

Abstract
Two putative haloalkane dehalogenases (HLDs) of the HLD‐I subfamily, DccA from Caulobacter crescentus and DsaA from Saccharomonospora azurea, have been identified based on sequence comparisons with functionally characterized HLD enzymes. The two genes were synthesized, functionally expressed in E. coli and shown to have activity toward a panel of haloalkane substrates. DsaA has a moderate activity level and a preference for long (greater than 3 carbons) brominated substrates, but little activity toward chlorinated alkanes. DccA shows high activity with both long brominated and chlorinated alkanes. The structure of DccA was determined by X‐ray crystallography and was refined to 1.5 Å resolution. The enzyme has a large and open binding pocket with two well‐defined access tunnels. A structural alignment of HLD‐I subfamily members suggests a possible basis for substrate specificity is due to access tunnel size.
Keywords: haloalkane dehalogenase, DccA, Caulobacter crescentus, HLD‐I subfamily, substrate specificity, Saccharomonospora azurea
Introduction
Halogenated alkanes have been used as herbicides, refrigerants, fire retardants, solvents, pesticides, degreasers, and as intermediates in organic synthesis. Unfortunately, the toxicity and limited biodegradability of these compounds went unrecognized until well after they were released into the environment, polluting many industrial sites and being found in all components of the biosphere.1 Haloalkanes are considered recalcitrant pollutants that are toxic to humans and are also potential carcinogens. The worst offenders of the haloalkanes are the small (C1—C4), chlorinated compounds such as chloroethane (CE), 1,2‐dichloroethane (DCE), 1,2‐dichloropropane (DCP), and 1,2,3‐trichloropropane (TCP), which are included in the EPA priority pollutant list.2 Significant effort toward bioremediation strategies, such as bacterial biodegradation pathways, led to the identification of the haloalkane dehalogenase (HLD) family of enzymes in 1985.3 These naturally occurring enzymes have evolved to detoxify organohalogens by replacing the halogen with a benign hydroxyl.
Because of their robust and versatile properties, HLDs have been exploited in a variety of industrial processes such as in biosensing of pollutants, production of chiral building blocks for industrial biocatalysis, and decontamination of chemical warfare agents.4 However, bioremediation remains one of the most promising potential uses for the HLD family.4 The haloalkane dehalogenase from Rhodococcus sp., DhaA, can slowly detoxify TCP, and mutagenesis and selection yielded a DhaA variant with 8‐fold improved activity. Though this finding was significant, the activity was not yet sufficient to enable a bioremediation process.5 DhlA, the HLD from Xanthrobacter autotrophicus, was isolated from a strain that degrades DCE and has been used in groundwater treatment plants.6 Bioremediation of 1‐chlorobutane (1CB) from waste gas, through the use of biofilters charged with dehydrated bacterial cells expressing either DhaA or DhlA, has been extensively studied and is considered a promising technology.7, 8, 9 Although these developments are exciting, highly active, robust HLDs with activity against these pollutants are still lacking. Such HLDs must be discovered or engineered to improve the feasibility and outcomes of an HLD‐centered bioremediation process.
Structural and phylogenetic analyses of HLDs reveal a conserved structure and mechanism that have been acted upon by natural selection to achieve broad substrate specificity profiles. The HLD family belongs to the α/β‐hydrolase superfamily and is comprised of two domains: the highly conserved α/β‐hydrolase main domain and the highly variable cap domain. The active sites are located at the interface of the main domain and the cap domain in a single subunit. The accessibility of the active site varies and it has been shown that the size and properties of the access tunnels are key determinants of substrate specificity and catalytic efficiency of the enzymes.10, 11 The five catalytic residues common to all HLDs comprise a nucleophile (Asp), general base (His), and catalytic acid (Asp or Glu), plus a pair of halide‐stabilizing residues (Trp, Tyr, or Asn).12 The composition and precise positioning of this “catalytic pentad” varies among three phylogenetically‐derived HLD subfamilies: Asp‐His‐Asp + Trp‐Trp for HLD‐I, Asp‐His‐Glu + Asn‐Trp for HLD‐II, and Asp‐His‐Asp + Asn‐Trp for HLD‐III.12 Another difference between the subfamilies is that the HLD‐I and HLD‐II members act as monomers, whereas the characterized members of HLD‐III function as large multimeric complexes. An alternative grouping of HLDs by substrate specificity profile produces four substrate specificity groups (SSG).13 HLDs grouped in SSG‐I display activity towards most of the tested substrates including poorly degraded compounds such as DCE and TCP. Those that are grouped in SSG‐II show more restricted substrate specificity profile than SSG‐I with lack of activity towards 1,3‐diiodopropane. SSG‐III enzymes display low to no activity towards all of the tested compounds whereas SSG‐IV prefers terminally substituted brominated and iodinated alkanes. Intriguingly, these SSG groupings do not significantly correlate with the phylogenetic subfamilies.13 This disagreement underscores the need to characterize more HLDs to clarify the sequence‐structure‐function relationship within this industrially important family of enzymes.
