50 nm/min prior to averaging and mathematical smoothing using software provided by the manufacturer. Samples were not degassed, nor were spectra collected under a vacuum; thus, the observations at shorter wavelengths should be considered unreliable.
Peptidoglycan degradation assay
Purified Bacillus subtilis peptidoglycan (Sigma) was resuspended in 50 mM NaH2PO4 (pH 6.0) to a concentration of 0.25 mg/mL and added to a 96‐well plate. Peptidoglycan was incubated with 32 μM of each recombinant protein as determined in pilot studies. Lysozyme, which served as the positive control, was used at a concentration of 4 μM. All reactions had a final volume of 200 μL and were performed in a 96‐well plate at room temperature with shaking at 200 rpm on a WVR microplate shaker. Absorbance was measured at 540 nm every 15 min using a Victor X3 plate reader (Perkin Elmer). Data were assembled and analyzed using GraphPad Prism 6.0.
Bacillus subtilis viability assay
An LB broth culture of B. subtilis was incubated overnight at 37°C with shaking at 200 rpm. Mid‐log phase B. subtlilis cells (OD600nm = 0.1) were added to tubes containing 20 µM of lysozyme or recombinant protein diluted in 0.1× phosphate‐buffered saline (PBS; 15mM NaCl, 1mM Na2HPO4; pH 7.3) as indicated. The IpaD protein from Shigella flexneri was used as a negative control.46 After 60 min, enzyme activity was quenched in a 5 min ice bath and serial dilutions were plated on LB broth and incubated overnight at 37°C to determine percent survival. Each survival assay was run in triplicate.
Peptidoglycan binding assay
Proteins were quantitated by BCA assay and loaded onto commercial SDS‐PAGE gels (BioRad) stained with GelCode blue (Thermo Scientific) to standardize between samples. The binding assay was performed essentially as described previously.30 Briefly, insoluble trichloroacetic acid treated peptidoglycan (1 mg/mL) from Staphylococcus aureus (Sigma chemicals) or Bacillus subtilis (Sigma chemicals) was suspended in binding buffer (0.15M NaCl and 25mM Na‐Hepes, pH 7.0) and incubated with 0.2 mg/mL of each recombinant protein or the positive and negative controls, hen egg white lysozyme (HEWL) and bovine serum albumin (BSA), all of which were dialyzed in binding buffer. The protein–peptidoglycan mixture was incubated for 5 min at 22°C to allow binding. The samples were centrifuged at 12,000g for 5 min and only the top half of the supernatant was collected. The remaining supernatant was then removed and discarded. Both the supernatant and pellet fractions were mixed with loading dye (1% SDS; 50mM Tris, pH 6.8; 1% 2‐mercaptoethanol; 10% glycerol) and then separated on 12% SDS‐PAGE gels followed by staining with GelCode blue.
Statistical analysis
Measurements of statistical significance regarding differences in B. subtilis survival were obtained by an unpaired t test using GraphPad Prism 6.0. The mean values of each treatment (±SD) were calculated and compared to that of the negative control (IpaD) by an unpaired t test to assess statistical significance. Notations: ****p ≤ 0.0001; ns, not significant.
Supporting information
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Figure 3.
Supporting Information Figure 4.
Supporting Information Figure 5.
Supporting Information Figure 6.
Supporting Information Figure 7.
Acknowledgment
The technical assistance of Janis K. Hansen (NADC) is gratefully acknowledged.
Importance: Johne's disease in ruminant livestock is caused by the bacterium Mycobacterium avium subspecies paratuberculosis. This research describes the functional aspects of two proteins that show promise in a subunit vaccine for Johne's disease. Through crystal structure determination and amino acid modification, we demonstrate that although both proteins have a similar structure, one of them lacked hydrolytic activity on peptidoglycan. We show that a specific amino acid is likely responsible for this lack of hydrolytic activity.
