

Significance
Translational control is a key component of gene regulation; therefore, it is important to understand alterations to the translational machinery that occur during development. We identified a unique mTOR (the mechanistic target of rapamycin)-independent mechanism of 4E-BP (a repressor of cap-dependent translation) regulation that is utilized by chondrocytes in response to FGF signaling. It is well established that FGF signaling leads to inhibition of chondrocyte proliferation, and is crucial for bone development. We found that FGF specifically suppresses protein synthesis in chondrocytes by activating 4E-BP. Importantly, FGF-induced changes in the translation apparatus observed in cell culture are similarly detected in vivo, and reflect the action of FGF signaling on downstream targets during bone development.
Keywords: 4E-BP, FGF, chondrocytes, translational control
Abstract
Regulation of protein synthesis plays a vital role in posttranscriptional modulation of gene expression. Translational control most commonly targets the initiation of protein synthesis: loading 40S ribosome complexes onto mRNA and AUG start codon recognition. This step is initiated by eukaryotic initiation factor 4E (eIF4E) (the m7GTP cap-binding protein), whose binding to eIF4G (a scaffolding subunit) and eIF4A (an ATP-dependent RNA helicase) leads to assembly of active eIF4F complex. The ability of eIF4E to recognize the cap is prevented by its binding to eIF4E binding protein (4E-BP), which thereby inhibits cap-dependent translation by sequestering eIF4E. The 4E-BP activity is, in turn, inhibited by mTORC1 [mTOR (the mechanistic target of rapamycin) complex 1] mediated phosphorylation. Here, we define a previously unidentified mechanism of mTOR-independent 4E-BP1 regulation that is used by chondrocytes upon FGF signaling. Chondrocytes are responsible for the formation of the skeleton long bones. Unlike the majority of cell types where FGF signaling triggers proliferation, chondrocytes respond to FGF with inhibition. We establish that FGF specifically suppresses protein synthesis in chondrocytes, but not in any other cells of mesenchymal origin. Furthermore, 4E-BP1 repressor activity is necessary not only for suppression of protein synthesis, but also for FGF-induced cell-cycle arrest. Importantly, FGF-induced changes in the 4E-BP1 activity observed in cell culture are likewise detected in vivo and reflect the action of FGF signaling on downstream targets during bone development. Thus, our findings demonstrate that FGF signaling differentially impacts protein synthesis through either stimulation or repression, in a cell-type–dependent manner, with 4E-BP1 being a key player.
Regulation of translation allows rapid and reversible adjustments of mRNA translation in response to various physiological and pathological conditions (1). The rate-limiting step in protein synthesis is initiation, during which the small ribosomal subunit, bound to several translation initiation factors and initiator MettRNA, is recruited to mRNA. Ribosomal loading onto mRNAs is mediated by eukaryotic initiation factor 4F (eIF4F), eIF4A, and eIF4B. By binding to the cap structure of mRNAs, eIF4F positions the ribosomal preinitiation complex onto the 5′ end of mRNA and facilitates scanning toward the 5′ proximal AUG codon. One of the key mechanisms controlling protein synthesis involves modulating the activity of the m7GTP cap-binding protein eIF4E by a family of eIF4E binding proteins (4E-BPs). The ability of 4E-BPs to sequester eIF4E from eIF4F complexes depends on their phosphorylation status (2). Hypophosphorylated 4E-BP binds eIF4E to repress cap-dependent mRNA translation, whereas phosphorylation results in its release from eIF4E, which can then form functional eIF4F complexes. Phosphorylation of 4E-BP is mediated by the serine/threonine kinase mTOR (the mechanistic target of rapamycin), which is activated by nutrients and numerous growth factors, including fibroblast growth factors (FGFs) (3).