The first identified and best characterized HLD is DhlA, from Xanthobacter autotrophicus.3, 14 DhlA's substrate preference is toward small (three carbons or less), chlorinated and brominated compounds including 1,2‐dichloroethane and 1,3‐dichloropropane. This is an unusual and propitious substrate specificity among known HLDs,14 making DhlA the most extensively used HLD for bioremediation. For example, a strain of X. autotrophicus containing DhlA is currently used in a German ground‐water treatment plant to treat water contaminated with 1,2‐dichloroethane15 and a transgenic tobacco plant expressing DhlA was found to degrade both 1,2‐dichloroethane and 1‐chlorobutane.16
DhlA is a member of the HLD‐I subfamily, whose other members do not appear to share this useful substrate specificity profile. HLD‐I subfamily members DpcA from Psychrobacter cryohalolentis, DmrA from Mycobacterium rhodesiae strain JS60, and DppA from Plesiocystis pacifica SIR‐1 all showed a strong preference for longer (greater than three carbons) bromoalkanes with little to no activity toward smaller or chlorinated compounds (Table 1).17, 18, 19 This substrate specificity profile is characteristic of substrate specificity group (SSG)‐IV; by contrast, DhlA is categorized in SSG‐I. Both DmbB from Mycobacterium bovis 18 and DhmA from Mycobacterium avium N85 21 were categorized in the HLD‐I subfamily but their substrate specificities were not reported due to expression and stability problems.
Table 1.
Characteristics of the HLD‐I Subfamily
| DhlA | DpcA | DppA | DmrA | DccA | DsaA | |
|---|---|---|---|---|---|---|
| Overall activity | ** | ** | ** | *** | ||
| Chlorinated substrates | ** | * | * | |||
| Brominated substrates | *** | *** | *** | *** | *** | ** |
| Iodinated substrates | *** | *** | *** | ** | * | |
| SSG class | I | IV | IV | IV | IV | IV |
| Catalytic efficiency | ||||||
| K m range (mM) | 0.01–48 | 4–6 | 1.9 | 1–7 | ||
| k cat range (s−1) | 0.09–4 | 1–3 | 3.1 | 3–29 | ||
| Structural information | ||||||
| Sample structure | 2DHC | 2XT0 | 4MJ3 | 5ESR | ||
| Volume of active‐site cavity (Å3) | 122 | 356 | 1063 | 874 |
The substrate specificity and activity of HLDs have been rationalized by comparing structural differences in the tunnel(s) that provide access to the active site from the surface of the enzyme.10, 11 Crystal structures of DhlA, DppA, and DmrA18, 19, 22 overlay with an RMSD of 1.5 Å or less. However, three key differences in DhlA distinguish it from DppA and DmrA: an insertion of 10 residues that reduces the size of the slot channel by folding into the channel entrance, a Trp blocking the main entrance tunnel, and a rotation of the halide‐stabilizing Trp175. It has been suggested that the 10 residue insertion, composed of two short‐sequence repeats, is responsible for DhlA's ability to work on 1,2‐dichloroethane.23 The short‐sequence repeats result in a dramatically smaller slot tunnel and a repositioning of the halide‐stabilizing Trp175.24 A DhlA variant in which one of the short‐sequence repeats is removed lost its activity towards 1,2‐dichloroethane, but retained activity towards 1,2‐dichlorobutane.24 Taken together, these differences create a small, solvent‐restricted active site in DhlA, compared to larger, more accessible active sites in DppA and DmrA (Fig. 1).
Figure 1.
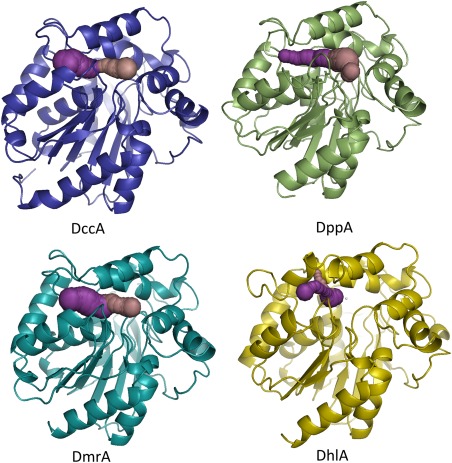
HLD‐I subfamily tunnels. CAVER was used to identify main and slot tunnels, pink and purple colored, respectively, in HLD‐I subfamily structures: DmrA(4MJ3), DppA(2XT0), DhlA(2DHC), and DppA(5ESR). The Asp123 of the catalytic pentad was used as the center for tunnel calculations. Figure made using PyMOL.25
To gain a greater understanding of the sequence‐structure‐function relationship within the HLD‐I subfamily, we identified two putative HLD sequences based on sequence identity with known HLD‐I members, DccA, from Caulobacter crescentus and DsaA, from Saccharomonospora azurea. Here we describe the cloning, functional expression, and biochemical characterization of both DccA and DsaA and describe the crystal structure of DccA.
Results
Sequence identification, cloning, recombinant expression, and purification of DccA and DsaA
The DccA protein sequence was identified as a putative HLD‐I subfamily member based on the phylogenetic analysis of Chovancova et al.12 It shares the catalytic pentad with other characterized HLD‐I subfamily members [Fig. 2(A)]. Among characterized HLDs, DpcA from Psychrobacter cryohalolentis has the greatest sequence identity to DccA (57.1%), with the next closest being DhmA from Mycobacterium avium N85 (54.6%). The other HLD‐I members are between 32 and 49% identical [Fig. 2(B)].
