

Abstract
Stem cell-like colon cancer cells (SCCs) pose a major challenge in colon cancer treatment because of their resistance to chemotherapy and radiotherapy. Oncolytic virus-based therapy has shown promising results in uncured cancer patients; however, its effects on SCCs are not well studied yet. Here, we engineered a cancer-favoring oncolytic vaccinia virus (CVV) as a potent biotherapeutic and investigated its therapeutic efficacy in terms of killing SCCs. CVV is an evolved Wyeth strain vaccinia virus (EVV) lacking the viral thymidine kinase. SCC models were established using human or mouse colon cancer spheres, which continuously expressed stemness markers. The cancer-favoring characteristics and different cytotoxic pathways for killing cancer cells successfully overrode general drug resistance, thereby killing colon cancer cells regardless of the presence of SCCs. Subcutaneously injected HT29 spheres showed lower growth in CVV-treated models than in 5-Fu-treated models. Intraperitoneally injected CT26 spheres induced tumor masses in the abdominal region. CVV-treated groups showed higher survival rates and smaller tumor mass formation, compared to 5-Fu-treated groups. Interestingly, the combined treatment of CVV with 5-Fu showed improved survival rates and complete suppression of tumor mass. The CVV developed in this study, thus, effectively suppresses SCCs, which can be synergistically enhanced by simultaneous treatment with the anticancer drug 5-Fu. Our novel CVV is highly advantageous as a next-generation therapeutic for treating colon cancer.
Keywords: stem cell-like colon cancer cells, oncolytic virus, cancer-favoring vaccinia virus, wyeth strain, resistance
INTRODUCTION
Cancer is one of the leading causes of death, with a mortality rate (deaths per 1,000 individuals per year) of approximately 14% worldwide. Among the various types of cancer, colorectal cancer (CRC) has a relatively poor prognosis and is diagnosed as the third most common cancer in men and the second in women in 2012 [1, 2]. Despite advances in anticancer drug development, most CRCs are resistant to conventional cancer therapy. This poor responsiveness to chemo- and/or radiotherapy might be attributed to stem cell-like colon cancer cells (SCCs) [3]. It is generally accepted that cancer stem cells (CSCs) are capable of self-renewal and have the exclusive ability to reproduce malignant tumors even though CSCs constitute a minor population in tumors. Drug resistance and tumor recurrence after the initial response to chemo- or radiotherapy may be due to the survival of CSCs within the original tumor. Therefore, SCCs pose a considerable challenge and are considered the next target in colon cancer treatment.
Genetic engineering of viruses provides unprecedented opportunities for various biomedical applications, including drug/gene delivery, tissue engineering, targeting cancer, and/or cancer imaging [4–11]. Oncolytic viruses (OVs) are unique biomaterials having merits over conventional anticancer reagents in terms of their tumor selectivity and ability to lyse cancer cells. Tumor selectivity is usually introduced by genetic engineering, which attenuates viral replication in normal cells. In our previous study, clinically applied JX-594 conferred tumor selectivity via viral thymidine kinase (vTK) inactivation because vaccinia virus has evolved to replicate in EGFR pathway-activated cells, which are usually cancer cells with high cellular TK levels [10, 12–14]. Thus, OVs can selectively infect and replicate in cancer cells. OVs are replication competent; thus, the infectious progeny generated by OV replication in tumor cells can expand to kill the tumor mass, whereas OV rarely harms normal cells. OV-based therapy in actual clinical settings began over a century ago, demonstrating the effectiveness of OVs in cancer treatment [13, 15–17]. Among them, vaccinia virus-based therapy is well tolerated and has shown relatively low side effects: minor and expected controllable toxicity and no evidence of uncontrolled or latent infection, or unexpected disease occurrence [18].
Despite the above proven efficacy of OVs in cancer cells/tissues in clinical settings, the effects of OVs on SCCs need to be investigated further. Herein, we engineered a cancer-favoring oncolytic vaccinia virus (CVV) and investigated its effects on CRC in terms of killing SCCs. We hypothesized that the cancer-favoring characteristics, cancer cell selectivity, and cancer cell infectivity mediated by vaccinia virus differ from those of conventional anti-cancer drugs; thus they may help suppress the growth of SCCs.
RESULTS
CVV selectively infects and kills various CRC cell lines better than VR1536
CVV was generated by replacing the vTK gene from a naturally evolved cancer-favoring Wyeth strain vaccinia virus (EVV) strain [19] with the green fluorescence protein gene (Figure 1A). EVV was constructed from the Wyeth strain of vaccinia virus to achieve the cancer-favoring property and then isolated and characterized by repeated replication and tumor tissue lysis [19]. EVV was isolated from the blood of a vaccinia virus-injected VX2 tumor animal model when the tumor size became reduced and started to release viruses into the serum. Previously, we found that EVV had superior tumor selectivity compared with the wild type (WT) virus and other engineered vaccinia viruses [19]. CVV may work highly effectively compared to other type of virus. Replication efficacy generally reflects the antitumor activity and was examined in CT26 cells (Figure 1B). Viral replication assay results showed that CVV deficient of vTk showed lower infection at 24 h, but showed higher replication rates subsequently, compared to EVV and the WT virus. A lower initial replication of CVV likely resulted from vTk deficiency, where higher replication rates of CVV in Tk-activated host cancer cell lines are attributable to its higher tumor selectivity. Enhanced suppression of colon tumors by CVV treatment, compared to PBS, WT, or EVV administration, was confirmed in an in vivo CT26 xenograft model (Figure 1C). We used 106 plaque-forming units (pfu) virus/mouse because CVV may have a higher replication rate than the WT virus or EVV. The infectious dose of the WT or JX594 viruses used in a previous in vivo study was more than 107 pfu [14]. As expected, CVV infection exhibited better results than WT or EVV, even with a single injection at the low dose of 106 pfu/mouse.