FGFs are critical regulators of many developmental processes, including bone development. Long bones and vertebrae are formed through endochondral ossification, a process that requires the proliferation and differentiation of chondrocytes (4). Chondrocyte homeostasis is regulated by a number of signaling molecules and transcription factors, and genetic studies identified FGF signaling as a crucial player in these processes (5). FGF molecules signal by activating FGF Receptors (FGFRs), and several FGFR mutations have been linked to autosomal, dominant bone morphogenetic disorders (6). Importantly, all of these mutations are “gain of function” mutations, resulting in excessive FGF signaling. It has been well established that the major effect of FGF signaling in chondrocytes is growth inhibition (7, 8), an effect which is in striking contrast to a universal proliferative response seen in nearly all cell types. We previously identified several downstream effectors of FGF signaling in chondrocytes at the transcriptional level (9). However, the effect of FGF signaling on protein synthesis has never been studied, and the mechanism of this unique chondrocyte-specific response remains to be elucidated.
Here, we investigated the impact of FGF signaling on protein synthesis in chondrocytes, where it causes growth inhibition. Unexpectedly, we report for the first time to our knowledge that FGF signaling can inhibit protein synthesis. This unique response is acquired de novo during chondrogenesis because mesenchymal stem cells (MSC) from which chondrocytes derive do not respond in a similar manner. FGF-induced inhibition of protein synthesis requires the activity of 4E-BP1, the canonical inhibitor of cap-dependent mRNA translation. We suggest a previously unidentified mechanism of 4E-BP1 activation that is implemented in the presence of active mTORC1 (mTOR complex 1). This mechanism would allow for selective and rapid adjustments in the activity of mTOR targets in response to extracellular cues such as FGF signaling.
Results
FGF Signaling Down-Regulates Protein Synthesis in Chondrocytes.
We first analyzed the effect of FGF on protein synthesis in chondrocytes using Rat ChondroSarcoma (RCS) cells. These cells exhibit most properties of proliferating chondrocytes, including FGF response, and allow genetic manipulations that cannot be performed in primary chondrocytes, whose life span in culture is limited. The changes in total protein synthesis in FGF-treated and untreated cells were measured after pulse-labeling with 35S Met/Cys mix. In parallel, we monitored FGF-induced changes in the cell cycle by FACScan analysis. Two additional cell lines with FGF proliferative response were included: OB1 cells (immortalized osteoblasts) (10) and C3H10T1/2 cells that are functionally similar to MSC (11) and are able to differentiate into multiple lineages. Relative S-phase levels of OB1 and C3H10T1/2 cells were increased following FGF treatment (1.5-fold and threefold, respectively, at 24 h), whereas proliferation of RCS cells was inhibited (Fig. S1A). The rate of total protein synthesis in OB1 cells was virtually unaffected and slightly up-regulated in C3H10T1/2 cells (1.3-fold). Importantly, FGF suppressed the rate of protein synthesis in RCS cells by 70–75% after 24 h treatment (Fig. 1A). We observed a similar decline (60%) in primary chondrocytes isolated from neonatal rat pups (Fig. 1B) and in the chondrocytes obtained using high-density micromass cultures derived from prechondrogenic limb bud mesenchyme from E12.5 embryos (Fig. 1C). These mesenchyme cultures form multiple condensations in which both chondrocyte differentiation and proliferation take place (12). Therefore, this response is not restricted to immortalized cells but is also observed in other models of proliferating chondrocytes. Interestingly, despite these striking differences, FGF similarly induced ERK1/2 activation in RCS, micromass, and OB1 cells (Fig. S1 B and C). This implies that canonically induced FGF-signaling pathways likely have chondrocyte-specific targets responsible for the inhibitory response. To further support this hypothesis, we induced differentiation of C3H10T1/2 cells into osteoblasts, chondrocytes, and adipocytes (Fig. S1D) (13, 14). After 11 d in differentiation media the cells were treated with FGF and total protein synthesis was analyzed. As shown in Fig. 1D, FGF treatment inhibited protein synthesis only in the cells committed to the chondrocyte lineage, supporting our hypothesis that FGF-induced down-regulation of protein synthesis is chondrocyte-specific and likely acquired during chondrocyte differentiation.
Fig. S1.