Figure 2.
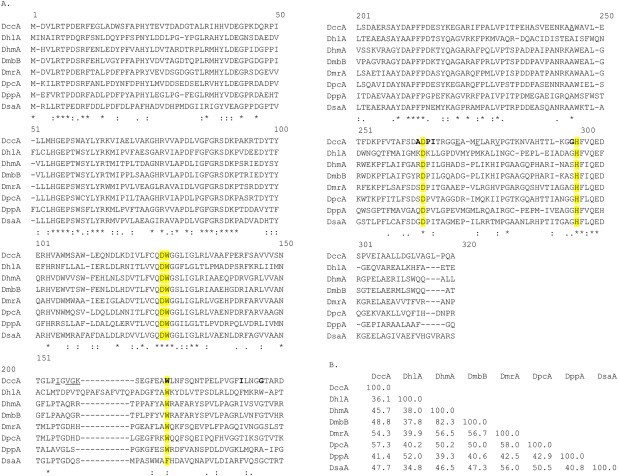
Comparison of DccA and DsaA with other HLD‐I family members. (A) Sequence alignment of HLD‐I subfamily members. CLUSTAL W (1.83) multiple sequence alignment. The pentad is highlighted in yellow. The DccA residues, which define the main tunnel. (B) Percent identity matrix of HLD‐I subfamily members.
The DsaA amino acid sequence was identified in a BLAST26 search using DhlA as an anchor. The DsaA putative HLD was selected for further study since it has an unusual catalytic pentad: instead of two halide‐stabilizing tryptophans it has Trp125 and Phe165 [Fig. 2(A)]. The closest HLD‐I member to DsaA is DmrA from Mycobacterium rhodesiae at 56.0% sequence identity. All other members are less than or equal to 50% [Fig. 2(B)]. Genes for DccA and DsaA were synthesized to be codon‐optimized for expression in Escherichia coli with a C‐terminal His‐tag in the PJ401 expression vector (DNA 2.0).27 Both genes were functionally expressed in E. coli BL21 and purified over a Ni‐NTA‐Sepharose affinity column to greater than 95% purity.
Specific activity and kinetic parameters
Both DsaA and DccA were tested against a panel of 18 halogenated substrates as shown in Figure 3. DsaA was found to have activity with eight of the tested substrates, showing highest activity toward 1‐bromohexane at 64.9 nmol s −1 mg−1. DsaA also displayed significant activity towards 1,3‐dibromopropane, 1‐bromobutane, 1‐bromo‐3‐chloropropane, 1‐iodobutane, and 1‐iodopropane. The only chlorinated compound for which DsaA showed activity was 1,5‐dichloropentane, with a specific activity of 7.8 nmol s−1 mg−1. DccA was found to be active against 12 of the 18 substrates tested and with much greater activity than found with DsaA. As with DsaA, the highest activity with DccA was found with 1‐bromohexane at 641 nmol s−1 mg−1. Similarly, DccA also shows significant activity with 1,3‐dibromopropane, 1‐bromobutane, 1‐bromo‐3‐chloropropane, and 1‐iodobutane. Where the activities diverged was on the brominated compounds 3‐bromo‐1‐propanol, 1,2‐dibromopropane, and 1,2,3‐tribromopropane where DccA demonstrated activity but DsaA did not. DccA also had activity towards chlorinated compounds such as 1,5‐dichloropentane (133 nmol s−1 mg−1) and 1‐chlorohexane (90 nmol s−1 mg−1). To establish that our pH‐based assay conformed with previously published results on HLDs, we tested DhlA (purified in the same manner as the other HLDs) against 1‐bromobutane, 1,2‐dichloroethane, and 1,2‐dibromoethane and found specific activities of 13.4, 25.5, and 25.1 nmol s−1 mg−1, respectively.
Figure 3.
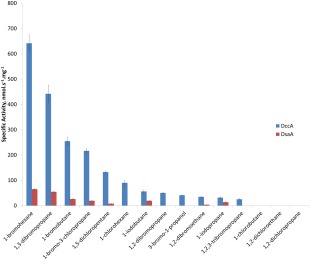
Specific activity graph for DccA and DsaA with a panel of haloalkanes. Initial reaction rates were measured following the absorbance at 540 nm, converted to [H+] standard curve, and normalized for enzyme concentration. Conditions: 8.1 mM substrate, 20 μg/mL phenol red, 20 mM Na2SO4, 1 mM EDTA, 1 mM HEPES pH 8.2.
To establish the basis for substrate specificity, the kinetic parameters for DccA were determined for a subset of substrates (Table 2). K m appears to be inversely correlated with chain length: the series 1‐bromohexane, 1‐bromobutane, and 1,3‐dibromopropane had K m values of 1.13, 6.58, 7.23 mM. By contrast, k cat was highest with 1,3‐dibromopropane with 28.7 followed by 1‐bromohexane and 1‐bromobutane with values of 23.6 and 13.0 s−1, respectively. The chlorinated compounds gave similar results to each other with k cat of 3.1 s−1 for 1‐chlorohexane and 5.0 s−1 for 1,5‐dichloropentane and K m values of 1.2 mM for each.
Table 2.