Figure 1. Schematic illustration of our approach to construct CVV and its higher cancer selectivity.

(A) We engineered a cancer-favoring virus (CVV) from the Wyeth strain of vaccinia virus. (B) Viral replication assay showing that CVV deficient of vTk replicated at lower levels at 24 h post-infection, but showed higher replication rates than EVV and WT. (C) CVV shows enhanced suppression of tumor size in CT26 xenograft mice compared to that observed in mice administered PBS, WT or EVV (n = 6, *p < 0.05 vs. PBS). (D) CVV-biodistribution results at 2 weeks post-intraperitoneal injection in HT29-bearing mice, showing that CVV selectively infected tumor mass. (E) CVV showed higher infectivity in colorectal cancer cell lines than in normal mouse embryonic fibroblasts.
The higher anti-cancer efficacy of CVV in colon cancer cells is due to its greater selectivity (cancer-favoring characteristics via evolution and TK deletion). To test the tumor selectivity and safety for normal tissues, tumor tissues and normal tissues (kidney, liver, spleen, lung, heart, brain, colon, stomach, and testis) were harvested at 2 weeks after intraperitoneal injection of HT29 cells into nude mice, and virus titers in each tissue (pfu/mg) confirmed that selective infection of tumor tissues occurred (Figure 1D). We found marginal viral replication (∼5 pfu/mg) in normal kidney and liver tissues, and no viral replication in normal spleen, lung, and colon tissues in the biodistribution data, which was is marked contrast with viral titers found in tumor tissues (∼106 pfu/mg). The higher toxicity of CVV in cancer cells than in mouse embryonic fibroblasts (MEFs), used as normal cells might be ascribed to its higher infectivity in CRC cell lines (Figure 1E), and the fact that the CVV infectivity was much lower than that in cancer cell lines. The reason why we found viral replication in MEF cell lines, but not in normal tissues, in the biodistribution data may be because selectivity can be generally measured only when a virus has the option to infect either normal or tumor tissues. Within MEF cells, viral replication should occur in normal cells (given no other choice) by utilizing the replication properties of the cell lines. Therefore, higher infectivity in cancer cell lines than that in normal cells can explain why CVV selectively distributed to tumor tissues. To demonstrate the cancer-favoring and oncolytic potency of CVV in colon cancer, a panel of CRC cell lines (HT-29, DLD-1, HCT-116, SNU4, SNU5, and/or Lovo) was tested. Following infection, CVV showed higher toxicity than the WT virus in the various CRC cell lines (Figure 2).
Figure 2. CVV selectively infected and killed various colorectal cancer cell lines more efficiently than did the WT vaccinia virus VR1536 (WT).
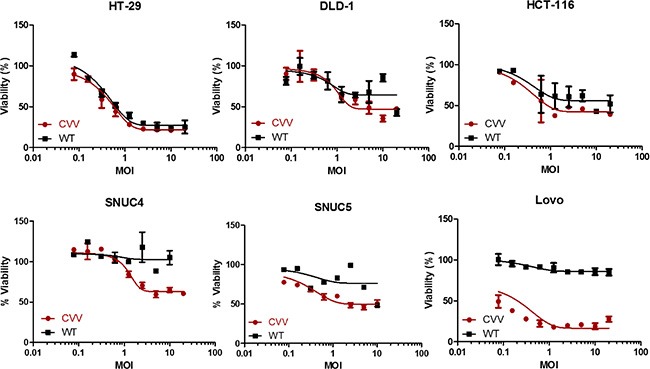
CVV infection showed higher cytotoxicity than did the WT virus in various colorectal cancer cells (CRCs) used in the WST assay.
CVV might overcome resistance observed in stem cell-like cancer cell populations
CVV infection showed greater toxicity than that of the anti-colon cancer drug CPT11 in CRCs (HT29, LOVO, DLD-1, and HCT-116), as shown in Figure 3A. The units used for CVV and CPT11 treatment are the multiplicity of infection (MOI) and the micromolar concentration, respectively. Although a direct comparison of the viral MOI and CPT11 concentration is impossible, it is safe to assume that the virus concentration used was much lower than CPT11 concentration used when considering their approximate molar concentrations. In addition, all of the cell lines tested showed resistance to CPT11, even up to doses of ∼10 μM (Figure 3A). We then analyzed the percentage of the CD133+/CD44+ double-positive cell population, which represents SCCs, and its relation to drug resistance. The population of SCCs is shown in Figure 3B (left panel). The percent viability of each CRC after CVV infection (MOI = 10) or CPT treatment (10 μM) is shown in the right panel of Figure 3B. Interestingly, the percentage of the CD133+/CD44+ population appeared to be related with CPT11 resistance, whereby the CD133+ population seemed to majorly confer the percentage of CD133+/CD44+ SCCs and, thus, drug resistance. In terms of the percent cell viability of CPT11-treated HCT-116 cells, we observed fluctuations in the WST-1 results, which exceeded 100% compared to the viability of untreated cells (Figure 3A). The observation of elevated cell viability of HCT-116 cells (over 100%) after the 72 h-treatment may have been because WST-1 measures dehydrogenase activity, which is generally elevated in cancer stem cells. We expect that higher SCC populations remaining after CPT11 treatment would give an enhanced signal, resulting in measured cell viabilities of over 100%. The cytotoxicity of CVV towards CPT11-resistant cells may overcome the stem cell-like cancer cell population. The relative expression of ABCG2 also demonstrated the relationship between drug resistance and the SCC population (Figure 3B bottom). Thus, CPT11 resistance might be related to the SCC population and ABCG2 expression, whereas CVV showed dose-dependent responses regardless of the SCC population, suggesting that our CVV might overcome drug resistance originating from the stem cell-like colon cancer cell population.