References
- 1. Lombard JE, Gardner IA, Jafarzadeh SR, Fossler CP, Harris B, Capsel RT, Wagner BA, Johnson WO (2013) Herd‐level prevalence of Mycobacterium avium subsp. paratuberculosis infection in United States dairy herds in 2007. Prev Vet Med 108:234–238. [DOI] [PubMed] [Google Scholar]
- 2. Stabel JR (1998) Johne's disease: a hidden threat. J Dairy Sci 81:283–288. [DOI] [PubMed] [Google Scholar]
- 3. Sorge US, Lissemore K, Godkin A, Jansen J, Hendrick S, Wells S, Kelton DF (2011) Changes in management practices and apparent prevalence on Canadian dairy farms participating in a voluntary risk assessment‐based Johne's disease control program. J Dairy Sci 94:5227–5237. [DOI] [PubMed] [Google Scholar]
- 4. Lu Z, Schukken YH, Smith RL, Grohn YT (2013) Using vaccination to prevent the invasion of Mycobacterium avium subsp. paratuberculosis in dairy herds: a stochastic simulation study. Prev Vet Med 110:335–345. [DOI] [PubMed] [Google Scholar]
- 5. Espejo LA, Godden S, Hartmann WL, Wells SJ (2012) Reduction in incidence of Johne's disease associated with implementation of a disease control program in Minnesota demonstration herds. J Dairy Sci 95:4141–4152. [DOI] [PubMed] [Google Scholar]
- 6. Abdallah AM, Gey van Pittius NC, Champion PA, Cox J, Luirink J, Vandenbroucke‐Grauls CM, Appelmelk BJ, Bitter W (2007) Type VII secretion–mycobacteria show the way. Nat Rev Microbiol 5:883–891. [DOI] [PubMed] [Google Scholar]
- 7. Donaghy J, Keyser M, Johnston J, Cilliers FP, Gouws PA, Rowe MT (2009) Inactivation of Mycobacterium avium ssp. paratuberculosis in milk by UV treatment. Lett Appl Microbiol 49:217–221. [DOI] [PubMed] [Google Scholar]
- 8. Whittington RJ, Marshall DJ, Nicholls PJ, Marsh IB, Reddacliff LA (2004) Survival and dormancy of Mycobacterium avium subsp. paratuberculosis in the environment. Appl Environ Microbiol 70:2989–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stabel JR, Lambertz A (2004) Efficacy of pasteurization conditions for the inactivation of Mycobacterium avium subsp. paratuberculosis in milk. J Food Prot 67:2719–2726. [DOI] [PubMed] [Google Scholar]
- 10. Eppleston J, Begg DJ, Dhand NK, Watt B, Whittington RJ (2014) Environmental survival of Mycobacterium avium subsp. paratuberculosis in different climatic zones of eastern Australia. Appl Environ Microbiol 80:2337–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Anantharaman V, Aravind L (2003) Evolutionary history, structural features and biochemical diversity of the NlpC/P60 superfamily of enzymes. Genome Biol 4:R11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Parthasarathy G, Lun S, Guo H, Ammerman NC, Geiman DE, Bishai WR (2012) Rv2190c, an NlpC/P60 family protein, is required for full virulence of Mycobacterium tuberculosis . PLoS One 7:e43429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gao LY, Pak M, Kish R, Kajihara K, Brown EJ (2006) A mycobacterial operon essential for virulence in vivo and invasion and intracellular persistence in macrophages. Infect Immun 74:1757–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wuenscher MD, Kohler S, Bubert A, Gerike U, Goebel W (1993) The iap gene of Listeria monocytogenes is essential for cell viability, and its gene product, p60, has bacteriolytic activity. J Bacteriol 175:3491–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tsuge Y, Ogino H, Teramoto H, Inui M, Yukawa H (2008) Deletion of cgR_1596 and cgR_2070, encoding NlpC/P60 proteins, causes a defect in cell separation in Corynebacterium glutamicum R. J Bacteriol 190:8204–8214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hett EC, Chao MC, Deng LL, Rubin EJ (2008) A mycobacterial enzyme essential for cell division synergizes with resuscitation‐promoting factor. PLoS Pathog 4:e1000001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li L, Bannantine JP, Zhang Q, Amonsin A, May BJ, Alt D, Banerji N, Kanjilal S, Kapur V (2005) The complete genome sequence of Mycobacterium avium subspecies paratuberculosis . Proc Natl Acad Sci USA 102:12344–12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Patel D, Danelishvili L, Yamazaki Y, Alonso M, Paustian ML, Bannantine JP, Meunier‐Goddik L, Bermudez LE (2006) The ability of Mycobacterium avium subsp. paratuberculosis to enter bovine epithelial cells is influenced by preexposure to a hyperosmolar environment and intracellular passage in bovine mammary epithelial cells. Infect Immun 74:2849–2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bannantine JP, Lingle CK, Stabel JR, Ramyar KX, Garcia BL, Raeber AJ, Schacher P, Kapur V, Geisbrecht BV (2012) MAP1272c encodes an NlpC/P60 protein, an antigen detected in cattle with Johne's disease. Clin Vaccine Immunol 19:1083–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li L, Munir S, Bannantine JP, Sreevatsan S, Kanjilal S, Kapur V (2007) Rapid expression of Mycobacterium avium subsp. paratuberculosis recombinant proteins for antigen discovery. Clin Vaccine Immunol 14:102–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stabel JR, Barnhill A, Bannantine JP, Chang YF, Osman MA (2012) Evaluation of protection in a mouse model after vaccination with Mycobacterium avium subsp. paratuberculois protein cocktails. Vaccine 31:127–134. [DOI] [PubMed] [Google Scholar]
- 22. Petersen TN, Brunak S, von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786. [DOI] [PubMed] [Google Scholar]
- 23. Both D, Steiner EM, Izumi A, Schneider G, Schnell R (2014) RipD (Rv1566c) from Mycobacterium tuberculosis: adaptation of an NlpC/p60 domain to a non‐catalytic peptidoglycan‐binding function. Biochem J 457:33–41. [DOI] [PubMed] [Google Scholar]
- 24. Xu Q, Abdubek P, Astakhova T, Axelrod HL, Bakolitsa C, Cai X, Carlton D, Chen C, Chiu HJ, Chiu M, et al (2010) Structure of the gamma‐D‐glutamyl‐L‐diamino acid endopeptidase YkfC from Bacillus cereus in complex with L‐Ala‐gamma‐D‐Glu: insights into substrate recognition by NlpC/P60 cysteine peptidases. Acta Cryst F66:1354–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aramini JM, Rossi P, Huang YJ, Zhao L, Jiang M, Maglaqui M, Xiao R, Locke J, Nair R, Rost B, et al (2008) Solution NMR structure of the NlpC/P60 domain of lipoprotein Spr from Escherichia coli: structural evidence for a novel cysteine peptidase catalytic triad. Biochemistry 47:9715–9717. [DOI] [PubMed] [Google Scholar]
- 26. Mahapatra S, Crick DC, McNeil MR, Brennan PJ (2008) Unique structural features of the peptidoglycan of Mycobacterium leprae . J Bacteriol 190:655–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Prigozhin DM, Mavrici D, Huizar JP, Vansell HJ, Alber T (2013) Structural and biochemical analyses of Mycobacterium tuberculosis N‐acetylmuramyl‐L‐alanine amidase Rv3717 point to a role in peptidoglycan fragment recycling. J Biol Chem 288:31549–31555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Whittington RJ, Whittington AM, Waldron A, Begg DJ, de Silva K, Purdie AC, Plain KM (2013) Development and validation of a liquid medium (M7H9C) for routine culture of Mycobacterium avium subsp. paratuberculosis to replace modified Bactec 12B medium. J Clin Microbiol 51:3993–4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shin SJ, Collins MT (2008) Thiopurine drugs azathioprine and 6‐mercaptopurine inhibit Mycobacterium paratuberculosis growth in vitro. Antimicrob Agents Chemother 52:418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Both D, Schneider G, Schnell R (2011) Peptidoglycan remodeling in Mycobacterium tuberculosis: comparison of structures and catalytic activities of RipA and RipB. J Mol Biol 413:247–260. [DOI] [PubMed] [Google Scholar]