Kinetic constants for DccA
| Substrate | k cat (s−1) | K m (mM) | k cat /K m (M−1s−1) |
|---|---|---|---|
| 1‐bromohexane | 23.6 ± 0.8 | 1.1 ± 0.1 | 21,000 ± 2000 |
| 1‐bromobutane | 13.0 ± 0.4 | 6.6 ± 0.6 | 2000 ± 200 |
| 1,3‐dibromopropane | 29 ± 1 | 7.2 ± 0.8 | 4000 ± 500 |
| 1‐chlorohexane | 3.1 ± 0.1 | 1.2 ± 0.1 | 2600 ± 200 |
| 1,5‐dichloropentane | 5.0 ± 0.1 | 1.2 ± 0.1 | 4200 ± 400 |
Analysis of the crystal structure of DccA
The overall structure of DccA does not deviate significantly from the other structurally characterized HLDs. The refinement statistics are shown in Table 3. The variable cap domain is composed of residues 158–213, and the rest of the protein folds into the core domain (Fig. 4). The active site contains a catalytic pentad consistent with subfamily HLD‐I (Fig. 5). The catalytic residues Asp123, His278, and Asp249 and the halide‐stabilizing residues of Trp 124 and Trp 163 lie within a hydrophobic cavity. A chloride ion is bound approximately equidistant between Trp 124 and Trp163 at a distance of 3.5 Å and 3.3 Å, respectively. Electron density was identified adjacent to the chloride ion. On the basis of the peak height on the electron density maps, the distances to adjacent residues and the anomalous signals of the possible metal ions in the crystallization conditions, the density was identified as most likely from a cobalt ion.
Table 3.
Data collection and refinement statistics for the DccA crystal structure
| PDB entry | 5ESR |
|---|---|
| Data Collection | |
| Resolution range (Å) | 20.0–1.48 |
| Wavelength (Å) | 0.979 |
| Space group | C2221 |
| Unit cell dimensions (Å) | a = 79.38 |
| b = 79.64 | |
| c = 94.77 | |
| α = β = γ = 90° | |
| Observed reflections | 739,115 |
| Unique reflections | 50,696 |
| Completeness (%)a | 100.0(100.0) |
| I/σI | 19.1(5.8) |
| R‐merge (I)b | 0.110(0.563) |
| Structure refinement | |
| R cryst (%)c | 0.126 |
| R free (%)c | 0.149 |
| Protein nonhydrogen atoms | 2,459 |
| Water molecules | 446 |
| Average B‐factor (Å2) | 8.7 |
| RMS deviations from | |
| Ideal value | |
| Bonds (Å) | 0.008 |
| Angles (°) | 1.26 |
| Torsion angles (°) | 12.3 |
| Overall coordinate error (maximum‐likelihood) | 0.10 |
| Ramachandran statistics (%) (for non‐Gly/Pro residues) | |
| Most favorable | 97.1 |
| Additional allowed | 2.9 |
Values in parentheses indicate statistics for the high resolution bin.
R merge = ΣΣ j∣Ij(hkl)−<I(hkl)>∣/ΣΣ j∣<I(hkl)>∣, where I j is the intensity measurement for reflection j and 〈I〉 is the mean intensity over j reflections.
R cryst/(R free) = ΣǁFo(hkl)∣–∣Fc(hkl)ǁ/Σ∣Fo(hkl)∣, where F o and F c are observed and calculated structure factors, respectively. No σ‐cutoff was applied. 5% of the reflections were excluded from refinement and used to calculate R free.
Figure 4.
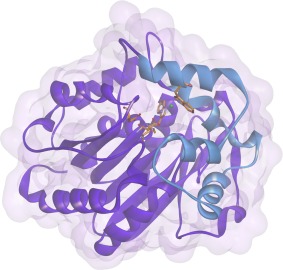
Tertiary structure of DccA. Catalytic pentad residues are shown in goldenrod. Chloride and cobalt ions shown as green and gray, respectively. Ribbon diagram of the core domain shown in purple and the cap domain in blue. This figure was made using Discovery Studio 4.0.28
Figure 5.