Figure 3. Cytotoxicity of CVV towards CPT11-resistant cells may overcome the stem cell-like cancer cell population.
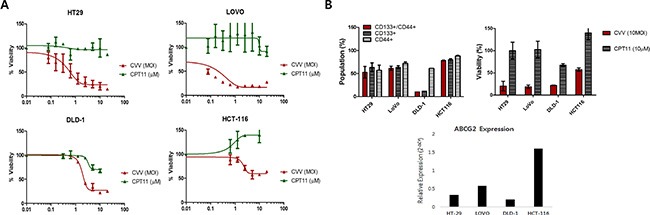
(A) The percent viability of 4 different colorectal cancer cells (CRCs) shows their resistance to CPT11 exposure, but not to CVV infection. (B) The percentage of the CD133+/CD44+ double-positive cell population, which represents SCCs, with CD133+ and CD44+ cells (left panel) and the percent viability of each CRC following CVV infection (MOI = 10) and CPT11 treatment (10 μM) (right panel). The relative expression of ABCG2 (bottom panel) shows its relationship to drug resistance and the SCC population.
Stem cell-like colon cancer cells have sphere-forming ability and highly tumorigenic, but are more susceptible to CVV than to CPT11
Three-dimensional sphere cultures of tumor cells lead to the enrichment of CSC populations [20]. To confirm the relationship between sphere-forming ability and cell stemness, CD133+/CD44+ and CD133−/CD44− HT29 cells were analyzed after fluorescence-activated cell sorting (FACS) and compared with unsorted HT29 cells. As expected, double-positive (CD133+/CD44+) HT29 cells showed better sphere-forming ability than did CD133−/CD44− cells (Figure 4A). The sphere size of CD133+/CD44+ cells increased compared to CD133−/CD44− over sequential days in culture. Immunostaining of HT29 spheres confirmed that they expressed stemness marker proteins (Figure 4B). When 5–7-day-old cultured spheres were dissociated and re-cultured in stem cell media, HT29 spheres could be formed repeatedly up to 6 or more times in sequential sphere cultures. In addition, repeated sphere formation/dissociation did not impair the sphere-forming ability or stemness gene expression for up to 6 days (Figure 4C), demonstrating that sphere formation contributes to the characteristics of stemness of cancer cells. Different numbers of monolayer-cultured and sphere-cultured HT29 cells (5 × 103, 1 × 104, and 5 × 104) were injected subcutaneously into Balb/c nude mice, after which their tumorigenicity (Figure S1A) and tumor volumes (Figure S1B) were examined As expected, HT29 spheres showed characteristics of SCCs in a mouse in vivo model, as earlier tumor formation and larger tumor volumes were observed with the group injected sphere-cultured cell than the group administered monolayer-cultured cell.
Figure 4. Stem cell-like cancer cells have sphere-forming ability.
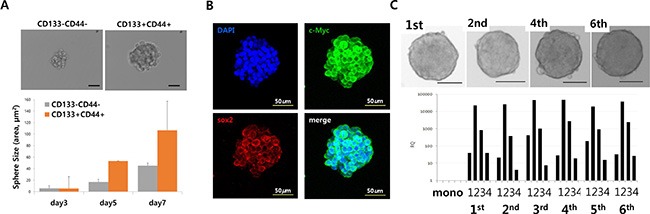
(A) Sphere-forming ability of CD133/CD44− and CD133+/CD44+ cells (upper panels). Sphere size of CD133−/CD44− and CD133+/CD44+ cells (bottom panels). (B) Immunostaining of stemness markers c-Myc and Sox2 in HT29 sphere cells. (C) Sequential sphere-forming ability and continuous stemness maker expression (Scale bar = 100 μm; upper panels). Relative expression of the stemness markers Nanog (1), Sox2 (2), Oct4 (3), and c-Myc (4) in CD133−/CD44− and CD133+/CD44+ cells (bottom panel).
Enhanced suppression of stem cell-like colon cancer cells by CVV may overcome anticancer drug-resistance in SCCs
We then cultured HT29 spheres for 5 days, treated them with 0.1 μM Fluorouracil (5-Fu) or CVV (MOI = 0.1), and harvested them after an additional 3 days in culture. Monolayer-cultured HT29 cells (mono HT29) and sphere-cultured HT29 cells (sphere HT29) were also harvested. Live cells (1 × 106), including mono HT29s, sphere HT29s, sphere HT29s treated with 5-Fu (sphere HT29s+5-Fu), and HT29s infected with CVV (sphere HT29s+CVV), were injected subcutaneously into Balb/c nude mice, and tumor growth from the injected surviving cells was examined after treatment (Figure 5). As expected, animals injected with HT29 spheres having the characteristics of SCCs showed earlier and larger tumor growth versus animals injected with monolayer-cultured cells. Animals with surviving HT29 sphere cells after CVV treatment showed attenuated tumor growth resulting from successful killing of stem cell-like cells by CVV, whereas surviving HT29 sphere cells following 5-Fu treatment did not (Figure 5A & 5B). FACS analysis (Figure 5C, left panel) and real-time PCR results (Figure 5C, right panel) showed that the CD133+/CD44+ double-positive cell population increased or decreased after 5-Fu or CVV treatment, respectively, suggesting that CVV infection could kill stem cell-like HT29 cells more so that 5-Fu treatment. Thus, surviving cells after 5-Fu treatment showed much higher levels of SCC marker expression. Therefore, cancer-favoring natural and engineered vaccinia viruses have more powerful cancer selectivity and higher cytotoxicity. The underlying mechanism, however, differs from that of anticancer drugs, thereby obviating the problem of resistance.