FGF-mediated suppression of protein synthesis is chondrocyte-specific. (A) Rat Chondrosarcoma (RCS), OB1, and C3H10T1/2 cells were treated with FGF1 for the times indicated and were analyzed by FACScan analysis. Numbers on the y axis indicate relative percentage of total cells in S-phase. (B) RCS, OB1, and micromass cultures were treated with FGF for times indicated and analyzed by immunoblotting using specific antibodies. Ten micrograms of total protein were used for immunodetection, and equal amount of protein loading was confirmed by immunodetection of α-actin. (C) BrdU incorporation in micromass cultures was examined by immunofluorescence in untreated (red, Top) and FGF-treated cells (red, Bottom). Nuclei were stained with DAPI (blue). Quantification of BrdU staining was performed on four different areas of three independent experiments (Right). Numbers on the y axis indicate relative percentage of cells positive for BrdU staining. (D) C3H10T1/2 cells were differentiated into chondrocytes (C), adipocytes (A), and osteoblasts (O) using lineage-specific differentiation medium. Osteogenic differentiation was detected using alkaline phosphatase staining, adipogenesis was detected using Oil Red O staining, and chondrogenesis was detected using Alcian blue staining. Days in the corresponding differentiation medium are indicated. Differentiation is analyzed by qPCR using known markers for adipogenesis [adipocyte protein 2 (AP2)], osteogenesis [alkaline phosphatase (ALP)], and chondrogenesis (Col2a1 and Col10a1) (Bottom).
Fig. 1.

Regulation of protein synthesis by FGF signaling in chondrocytes. (A–C) Indicated cells were treated with FGF1, and protein synthesis activity was measured by S35-Met/Cys incorporation. Relative levels of radioactivity incorporated are compared with untreated cells. Lysates were normalized to the amount of total protein. The data are representative of three independent experiments, error bars represent mean ± SD, and P value is indicated when significant. (D) C3H10T1/2 cells were differentiated into chondrocytes (C), adipocytes (A), and osteoblasts (O). After 11 d in differentiation medium, cells were treated with FGF1, and protein synthesis activity was measured as described above.
FGF Signaling in Chondrocytes Activates 4E-BP1.
To elucidate the mechanism of this chondrocyte-specific effect of FGF on protein synthesis we analyzed overall abundance and phosphorylation status of 4E-BP1, the repressor of cap-dependent translation. There are four well-described 4E-BP phosphorylation sites: Thr37 and Thr46 are phosphorylated first, followed by Thr70 and Ser65. mTORC1 complexes, comprised of mTOR kinase, Raptor, and GβL, mediate 4E-BP inactivation (15). Differentially phosphorylated 4E-BP isoforms can be separated on high-density SDS PAGE according to their phosphorylation marks with the uppermost band representing fully phosphorylated, Ser65 positive, incapable of eIF4E binding, 4E-BP. As shown in Fig. 2A, dephosphorylated 4E-BP1 was detected as early as after 4 h of FGF treatment, and the protein was completely dephosphorylated by 16 h. Accordantly, Ser65 phosphorylation declined significantly at 4 h of treatment and was not detectable after 8 h. The 4E-BP dephosphorylation was induced in a similar manner in primary and micromass chondrocytes treated with FGF (Fig. 2A).
Fig. 2.

FGF signaling activates 4E-BP1 in chondrocytes. (A) RCS, primary chondrocytes, and micromass cultures were treated with FGF1. Five micrograms of total protein were analyzed by SDS/PAGE followed by immunoblotting. The upper bands correspond to inactive 4E-BP1 incapable of binding eIF4E. (B) Ten-micron sections of paraformaldehyde (PFA)-fixed tissue from the growth plate of tibia of newborn mice were stained with 4E-BP1(Ser65) and 4E-BP1 antibodies. The proliferating (P) and hypertrophic (H) regions are indicated by arrows. Quantification was carried out by counting the number of positively stained cells in six fields for each cell type in three different slides. (C) Metatarsal bone rudiments were isolated from E15.5 embryos and cultivated in vitro for 24 or 48 h with or without FGF1. BrdU was added during the last 6 h of FGF treatment. After fixation, rudiments were analyzed for immunodetection of BrdU, 4E-BP1(S65), and 4E-BP1. Staining with Alcian blue confirmed the presence of cartilage-specific sulfate proteoglycans in the extracellular matrix, and the reserve (R), proliferating (P), and hypertrophic (H) zones are shown. Representative pictures of immunostaining in the proliferating zone are shown. Quantification of staining was carried out by counting the number of positively highly stained cells in four fields for three different rudiments. (D) FGF signaling stimulates association of 4E-BP1 with eIF4E. Extracts from FGF-treated RCS cells were incubated with agarose-conjugated eIF4E antibody. As a reference, 5% of the immunoprecipitated (IP) whole-cell lysate (“Input”) was loaded.