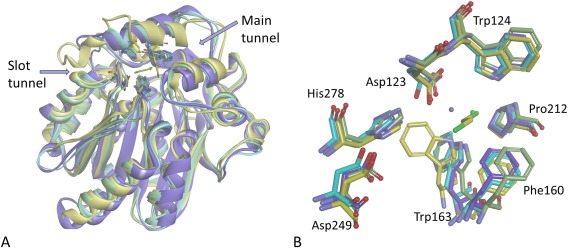
Overlay of HLD‐I structures. (A) Overlay of the structures of DccA (purple), DppA (green), DmrA(turquoise), and DhlA(yellow) with its cocrystallized 1,2‐dichloroethane with the backbones shown as ribbon diagrams. (B) Active site residues (Asp123: nucleophile, His278: general base, Asp249: catalytic acid, Trp124 and Trp 163: halide‐stabilizing residues) are shown as stick models. This figure was made using Discovery Studio 4.0.28
Two tunnels in DccA provide access from the solvent to the buried active site as analyzed by CAVER tunnel calculation software.29 The main tunnel, whose entrance is comprised of Trp163, Ile178, Gly182, Asp249, Pro250, Ile251, and Gly277, is a wide and deep tunnel with unrestricted access to the catalytic pentad. The slot tunnel entrance is comprised of Ile152, Val154, Gly155, Gly254, and Met258, and also provides access between the solvent and the active site. It is unclear if the slot and main tunnels have different roles to play in the mechanism; however, the two tunnels generate a highly accessible active site. The tunnel size as calculated by CASTp20 is 874 Å3 somewhat smaller than DmrA's volume of 1063 Å3, and much larger than DppA and DhlA with volumes of 356 Å3 and 122 Å3, respectively. As seen in Figure 1, the tunnels as defined by CAVER are similar between DmrA and DccA with DccA having a larger calculated bottleneck radius at 1.5 Å vs. 1.2 Å for DmrA. DppA tunnels are open, but with different shapes as that of DccA, but with a similar bottleneck at 1.4 Å. DhlA tunnels are smaller and more restrictive (bottleneck radius of 0.8 Å) as compared to those of the other members of the HLD‐I subfamily. DhlA has a 10 residue insertion in this loop that effectively blocks solvent access at this point and instead has a slot tunnel between α4′ and α4 rather than between β6 and α4 in DccA. The main tunnel in DhlA is blocked by a Trp residue and the tunnel repositions to behind this residue.
Discussion
We have demonstrated that both DsaA and DccA are active HLD enzymes. The different halide‐stabilizing pair did not appear to provide DsaA with a substantially different substrate specificity profile than other HLD‐I family members such as DppA, DpcA, and DmrA. Like these HLD‐I enzymes, DsaA vastly prefers brominated or iodinated substrates and showed only slight activity with 1,5‐dichloropentane. Of this set of enzymes, the only one with a reported specific activity was DpcA. For both DpcA and DsaA, 1,3‐dibromopropane was a preferred substrate with DsaA having approximately half the specific activity of DpcA (55 nmol s−1 mg−1 and 118 nmol s−1 mg−1, respectively).
DccA exhibited substantially higher activity than the other HLD‐I family members and accepts a broader range of substrates. It has very high specific activity with 1‐bromohexane and 1,3‐dibromopropane, but also has significant activity with 1,5‐dichloropropane (132 nmol s−1 mg−1) and 1‐chlorohexane (90 nmol s−1 mg−1) demonstrating activity with chlorinated compounds, unlike DppA, DpcA, and DmrA.17, 18, 19 The K m values for DccA are on par with reported values, whereas the k cat values are up to 10‐fold higher than reported for other HLD‐I subfamily members (Table 1).
Our structure of DccA adds to our structural understanding of the HLD‐I enzymes, with structures for DmrA, DppA, and DhlA having been previously reported.18, 19, 30 DccA shows the greatest similarity to DmrA followed by DhlA and DppA with a main‐chain RMSDs of 1.01 Å, 1.57 Å, and 1.64 Å, respectively. DccA and DmrA are 53% identical by sequence and have virtually identical active sites (Fig. 5). However, there are a number of changes in the entrance to the slot tunnels. Among the residues that define the slot tunnel entrance (Ile152, Val154, Gly155, Gly254, and Met258 in DccA), none are conserved between DccA, DppA, or DmrA, whereas the main tunnel entrance (Trp163, Ile178, Gly182, Asp249, Pro250, Ile251, and Gly277) is conserved for four out of seven positions. Access tunnel mutations have been shown to have substantial effect on HLD activity in other HLD members.10, 11 Comparison of DhlA to the other HLD‐I structures identifies three key differences unique to DhlA: a 10 amino acid insertion in the loop comprising the entrance to the slot tunnel, the repositioning the side‐chain torsion angle of the highly conserved halide‐stabilizing Trp175, and the blocking of the main tunnel through the position of Trp194 in DhlA, all of which result in DhlA’s restricted binding pocket unlike other HLD‐I structures. The insertion has been cited as the key reason that DhlA has activity on chlorinated compounds.24 However, DccA does not have this insertion and yet is still capable of efficiently dehalogenating some chlorinated alkanes, albeit longer ones than DhlA. The long and wide tunnels of DmrA, DppA, and DccA appear to be important for preferential activity with longer haloalkanes, as compared to the restricted tunnels of DhlA resulting in preference for smaller substrates. The ability to efficiently dechlorinate alkanes does not appear to be related to the access tunnel or binding pocket sizes in these enzymes.
Figure 6 is a comparison of the substrate specificity profile of the transformed data from HLDs from all subfamilies as described in Koudelakova et al.13 As is apparent from this graph, DccA, and DsaA both have similar substrate specificity profiles as DbeA from B. elkani, DatA from A. tumefaciens, and DmbC from M. bovis, all classified as SSG‐IV.13 DmbC is a member of the HLD‐III subfamily, forms large oligomeric structures and has poor activity towards the tested haloalkane substrates.31 Both DatA and DbeA are members of the HLD‐II subfamily. DbeA is unusual in that it has a second halide‐binding site, and mutation to remove the second site shifted the mutant from SSG‐IV to SSG‐I.32 DatA is unusual in that it has a Tyr for halide stabilization instead of a Trp, although mutation of Trp for Tyr did not significantly alter its substrate specificity profile.33
Figure 6.