Figure 5. Enhanced suppression of stem cell-like HT29 human colon cancer cells by CVV.
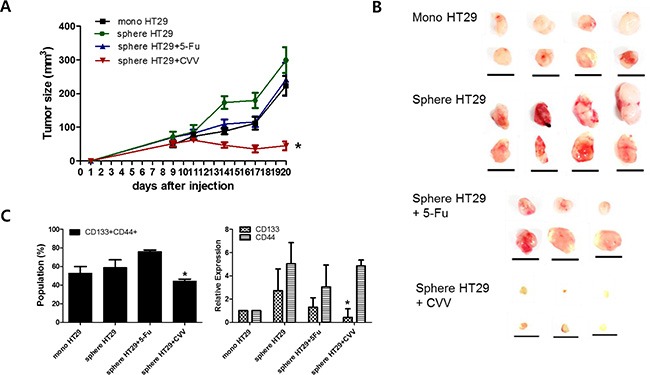
(A, B) Tumor sizes measured in xenograft models of 106 live monolayer-cultured HT29 cells (mono HT29), sphere-cultured HT29 cells (sphere HT29), sphere HT29 cells treated with 5-Fu, and HT29 cells treated with CVV showed that CVV can attenuate tumor growth and successfully kill stem cell-like cells (n = 6–8; *p < 0.05 vs. mono HT29, t test; scale bar = 1 cm). (C) The CD133+/CD44+ double-positive SCCs population (left panel) and real-time PCR results (right panel) show that CVV can highly suppress stem cell-like HT29 cells compared to 5-Fu treatment (n = 3, *p < 0.05).
Enhanced suppression of stem cell-like CT26 mouse colon cancer cells by CVV in a syngeneic model of CRC peritoneal carcinomatosis
The CT26 mouse colon cancer cell line was chosen for an in vivo syngeneic carcinomatosis mouse model with SCCs. Percent survival rates of the models intraperitoneally injected with CT26 spheres and treated with PBS, CVV, 5-Fu, or CVV+5-Fu were in line with our expectations (Figure 6A). Comparison of the efficacy of 5-Fu and CVV treatment on the survival showed that the 5-Fu group had good survival until 24 days post-treatment; however, CVV-treated mice showed a longer survival period. Interestingly, combined treatment with CVV and 5-Fu improved the survival rate compared to the 3 other groups. At 30 days post-treatment, the surviving mice were sacrificed, and the internal peritoneal regions were examined. Intraperitoneally injected sphere-cultured CT26 cells spread into the liver and also connected the tumor mass with colon tissues in the abdomen (in the PBS model). Interestingly, no tumor masses were found in the abdomens of the co-treatment group (CVV+5-Fu) (Figure 6A, bottom). Hematoxylin and eosin (H & E) staining results of colon tissues and tumor masses of CT26 sphere-injected models treated with PBS, CVV, 5-Fu, or CVV+5-Fu revealed that fewer inflammatory and tumor regions were found in the CVV-treatment and co-treatment groups than in the PBS- and 5-Fu-treatment groups (Figure 6B). Importantly, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining results of colon tissues and tumor masses of CT26 sphere-injected models treated with PBS, CVV, 5-Fu, or CVV+5-Fu indicated that CVV effectively induced apoptosis only in the tumor mass (Figure 6C). The results showed that SCCs can respond better to CVV than the anticancer drugs CPT11 or 5-Fu, particularly for longer periods. We propose that this might be because anticancer drugs are sufficiently cytotoxic to kill general cancer cells, and thus, better results in the short-term are exhibited. However, resistant SCCs can eventually emerge, although the effect of CVV infection can override the effect of resistance to anticancer drugs (Figure 7).
Figure 6. Enhanced suppression of stem cell-like CT26 mouse colon cancer cells by CVV.
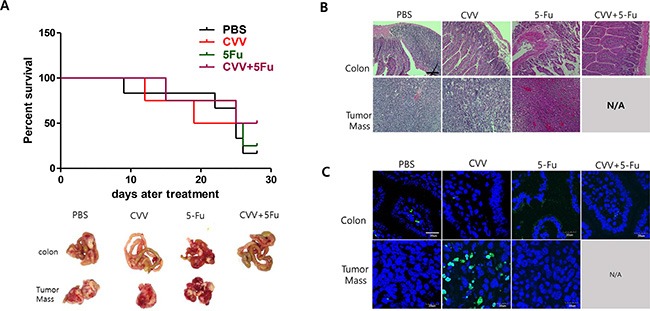
(A) Percent survival rates of mice intraperitoneally injected with CT26 spheres and treated with PBS, CVV, 5-Fu, or CVV+5-Fu (n = 4–6). (B) H & E staining results of colon tissues and tumor masses of mice intraperitoneally injected with CT26 spheres and treated with PBS, CVV, 5-Fu, or CVV+5-Fu. (C) TUNEL staining results of colon tissues and tumor masses of mice intraperitoneally injected with CT26 spheres and treated with PBS, CVV, 5-Fu, or CVV+5-Fu (Scale bar = 20 μm).
Figure 7. Schematic illustration of how CVV can kill colon cancer cells by overcoming SCC issues.
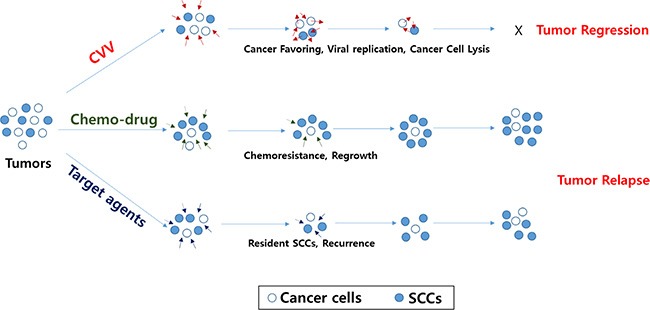
The CVV has enhanced cancer-favoring selectivity and cytotoxicity, thereby enabling it to successfully override drug resistance and/or other limitations of currently developed targeted therapies in terms of killing colon cancer cells, regardless of the presence of SCCs, because of its cancer-favoring characteristics and the different cytotoxic pathway used to kill cancer cells.
DISCUSSION
SCCs are tumorigenic and are responsible for the malignancy and drug resistance of colon cancer cells even though SCC populations constitute only 0.1–10% of all tumor cells [21]. The rare subset of cancer cells having “stem-like” features, namely, CSCs, possess natural quiescence and a high level of drug efflux transporters, leading to their low sensitivity to chemotherapy and radiotherapy, compared with normal tumor cells [21–24]. Therefore, the concept has emerged that CSCs represent an important challenge in the understanding of anticancer drug resistance, cancer recurrence, and metastasis [25], as well as CRC treatment. The available conventional therapeutic agents primarily eliminate the bulk of tumors, but do not affect CSCs [26, 27]. Therefore, the ability to target and eradicate CSCs is expected to markedly improve clinical outcomes. The most common way of identifying and isolating CSCs is based on cell surface marker expression using cell-separation methods, such as flow cytometry, magnetic sorting, or FACS. Surface markers for different types of CSCs in brain [28], breast [29], ovarian [30], colon [20, 31], lung [32], head and neck [33], pancreas [34], and liver cancers [35] have been identified. Other ways to identify CSCs include detecting the expression of transcription factors and various cytoplasmic and nuclear proteins, and/or determining functional properties such as DNA-label retention, dye-efflux properties, or enzymatic metabolism. However, no single marker can define CSCs, but the combined use of multiple markers now offers the potential to identify and define CSCs. Indeed, CSCs share many characteristics with normal stem cells [36]; therefore, developing specific agents for targeting and killing CSCs warrants further research.
Recently, OV-based therapy has attracted attention because it shows promising results in terms of cancer selectivity and safety in clinical trials [37–40]. OVs possess cancer selectivity and efficiently kill cancers because (i) OVs utilize the EGFR pathway, which is activated in cancerous cells; (ii) elevated TK in cancer cells promotes enhanced replication of vTk-deleted OVs; (iii) OV kills cancer by replicating within the cancer, so that it can be effectively used at lower concentrations to stop replication when all the host cancer cells are lysed; and (iv) cancer cells lysed by an OV can release debris containing tumor antigens, which triggers the immune system and leads to cytotoxic T-cell activation and cell-mediated immunity [41]. Although encouraging results from clinical trials are beginning to accumulate, the efficacy of using OVs for SCCs has not been well understood thus far. OVs can be ideal candidates, as opposed to directly targeting agents for CSCs, because they are cytotoxic and are not subject to the typical mechanisms of drug resistance, such as drug-efflux pumps and defective apoptotic signaling, which suggests that OVs might be effective against CSCs, including SCCs [42, 43].
In this study, efforts were made to develop CVV, a cancer-favoring oncolytic vaccinia virus, by first evolving vaccinia virus from the tumor mass and then inactivating the vTk gene. We next investigated its therapeutic efficacy in terms of killing SCCs. When applied to various CRC cell lines, CVV could selectively infect and kill host cells more efficiently than the wild-type vaccinia virus VR1536. However, most cells showed anticancer drug resistance, which is associated with SCC populations and are related with ABCG2 expression. SCC models were established by constructing colon cancer sphere cell lines, which have higher tumorigenicity and can continuously express stemness markers. As expected, sphere cells remained susceptible to CVV, but not to chemodrugs. Our CVV also selectively killed tumor cells in an in vivo mouse model. Tumor xenografts induced by subcutaneous injection with HT26 spheres that survived after pretreatment with PBS, CVV, or 5-Fu showed significantly suppressed tumor growth, only with CVV treatment. Interestingly, 5-Fu showed an early response in intraperitoneally injected CT26 mice in terms of suppressing tumor masses and increasing the survival rates of treated mice, but it was surpassed by CVV 24 at days post-treatment. Our data also showed that CVV eventually exhibited greater anti-tumor efficacy, even after a single, low-dose injection (106 pfu/mouse). Combined treatment with CVV and 5-Fu further improved survival rates and led to complete suppression of tumor mass production in the abdomen. We ascribe this finding to the early effect of 5-Fu, followed by the late effect of CVV owing to its replication and mild effect in cancerous tissue. The early effect of 5-Fu can kill bulk cancer cells, but SCCs can retain resistance. This issue, however, was counteracted by CVV. Furthermore, the immune reaction conferred by cancer cells lysed by CVV may also contribute to the relatively long-lasting effect of suppressing CSCs.
CONCLUSIONS
The CVV developed in this study can effectively and selectively infect and replicate in SCCs because it favors cancer cells and is not affected by drug-resistance pathways. We conclude that this CVV effectively suppressed SCCs, which can be synergistically enhanced by co-treatment with 5-Fu.