Endochondral bone formation is driven by changes in the growth plate, where proliferative chondrocytes differentiate into hypertrophic, directing the formation of mineralized matrix. It is proposed that FGF signaling induces not only growth arrest in chondrocytes but also some aspects of differentiation (16). FGFR3 is primarily expressed in proliferating chondrocytes, where it regulates proliferation and the transition to terminal (hypertrophic) differentiation, with no protein detected in hypertrophic cells (17). This implies that FGF-treated cells would represent more differentiated chondrocytes compared with untreated ones. We therefore examined the distribution of 4E-BP1 (Ser65) in the growth plate of newborn mice by immunohistochemistry (IHC). The hypertrophic region is defined by enlarged cells, whereas the proliferative chondrocytes are smaller in size (Fig. 2B). The majority of chondrocytes in the proliferative zone (80%) were Ser65 positive, whereas the hypertrophic region had fewer positive cells (15%). At the same time most of the cells in both regions were stained positive for 4E-BP1. This demonstrates that FGF-induced changes in the 4E-BP1 phosphorylation observed in cell culture are detected in vivo and reflect the action of FGF signaling on downstream targets during the endochondral ossification. Next we determined whether FGF-induced dephosphorylation of 4E-BP1 is similar in chondrocytes that are within developing bones that have normal cartilage architecture. For this purpose, we used organ culture of metatarsal bone rudiments (18). The proliferating chondrocytes responded to FGF with inhibition, as confirmed by BrdU incorporation (Fig. 2C), and the amount of S65 positive cells also declined with FGF treatment (Fig. 2C).
A functionally important reduction in 4E-BP phosphorylation should result in accumulation of 4E-BP/eIF4E complexes. We therefore immunoprecipitated eIF4E from FGF-treated and untreated lysates using eIF4E agarose-conjugated antibodies and analyzed proteins associated with eIF4E (Fig. 2D). In agreement with the kinetics of 4E-BP1 dephosphorylation, significantly more 4E-BP1 was detected in IPs after 4 h of FGF treatment. Accordingly, levels of eIF4G and eIF4A bound to eIF4E gradually decreased. These data, to our knowledge, provide the first evidence that 4E-BP activation contributes to FGF-induced inhibition of protein synthesis.
The 4E-BP Activation Precedes Down-Regulation of mTOR Kinase Activity.
It is well established that mTOR kinase phosphorylates 4E-BP on Thr37 and Thr46 (2). However, phosphorylation of Ser65 and Thr70, residues that determine the ability of 4E-BP to bind eIF4E, has also been attributed to different enzymes depending on the cell type (19, 20). We therefore confirmed that mTOR plays a role in maintaining the hyperphosphorylated status of 4E-BP1 in chondrocytes using the specific mTOR inhibitors rapamycin and Ku-63794. Rapamycin inhibits mTORC1 complexes, and Ku-63794 inhibits both mTORC1 and mTORC2. The latter is comprised of mTOR kinase, Rictor, SIN1, and GβL and has distinct biological substrates compared with mTORC1 (21). Both inhibitors increased the proportion of dephosphorylated 4E-BP, confirming that mTOR kinase is responsible for inactivating 4E-BP in unchallenged chondrocytes (Fig. 3A). To investigate how mTOR activity is modulated by FGF signaling, phosphorylation of mTOR Ser2448 and 2481 was monitored because mTOR activation is accompanied by phosphorylation of these residues (3). Surprisingly, as shown in Fig. 3B, phospho-S2481 mTOR was elevated at 2 h and