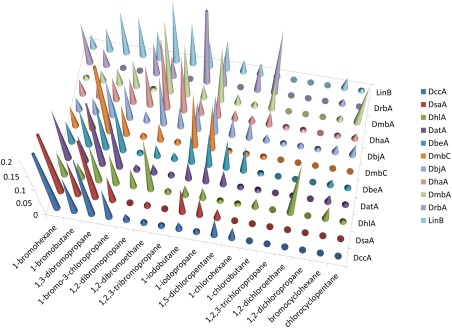
Substrate specificity profile. Transformation of the specific activity data. Data from Koudelkova et al. 13 was collated with the DsaA and DccA data for those substrates tested in both sets. The total values were normalized to total activity. The values that are higher than 0.2 were truncated for clarity.
DhlA has activity on smaller, chlorinated compounds whereas the other reported HLD‐I subfamily members, as well as DsaA, are highly specific for longer, brominated compounds. The highly active DccA displays a new substrate specificity profile, as it prefers longer, brominated compounds but still has significant activity toward longer, chlorinated haloalkanes—effectively bridging the two HLD‐I substrate specificity profiles. The high activity of DccA, as compared to other reported HLDs, also makes it an interesting target for biotechnological applications.
Materials and Methods
Materials
All chemicals were purchased from Sigma‐Aldrich (St. Louis, MO) at reagent grade or higher.
Cloning, expression, and purification
The genes for the DccA and DsaA were purchased from DNA2.0 in the PJ401express vector with a C‐terminal His‐6 tag. Both genes were codon optimized for expression in E. coli. The genes were transformed into chemically competent BL21 E. coli cells (Sigma, St. Louis, MO). To express the enzymes, 5 mL of an overnight growth was used to inoculate 1 L of LB broth with kanamycin (30 μg/mL). The cultures were incubated at 37°C while shaking at 250 rpm. Expression of DccA and DsaA were induced by the addition of 1 mM β‐d‐isopropylthiogalactoside (IPTG) at a culture density (OD600) of 0.6–0.9. Cells were harvested by centrifugation at 5000g 3 h later. The harvested bacteria were lysed by sonication and the lysate was clarified by centrifugation at 10,000g. All enzymes were purified by batch purification with Ni‐NTA. (Sambrook J, Russell DW. (2006) Purification of Histidine‐tagged Proteins by Immobilized Ni2+ Absorption Chromatography. CSH Protoc. 2006) After binding for 1 h at 4°C, the protein‐bound resin was poured into an empty column and was washed successively with 10 column volumes of 20 mM imidazole; the protein was eluted with 250 mM imidazole. Purity of the collected enzyme‐containing fractions was determined by SDS–PAGE (10% acrylamide, Coomassie stained). The enzyme concentration was determined by UV spectroscopy (ε280 = 48,930 M−1 cm−1 for DccA and 41,940 M−1 cm−1 for DsaA). Pure enzymes were stored in 1 mM Hepes, pH7.8, 20 mM Na2SO4, and 1 mM EDTA at 4°C and were used within three days of purification. For long term storage, glycerol (25%) was added as a cryoprotectant for storage at −80°C, and thawed samples were extensively dialyzed against storage buffer prior to assay.
Crystallization, data collection, and structure determination
Diffraction quality crystals for DccA were grown by sitting drop vapor diffusion by mixing 1 µL of protein (concentration was 20 mg/mL in 1 mM Hepes, pH 7.8, 20 mM Na2SO4, 1 mM EDTA) with 1 µL of reservoir solution and equilibrating the samples against the corresponding reservoir solution. The reservoir solution contained 0.005M Cobalt chloride, 0.005M Magnesium chloride, 0.005M Cadmium chloride, 0.005M Nickel chloride, 0.1M HEPES:NaOH, pH 7.5, 12% PEG 3350. Crystals with dimensions 0.1 × 0.1 × 0.2 mm3, were mounted in cryo‐loops directly from the crystallization droplet and flash‐cooled in liquid nitrogen. Prior to freezing, 20% glycerol was added to the drop as cryo‐protectant. Diffraction data were recorded on a Rayonix 225 HE CCD detector (Ryonyx, L.L.C., Evanston, IL) with 0.979 Å wavelength radiation on the LRL‐CAT beamline (Advanced Photon Source, Argone, IL). Intensities were integrated using the HKL2000 program and reduced to amplitudes using the SCALEPACK2MTZ program (Table 3 for statistics).34, 35 The structure was solved using molecular replacement with PHASER36 and PDB 2XT0 structure as a starting model. Final model building and refinement was performed with the programs COOT, REFMAC, and PHENIX, respectively.37, 38, 39 The quality of the final structures was verified with composite omit maps, and stereochemistry was checked with the program MOLPROBITY.40 LSQKAB and SSM algorithms were used for structural superpositions.41, 42 All other calculations were conducted using CCP4 program suite.34
Enzyme assay and kinetic studies
The kinetic properties were measured using a pH indicator dye‐based colorimetric method in a Spectromax384 Plus UV/Vis plate reader (Molecular Devices). For the specific activity determinations, 8.1 mM substrate was prepared in an indicator solution (20 μg/mL phenol red, 20 mM Na2SO4, 1 mM EDTA, 1 mM Hepes pH 8.2) and 170 μL was added to each well. The reaction was initiated by addition of 40 μL of enzyme solution (0.4–4.8 μg of enzyme) and mixed for 5 s in the plate reader before the initial absorbance reading. The absorbance was monitored at 540 nm at 13 s intervals with 1 s mixing between readings at 25°C. The reaction was monitored for 10 min and the initial rate of reaction was recorded with a parallel negative control run without enzyme. A standard curve relating absorbance to [H+] was used to convert the change in absorbance to [H+] produced. For determination of Michaelis–Menten parameters, the substrate concentration was varied using seven values that spanned a 10‐fold concentration range. Reactions were monitored for 10 min and the initial rate of reaction was recorded. Averaged data from at least three trials were fit to the Michaelis–Menten equation (v = k cat[E][S]/(K m + [S])43 to estimate k cat and K m using Kaleidagraph (Synergy Software, Reading, PA).