MATERIALS AND METHODS
Cell culture and reagents
The human colon cancer cells lines HT29, Lovo, DLD-1, and HCT116, and the mouse colon cancer cell line CT26 were obtained from the Korean Cell Line Bank (Seoul, Korea) and the ATCC (Manassas, VA). Cells were cultured in RPMI-1640 supplemented with 10% FBS and 100 U/mL penicillin and streptomycin under standard conditions of 37°C, 5% CO2, and a humidified atmosphere. All culture media and supplements were from Welgene (Daegu, Korea). The anticancer drugs CPT11 and 5-Fu were purchased from Sigma Korea (Yongin, Korea).
Sphere formation
Single cells were resuspended in serum-free DMEM/F12 (Welgene) containing 1 × B27 supplement (Invitrogen, Waltham, MA), 20 ng/mL epidermal growth factor, and 10 ng/mL basic fibroblast growth factor (PeproTech, Rocky Hill, NJ). Primary spheres were derived by plating 50,000 to 100,000 single cells per well into 6-well ultra-low attachment dishes (Corning, Corning, NY). Dishes were cultivated for 7 days prior to in vivo cell injection.
Cell proliferation (cytotoxicity) assay
Cells were seeded at 10,000 cells per well in 96-well plates. After 1 day, cells were treated with an anticancer drug or OV at the desired concentration in serum-free media for 2 h. Then, the media were replaced with normal culture media. After a 72-h treatment, cell viability was assessed using the WST Cell Proliferation Assay (EzCytotox, ITSBIO, Seoul, Korea) according to the manufacturer's instructions. The absorbance of each sample at 450 nm was measured using a microplate reader. The reference wavelength used was 680 nm.
Replication assay
Viral replication was quantified using a standard plaque assay. Cells were plated at 40,000 cells in 24-well flat-bottom plates in 1 ml of media and incubated at 37°C. Cells were infected with the virus of interest (WT, EVV, or CVV) at an MOI of 0.1. Cell lysates were harvested daily for 3 days and viruses were released by 3 cycles of freezing and thawing of the cell lysates. Serial dilutions of each sample were used to infect U2OS cells at 80% confluency in 6-well plates, and viral titers were determined by counting viral plaques.
Real-time PCR
Total RNA was extracted using the TRIzol reagent (Invitrogen). RNA purity was verified by measuring the ratio of absorbance at 260 and 280 nm (A260/280). The first strand of cDNA was synthesized with 2 μg of total RNA using the PrimeScript 1st Strand cDNA Synthesis Kit (Takara Korea, Seoul, Korea), and 1 μL of the cDNA was used for each PCR mixture containing SYBR-Green qPCR mix (Roche, BASEL, Switzerland). Real-time PCR was performed using a LightCycler 96 Real-Time PCR System (Roche). The reaction mixtures were subjected to a 40-cycle amplification at 95°C for 20 s, 60°C for 20 s, and 72°C for 25 s. Relative mRNA expression levels of the selected genes were normalized to beta-actin mRNA expression and quantified using the 2−ΔΔCt method. The sequences of the primers used are shown in Table 1.
Table 1. Sequences of primers used in this study.
| Name | Sequences (5′→3′) | Product size (bp) |
|---|---|---|
| Nanog Forward | CCT GAT TCT TCT ACC AGT CCC A | 123 |
| Nanog Reverse | GGC CTG AGA GAA CAC AGT CC | |
| Sox2 Forward | GCA CAT GAA CGG CTG GAG CAA CG | 207 |
| Sox2 Reverse | TGC TGC GAG TAG GAC ATG CTG TAG G | |
| Oct4 Forward | ATG TTT CTG AAG TGC CCG AA | 85 |
| Oct4 Reverse | AGA GAA GGA TGT GGT TCG AG | |
| cMyc Forward | TGA CCT AAC TCG AGG AGG AGC TGG AAT C | 170 |
| cMyc Reverse | AAG TTT GAG GCA GTT AAA ATT ATG GCT GAA GC | |
| β-Actin 184 Forward | AGA GCT ACG AGC TGC CTG AC | 184 |
| β-Actin 184 Reverse | AGC ACT GTG TTG GCG TAC AG |
Flow cytometry
FACS analysis and cell sorting were performed using a FACSCanto II and FACSAria II (Becton Dickinson, Franklin Lakes, NJ), respectively. FACS data were analyzed using FACS Diva software (BD). Antibodies to the following proteins were used: FITC-conjugated CD44 (MACS, Miltenyi Biotech Korea, Seoul, Korea) and PE-conjugated CD133 (BD). FACS gates were established by staining with isotype-matched PE or FITC conjugated antibodies (BD).
Immunofluorescence staining
Samples were fixed with 4% paraformaldehyde for fluorescence staining. Samples were permeabilized with 0.1% Triton X-100, and nonspecific binding was blocked with 5% normal goat serum (Invitrogen). Staining was performed using unlabeled primary antibodies and fluorophore-conjugated secondary antibodies. Stained samples were examined by fluorescence confocal microscopy (Olympus IX7; Hicksville, NY).
Animal study
All mice were maintained according to protocols approved by the Institutional Animal Care and Use Committee of the Pusan National University (PNU-2014-06850). Nude mice and BALB/c mice were purchased from Orient (Gapyeong, Korea) and HANA (Pusan, Korea). Colon cancer cell lines (105−106 cells) were injected into 6-week-old male BALB/c mice via subcutaneous or intraperitoneal injection. Tumor sizes were measured twice per week in the subcutaneous injection model. The tumor volume was calculated according to the following formula: tumor volume (mm3) = L × W2/2, where L is the tumor length and W is the tumor width. The animals in the intraperitoneal injection model were divided into 4 groups, which received intraperitoneal injection of PBS, 5-Fu (25 mg/kg, 3 times a week), CVV (1 × 106 pfu/mouse, once a week), and 5-Fu (25 mg/kg, 3 times a week) plus CVV (106 pfu/mouse, once a week). Mouse survival was monitored daily during the experimental period. At 4 weeks post-injection, the mice were sacrificed and tissues were immediately fixed in 4% paraformaldehyde.