started to decline only after 16 h. These kinetics mirrored the kinetics of p70S6K phosphorylation, a direct mTORC1 substrate and an accepted readout of activated mTORC1 (Fig. 3B). Importantly, distribution of phosphorylated p70S6K in the growth plate of newborn mice correlated with FGF treatment (Fig. 3C). Paradoxically, an increase in mTOR activity coincided with 4E-BP1 dephosphorylation (Fig. 3B). This, however, was not the case when OB1 cells were treated with FGF. Whereas phosphorylation of p70S6K was identical for both cell types at 16 h, 4E-BP1 was dephosphorylated in RCS cells but not in OB1 cells, where 4E-BP1(S65) was detected up to 24 h (Fig. S2B). The reason for this discrepancy might be a selective mTORC1 inhibition toward 4E-BP1. We therefore examined whether FGF induced any changes in the abundance/activity of known components and effectors of the mTOR pathway. No changes were detected in the expression of Raptor, Rictor, DEPTOR, and GβL upon FGF treatment or in their binding to mTOR (Fig. S2A). We also did not find any evidence that known negative regulators of mTOR, PRAS40 (22), and TSC2 (tuberous sclerosis 2) (23) might cause any inhibition of mTOR in chondrocytes (Fig. S2 A, C, and D). Moreover, the level of mTOR activity in FGF-treated RCS and OB1 cells was comparable up to 16 h, despite prominent difference in 4E-BP1 phosphorylation (Fig. 3B).
Fig. 3.

The 4E-BP1 activation precedes down-regulation of mTOR kinase activity. (A) WB of total cell lysates from RCS cells treated with Rapamycin (Rap) or Ku-63794. DMSO was used as a vehicle. (B) RCS and OB1 cells were treated with FGF1. Twenty micrograms of total protein was analyzed by WB. (C) Ten-micron sections of PFA-fixed tissue from the growth plate of tibia of newborn mice were analyzed by IHC using indicated antibodies. The proliferating (P) and hypertrophic (H) regions are indicated by arrows. Quantification was carried out by counting the number of positively stained proliferating and hypertrophic cells in six separate fields for each cell type. (D) In vitro mTOR kinase assay was done as described in SI Materials and Methods. The lysates were analyzed by immunoblotting using indicated antibodies.
Fig. S2.

Expression of mTOR pathway components in FGF-treated RCS cells. (A) RCS cells were treated with FGF1 for indicated periods of time and analyzed by WB using indicated antibodies. Twenty micrograms of total protein were used for immunodetection, and equal amount of protein loading was confirmed by either α-tubulin or α-actin immunodetection. (B) RCS and OB1 cells were treated with FGF1 for indicated periods of time and analyzed by WB using indicated antibodies. Twenty micrograms of total protein were used for immunodetection, and equal amount of protein loading was confirmed by α-tubulin immunodetection. (C) RCS cells were transfected with either siRNAs against DEPTOR or against TSC2 to evaluate their importance for FGF-induced growth arrest. Negative siRNA was used as a control. Down-regulation of DEPTOR expression did not change inhibitory response in RCS cells. siTSC2 transfected cells showed slightly delayed suppression of protein synthesis upon FGF treatment. Fifteen micrograms of total protein were used for immunodetection, and equal amount of protein loading was confirmed by β-catenin and α-actin immunodetection. (D) Protein synthesis activity was measured by S35-Met/Cys incorporation. Relative levels of radioactivity incorporated are compared with untreated cells. Lysates from S35-labeled samples were normalized to the amount of total protein.