References
- 1. Belkin S (1992) Microorganisms to Combat Pollution. Netherlands: 175–189. [Google Scholar]
- 2. US EPA, O. Priority Pollutants . http://water.epa.gov/scitech/methods/cwa/pollutants.cfm.
- 3. Janssen DB, Scheper A, Dijkhuizen L, Witholt B (1985) Degradation of halogenated aliphatic compounds by Xanthobacter autotrophicus GJ10. Appl Environ Microbiol 49:673–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Koudelakova T, Bidmanova S, Dvorak P, Pavelka A, Chaloupkova R, Prokop Z, Damborsky J (2013) Haloalkane dehalogenases: biotechnological applications. Biotechnol J 8:32–45. [DOI] [PubMed] [Google Scholar]
- 5. Bosma T, Damborskỳ J, Stucki G, Janssen DB (2002) Biodegradation of 1, 2, 3‐trichloropropane through directed evolution and heterologous expression of a haloalkane dehalogenase gene. Appl Environ Microbiol 68:3582–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fetzner S (1998) Bacterial dehalogenation. Appl Microbiol Biotechnol 50:633–657. [DOI] [PubMed] [Google Scholar]
- 7. Erable B, Goubet I, Lamare S, Seltana A, Legoy M, Maugard T (2005) Nonconventional hydrolytic dehalogenation of 1‐chlorobutane by dehydrated bacteria in a continuous solid‐gas biofilter. Biotechnol Bioeng 91:304–313. [DOI] [PubMed] [Google Scholar]
- 8. Erable B, Goubet I, Lamare S, Legoy MD, Maugard T (2004) Haloalkane hydrolysis by Rhodococcus erythropolis cells: comparison of conventional aqueous phase dehalogenation and nonconventional gas phase dehalogenation. Biotechnol Bioeng 86:47–54. [DOI] [PubMed] [Google Scholar]
- 9. Erable B, Goubet I, Seltana A, Maugard T (2009) Non‐conventional gas phase remediation of volatile halogenated compounds by dehydrated bacteria. J Environ Manage 90:2841–2844. [DOI] [PubMed] [Google Scholar]
- 10. Chaloupková R, Sykorova J, Prokop Z, Jesenska A, Monincova M, Pavlova M, Tsuda M, Nagata Y, Damborsky J (2003) Modification of activity and specificity of haloalkane dehalogenase from Sphingomonas paucimobilis UT26 by engineering of its entrance tunnel. J Biol Chem 278:52622–52628. [DOI] [PubMed] [Google Scholar]
- 11. Pavlova M, Klvana M, Prokop Z, Chaloupkova R, Banas P, Otyepka M, Wade R, Tsuda M, Nagata Y, Damborsky J (2009) Redesigning dehalogenase access tunnels as a strategy for degrading an anthropogenic substrate. Nat Chem Biol 5:727–733. [DOI] [PubMed] [Google Scholar]
- 12. Chovancová E, Kosinski J, Bujnicki JM, Damborskỳ J (2007) Phylogenetic analysis of haloalkane dehalogenases. Prot Struct Funct Bioinform 67:305–316. [DOI] [PubMed] [Google Scholar]
- 13. Koudelakova T, Chovancova E, Brezovsky J, Monicova M, Fortova A, Jarkovsky J, Damborsky J (2011) Substrate specificity of haloalkane dehalogenases. Biochem J 435:345–354. [DOI] [PubMed] [Google Scholar]
- 14. Keuning S, Janssen DB, Witholt B (1985) Purification and characterization of hydrolytic haloalkane dehalogenase from Xanthobacter autotrophicus GJ10. J Bacterial 163:635–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stucki G, Thueer M (1995) Experiences of a large‐scale application of 1,2‐dichloroethane degrading microorganisms for groundwater treatment. Environ Sci Technol 29:2339–2345. [DOI] [PubMed] [Google Scholar]
- 16. Mena‐Benitez GL, Gandia‐Herrero F, Graham S, Larson TR, McQueen‐Mason SJ, French CE, Rylott EL, Bruce NC (2008) Engineering a catabolic pathway in plants for the degradation of 1,2‐dichloroethane. Plant Physiol 147:1192–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Drienovska I, Chovancova E, Koudelakova T, Damborsky J, Chaloupkova R (2012) Biochemical characterization of a novel haloalkane dehalogenase from a cold‐adapted bacterium. Appl Environ Microbiol 78:4995–4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fung HKH, Gadd MS, Drury TA, Cheung S, Guss JM, Coleman NV, Matthews JM (2015) Biochemical and biophysical characterization of haloalkane dehalogenases DmrA and DmrB in Mycobacterium strain JS60 and their role in growth on haloalkanes. Mol Microbiol 97:439–353. [DOI] [PubMed] [Google Scholar]
- 19. Hesseler M, Bogdanovic X, Hidalgo A, Berenguer J, Palm GJ, Hinrichs W, Bornscheuer UT (2011) Cloning, functional expression, biochemical characterization, and structural analysis of a haloalkane dehalogenase from Plesiocystis pacifica SIR‐1. Appl Microbiol Biotechnol 91:1049–1060. [DOI] [PubMed] [Google Scholar]