H & E staining and TUNEL assay
To assay tumor generation and morphological characteristics, frozen tumor sections were used for H&E staining. Images were acquired using an Olympus confocal microscope. The TUNEL assay was performed on paraffin-embedded sections. Commercially available reagents (DeadEnd Fluorometric TUNEL System; Promega, Madison, WI) were used to perform the TUNEL analysis.
SUPPLEMENTARY MATERIALS FIGURE
Acknowledgments
This study was supported by a grant of the Korean Health Technology R & D Project, Ministry of Health & Welfare, Republic of Korea (HI13C0259) and by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT, and Future Planning (NRF-2015R1A2A2A01004489).
Footnotes
FUNDING
This study was supported by a grant of the Korean Health Technology R & D Project, Ministry of Health & Welfare, Republic of Korea (HI13C0259) and by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT, and Future Planning (NRF-2015R1A2A2A01004489).
CONFLICTS OF INTEREST
There are no conflicts of interest to declare.
REFERENCES
- 1.Stewart BW, Wild CP. World Cancer Report 2014. IARC Nonserial Publication; 2014. [Google Scholar]
- 2.Thomassen I, van Gestel YR, Lemmens VE, de Hingh IH. Incidence, prognosis, and treatment options for patients with synchronous peritoneal carcinomatosis and liver metastases from colorectal origin. Dis Colon Rectum. 2013;56:1373–1380. doi: 10.1097/DCR.0b013e3182a62d9d. [DOI] [PubMed] [Google Scholar]
- 3.Puglisi MA Tesori V, Lattanzi W, Gasbarrini GB, Gasbarrini A. Colon cancer stem cells: controversies and perspectives. World J Gastroenterol. 2013;19:2997–3006. doi: 10.3748/wjg.v19.i20.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoo SY, Merzlyak A, Lee SW. Facile growth factor immobilization platform based on engineered phage matrices. Soft Matter. 2011;7:1660–1666. [Google Scholar]
- 5.Yoo SY, Kobayashi M, Lee PP, Lee SW. Early osteogenic differentiation of mouse preosteoblasts induced by collagen-derived DGEA-peptide on nanofibrous phage tissue matrices. Biomacromolecules. 2011;12:987–996. doi: 10.1021/bm1013475. [DOI] [PubMed] [Google Scholar]
- 6.Yoo SY, Oh JW, Lee SW. Phage-chips for novel optically readable tissue engineering assays. Langmuir. 2012;28:2166–2172. doi: 10.1021/la203840n. [DOI] [PubMed] [Google Scholar]
- 7.Bhattarai SR, Yoo SY, Lee SW, Dean D. Engineered phage-based therapeutic materials inhibit Chlamydia trachomatis intracellular infection. Biomaterials. 2012;33:5166–5174. doi: 10.1016/j.biomaterials.2012.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoo SY, Merzlyak A, Lee SW. Synthetic phage for tissue regeneration. Mediators Inflamm. 2014;2014:192790. doi: 10.1155/2014/192790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chung WJ, Lee DY, Yoo SY. Chemical modulation of M13 bacteriophage and its functional opportunities for nanomedicine. Int J Nanomed. 2014;9:5825–5836. doi: 10.2147/IJN.S73883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parato KA, Breitbach CJ, Le Boeuf F, Wang J, Storbeck C, Ilkow C, Diallo JS, Falls T, Burns J, Garcia V, Kanji F, Evgin L, Hu K, et al. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol Ther. 2012;20:749–758. doi: 10.1038/mt.2011.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jefferson A, Cadet VE, Hielscher A. The mechanisms of genetically modified vaccinia viruses for the treatment of cancer. Crit Rev Oncol Hematol. 2015;95:407–416. doi: 10.1016/j.critrevonc.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 12.Guo ZS, Thorne SH, Bartlett DL. Oncolytic virotherapy: molecular targets in tumor-selective replication and carrier cell-mediated delivery of oncolytic viruses. Biochim Biophys Acta. 2008;1785:217–231. doi: 10.1016/j.bbcan.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu TC, Hwang T, Park BH, Bell J, Kirn DH. The targeted oncolytic poxvirus JX-594 demonstrates antitumoral, antivascular, and anti-HBV activities in patients with hepatocellular carcinoma. Mol Ther. 2008;16:1637–1642. doi: 10.1038/mt.2008.143. [DOI] [PubMed] [Google Scholar]
- 14.Breitbach CJ, Arulanandam R, De Silva N, Thorne SH, Patt R, Daneshmand M, Moon A, Ilkow C, Burke J, Hwang TH, Heo J, Cho M, Chen H, et al. Oncolytic vaccinia virus disrupts tumor-associated vasculature in humans. Cancer Res. 2013;73:1265–1275. doi: 10.1158/0008-5472.CAN-12-2687. [DOI] [PubMed] [Google Scholar]
- 15.Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotech. 2012;30:658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Workenhe ST, Mossman KL. Oncolytic virotherapy and immunogenic cancer cell death: sharpening the sword for improved cancer treatment strategies. Mol Ther. 2014;22:251–256. doi: 10.1038/mt.2013.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sampath P, Li J, Hou W, Chen H, Bartlett DL, Thorne SH. Crosstalk between immune cell and oncolytic vaccinia therapy enhances tumor trafficking and antitumor effects. Mol Ther. 2013;21:620–628. doi: 10.1038/mt.2012.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M, Cho M, Lim HY, Chung HC, Kim CW, Burke J, Lencioni R, Hickman T, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. 2013;19:329–336. doi: 10.1038/nm.3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hwang T, Lee HJ. Novel oncolytic virus derived from vaccinia virus. Republic of Korea: Patent No. 10-2012-0004060. 2012