- 31. Squeglia F, Ruggiero A, Romano M, Vitagliano L, Berisio R (2014) Mutational and structural study of RipA, a key enzyme in Mycobacterium tuberculosis cell division: evidence for the L‐to‐D inversion of configuration of the catalytic cysteine. Acta Cryst D70:2295–2300. [DOI] [PubMed] [Google Scholar]
- 32. Eberhardt RY, Chang Y, Bateman A, Murzin AG, Axelrod HL, Hwang WC, Aravind L (2013) Filling out the structural map of the NTF2‐like superfamily. BMC Bioinf 14:327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Korkmaz B, Horwitz MS, Jenne DE, Gauthier F (2010) Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol Rev 62:726–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Perera NC, Schilling O, Kittel H, Back W, Kremmer E, Jenne DE (2012) NSP4, an elastase‐related protease in human neutrophils with arginine specificity. Proc Natl Acad Sci USA 109:6229–6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Soehnlein O, Lindbom L (2009) Neutrophil‐derived azurocidin alarms the immune system. J Leukoc Biol 85:344–351. [DOI] [PubMed] [Google Scholar]
- 36. Iversen LF, Kastrup JS, Bjorn SE, Rasmussen PB, Wiberg FC, Flodgaard HJ, Larsen IK (1997) Structure of HBP, a multifunctional protein with a serine proteinase fold. Nat Struct Biol 4:265–268. [DOI] [PubMed] [Google Scholar]
- 37. Bannantine JP, Stabel JR, Bayles DO, Geisbrecht BV (2010) Characteristics of an extensive Mycobacterium avium subspecies paratuberculosis recombinant protein set. Protein Expr Purif 72:223–233. [DOI] [PubMed] [Google Scholar]
- 38. Bannantine JP, Waters WR, Stabel JR, Palmer MV, Li L, Kapur V, Paustian ML (2008) Development and use of a partial Mycobacterium avium subspecies paratuberculosis protein array. Proteomics 8:463–474. [DOI] [PubMed] [Google Scholar]
- 39. Bannantine JP, Paustian ML, Waters WR, Stabel JR, Palmer MV, Li L, Kapur V (2008) Profiling bovine antibody responses to Mycobacterium avium subsp. paratuberculosis infection by using protein arrays. Infect Immun 76:739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Geisbrecht BV, Bouyain S, Pop M (2006) An optimized system for expression and purification of secreted bacterial proteins. Protein Expr Purif 46:23–32. [DOI] [PubMed] [Google Scholar]
- 41. Otwinowski Z (1997) Processing of X‐ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. [DOI] [PubMed] [Google Scholar]
- 42. Adams PD, Grosse‐Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Cryst D58:1948–1954. [DOI] [PubMed] [Google Scholar]
- 43. Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Cryst D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 44. Evans PR (1999) Some notes on choices in data collection. Acta Cryst D55:1771–1772. [DOI] [PubMed] [Google Scholar]
- 45. Dauter Z (1999) Data‐collection strategies. Acta Cryst D55:1703–1717. [DOI] [PubMed] [Google Scholar]
- 46. Barta ML, Guragain M, Adam P, Dickenson NE, Patil M, Geisbrecht BV, Picking WL, Picking WD (YEAR) Identification of the bile salt binding site on IpaD from Shigella flexneri and the influence of ligand binding on IpaD structure. Proteins 80:935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Figure 3.
Supporting Information Figure 4.
Supporting Information Figure 5.
Supporting Information Figure 6.
Supporting Information Figure 7.