- 20. Dundas J, Ouyang Z, Tseng J, Binkovski A, Turpaz Y, Liang J (2006) CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res 34:W116–W118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jesenská A, Bartos M, Czernekova V, Rychlik I, Pavlik I, Damborsky J (2002) Cloning and expression of the haloalkane dehalogenase gene dhmA from Mycobacterium avium N85 and preliminary characterization of DhmA. Appl Environ Microbiol 68:3724–3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Verschueren KHG, Seljée F, Rozeboom HJ, Kalk KH, Dijkstra BW (1993) Crystallographic analysis of the catalytic mechanism of haloalkane dehalogenase. Nature 363:693–698. [DOI] [PubMed] [Google Scholar]
- 23. Pries F, van den Wijngaard AJ, Bos R, Pentenga M, Janssen DB (1994) The role of spontaneous cap domain mutations in haloalkane dehalogenase specificity and evolution. J Biol Chem 269:17490–17494. [PubMed] [Google Scholar]
- 24. Pikkemaat MG, Janssen DB (2002) Generating segmental mutations in haloalkane dehalogenase: a novel part in the directed evolution toolbox. Nucleic Acids Res 30:e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. The PyMOL Molecular Graphics System . (Schrodinger, LLC).
- 26. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410. [DOI] [PubMed] [Google Scholar]
- 27. Welch M, Govindarajan S, Ness JE, Villalobos A, Gurney A, Minshull J, Gustafsson C (2009) Design parameters to control synthetic gene expression in Escherichia coli . PLoS One 4:e7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Discovery Studio Modeling Environment . (Accelrys Software Inc., 2012).
- 29. Chovancova E, Pavelka A, Benes P, Strnad O, Brezovsky J, Kozlikova B, Gora A, Sustr V, Klvana M, Medek P, Biedermannova L, Sochor J, Damborsky J (2012) CAVER 3.0: a tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput Biol 8:e1002708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Silberstein M, Damborsky J, Vajda S (2007) Exploring the binding sites of the haloalkane dehalogenase DhlA from Xanthobacter autotrophicus GJ10. Biochemistry 46:9239–9249. [DOI] [PubMed] [Google Scholar]
- 31. Jesenská A, Monincová M, Koudeláková T, Hasan K, Chaloupková R, Prokop Z, Geerlof A, Damborsky J. (2009) Biochemical characterization of haloalkane dehalogenases DrbA and DmbC, Representatives of a Novel Subfamily. Appl Environ Microbiol. 75:5157–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Prudnikova T, Mozga T, Rezacova P, Chaloupkova R, Sato Y, Nagata Y, Brynda J, Kuty M, Damborsky J, Smatanova IK (2009) Crystallization and preliminary X‐ray analysis of a novel haloalkane dehalogenase DbeA from Bradyrhizobium elkani USDA94. Acta Cryst F65:353–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guan L, Yabuki H, Okai M, Ohtsuka J, Tanokura M (2014) Crystal structure of the novel haloalkane dehalogenase DatA from Agrobacterium tumefaciens C58 reveals a special halide‐stabilizing pair and enantioselectivity mechanism. Appl Microbiol Biotechnol 98:8573–8582. [DOI] [PubMed] [Google Scholar]
- 34. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Drissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Cryst D67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Otwinowski ZM, Minor W (1997) Processing of X‐ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. [DOI] [PubMed] [Google Scholar]
- 36. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Cryst 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Cryst D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 38. Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum‐likelihood method. Acta Cryst D53:240–255. [DOI] [PubMed] [Google Scholar]
- 39. Adams PD, Afonine PF, Bunkoczi G, Chen VB, Echols N, Headd JJ, Hung L‐W, Jain S, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriary NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2011) The Phenix software for automated determination of macromolecular structures. Methods 55:94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Richardson DC, Chen VB, Arendall WB, Headd JJ, Deedy DA (2010) MolProbity: all‐atom structure validation for macromolecular crystallography. Acta Cryst D66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kabsch W (1976) A solution for the best rotation to relate two sets of vectors. Acta Cryst A32:922–923. [Google Scholar]
- 42. Krissinel E, Henrick K (2004) Secondary‐structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Cryst D60:2256–2268. [DOI] [PubMed] [Google Scholar]
- 43. Cleland WW (1979) Statistical analysis of enzyme kinetic data. Methods Enzymol 63:103–138. [DOI] [PubMed] [Google Scholar]