- 20.Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 21.Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer. 2012;12:133–143. doi: 10.1038/nrc3184. [DOI] [PubMed] [Google Scholar]
- 22.Wang SC, Hong JH, Hsueh C, Chiang CS. Tumor-secreted SDF-1 promotes glioma invasiveness and TAM tropism toward hypoxia in a murine astrocytoma model. Lab Invest. 2012;92:151–162. doi: 10.1038/labinvest.2011.128. [DOI] [PubMed] [Google Scholar]
- 23.Gelmini S, Mangoni M, Serio M, Romagnani P, Lazzeri E. The critical role of SDF-1/CXCR4 axis in cancer and cancer stem cells metastasis. J Endocrinol Invest. 2008;31:809–819. doi: 10.1007/BF03349262. [DOI] [PubMed] [Google Scholar]
- 24.Lathia JD, Heddleston JM, Venere M, Rich JN. Deadly teamwork: neural cancer stem cells and the tumor microenvironment. Cell Stem Cell. 2011;8:482–485. doi: 10.1016/j.stem.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hanahan D, Weinberg RA. Hallmarks of Cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 26.Zhang M, Rosen JM. Stem cells in the etiology and treatment of cancer. Curr Opin Genet Dev. 2006;16:60–64. doi: 10.1016/j.gde.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 27.Fillmore C, Kuperwasser C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008;10:R25. doi: 10.1186/bcr1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 29.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stewart JM, Shaw PA, Gedye C, Bernardini MQ, Neel BG, Ailles LE, Mak TW. Phenotypic heterogeneity and instability of human ovarian tumor-initiating cells. Proc Natl Acad Sci U S A. 2011;108:6468–6473. doi: 10.1073/pnas.1005529108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haraguchi N, Ohkuma M, Sakashita H, Matsuzaki S, Tanaka F, Mimori K, Kamohara Y, Inoue H, Mori M. CD133+CD44+ population efficiently enriches colon cancer initiating cells. Ann Surg Oncol. 2008;15:2927–2933. doi: 10.1245/s10434-008-0074-0. [DOI] [PubMed] [Google Scholar]
- 32.Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A, Conticello C, Ruco L, Peschle C, De Maria R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2007;15:504–514. doi: 10.1038/sj.cdd.4402283. [DOI] [PubMed] [Google Scholar]
- 33.Prince ME, Sivanandan R, Kaczorowski A, Wolf GT, Kaplan MJ, Dalerba P, Weissman IL, Clarke MF, Ailles LE. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci U S A. 2007;104:973–978. doi: 10.1073/pnas.0610117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–1037. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 35.Yamashita T, Wang XW. Cancer stem cells in the development of liver cancer. The J Clin Invest. 2013;123:1911–1918. doi: 10.1172/JCI66024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tirino V, Desiderio V, Paino F, De Rosa A, Papaccio F, La Noce M, Laino L, De Francesco F, Papaccio G. Cancer stem cells in solid tumors: an overview and new approaches for their isolation and characterization. FASEB J. 2013;27:13–24. doi: 10.1096/fj.12-218222. [DOI] [PubMed] [Google Scholar]
- 37.Newman W, Southam CM. Virus treatment in advanced cancer. A pathological study of fifty-seven cases. Cancer. 1954;7:106–118. doi: 10.1002/1097-0142(195401)7:1<106::aid-cncr2820070112>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 38.Southam CM, Noyes WF, Mellors R. Virus in human cancer cells in vivo. Virology. 1958;5:395–398. doi: 10.1016/0042-6822(58)90033-3. [DOI] [PubMed] [Google Scholar]
- 39.Southam CM, Moore AE. Induced Virus Infections in Man by the Egypt Isolates of West Nile Virus. Am J Trop Med Hyg. 1954;3:19–50. doi: 10.4269/ajtmh.1954.3.19. [DOI] [PubMed] [Google Scholar]
- 40.Kirn DH, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer. 2009;9:64–71. doi: 10.1038/nrc2545. [DOI] [PubMed] [Google Scholar]
- 41.Kim MK, Breitbach CJ, Moon A, Heo J, Lee YK, Cho M, Lee JW, Kim SG, Kang DH, Bell JC, Park BH, Kirn DH, Hwang TH. Oncolytic and immunotherapeutic vaccinia induces antibody-mediated complement-dependent cancer cell lysis in humans. Sci Transl Med. 2013;5:185ra163. doi: 10.1126/scitranslmed.3005361. [DOI] [PubMed] [Google Scholar]
- 42.Coukos G, Makrigiannakis A, Kang EH, Rubin SC, Albelda SM, Molnar-Kimber KL. Oncolytic herpes simplex virus-1 lacking ICP34.5 induces p53-independent death and is efficacious against chemotherapy-resistant ovarian cancer. Clin Cancer Res. 2000;6:3342–3353. [PubMed] [Google Scholar]
- 43.Mahller YY, Williams JP, Baird WH, Mitton B, Grossheim J, Saeki Y, Cancelas JA, Ratner N, Cripe TP. Neuroblastoma cell lines contain pluripotent tumor initiating cells that are susceptible to a targeted oncolytic virus. PLoS One. 2009;4:e4235. doi: 10.1371/journal.pone.0004235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.