We next performed an in vitro mTOR kinase assay using recombinant 4E-BP1 and observed that 4E-BP1 was robustly phosphorylated at authentic sites in untreated lysates or lysates that had been treated for 8 h (Fig. 3D). This activity was inhibited in the presence of Ku63794, indicating that it is mTOR-specific. Lysates from the cells treated with FGF for 16 and 24 h did not exhibit significant kinase activity. Therefore, delayed decline in mTORC1 activity cannot account for 4E-BP1 activation at the early time points of FGF treatment. One possible explanation could be that 4E-BP1 activity is controlled by FGF-activated protein phosphatase. It has been reported that 4E-BP1 phosphorylation is sensitive to Calyculin, a chemical inhibitor of PP2A (24); we therefore examined whether PP2A plays a role in FGF-induced 4E-BP dephosphorylation. PP2A activity can be blocked by small T-antigen (ST) of SV40 virus, which forms a stable complex with PP2A (25). As shown in Fig. S3, following transduction with an adenovirus (Ad) vector, the majority of the cells expressed either GFP or SV40 ST, which was also detected by WB (Fig. 4A). Expression of SV40ST inhibited 4E-BP1 activation in FGF-treated cells, whereas expression of GFP had no effect on kinetics of 4E-BP1 dephosphorylation. Accordingly, the cells transduced with ST-expressing virus continued to cycle upon FGF treatment, and the protein synthesis level was decreased only by 15% compared with 75% in GFP-Ad transduced cells (Fig. 4B). These data indicate that PP2A (or PP2A-like phosphatase) might play a role in FGF-induced 4E-BP1 activation.
Fig. S3.
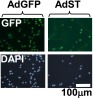
Activation of 4E-BP1 requires SV40ST sensitive phosphatase activity. RCS cells were transduced with adenoviruses expressing GFP or SV40 ST following FGF1 treatment. Expression of GFP and SV40 ST was examined by immunofluorescence before FGF treatment. Nuclei were stained with DAPI.
Fig. 4.

The 4E-BP1 activation requires SV40ST sensitive phosphatase activity. (A) RCS cells were transduced with adenoviruses expressing GFP or SV40 ST following FGF treatment. Ten micrograms of total protein were used for WB. (B) Protein synthesis was measured by S35-Met/Cys incorporation. Relative levels of radioactivity incorporated are compared with untreated cells. The data are representative of three independent experiments. (C) De novo RNA and protein are needed for FGF-induced 4E-BP1 activation in chondrocytes. RCS cells were pretreated either with cycloheximide (CHX) 10 μg/mL or with actinomycin D (1 μg/mL) following FGF1 treatment. Ten micrograms of total protein were used for WB.
We next treated cells with FGF in the presence of either actinomycin D (transcription inhibitor) or cycloheximide (inhibitor of protein synthesis) to test whether dephosphorylation of 4E-BP1 requires de novo protein and/or RNA (Fig. 4C). Both treatments abolished the ability of FGF to activate 4E-BP1, meaning that 4E-BP1 dephosphorylation requires both RNA and protein synthesis.
FGF-Induced 4E-BP1 Activation Is Crucial for Mediating Growth Arrest in Chondrocytes.
The most abundant out of three 4E-BP isoforms is generally considered to be 4E-BP1; however, isoform levels have not been previously characterized in chondrocytes. We therefore determined the expression of each isoform by RT-qPCR. We demonstrated that 4E-BP1 mRNA level in RCS cells and primary chondrocytes is threefold higher than 4E-BP2 and about 1000-fold higher than 4E-BP3 (Fig. 5A and Table S1). As mRNA levels of 4E-BP1 and 4E-BP2 were comparable, we examined their protein levels. FLAG-tagged 4E-BP1 (26) and 4E-BP2 were transiently overexpressed in RCS cells at comparable levels as evaluated by FLAG antibodies (Fig. S3A). Using isoform-specific antibodies for the same samples we estimated that 4E-BP1 expression is at least 10-fold higher than 4E-BP2 expression. Kinetics of 4E-BP2 and 4E-BP1dephosphorylation were identical upon FGF treatment (inputs in Fig. 5B), and 4E-BP(S65) IPs were recognized by 4E-BP2 antibodies when present (Fig. S4), indicating that both isoforms are activated in a similar manner. Accordingly, 4E-BP2 was able to sequester eIF4E upon FGF treatment; however, the portion of 4E-BP1/eIF4E complexes was considerably higher compared with 4E-BP2/eIF4E complexes (Fig. 5B). Therefore, to determine whether 4E-BP activation contributes to FGF-induced growth arrest in chondrocytes, we knocked down only 4E-BP1 in RCS cells using a shRNA (Fig. 5C). Although FGF treatment decreased the fraction of BrdU positive cells in cultures expressing a nonsilencing (ns) scrambled shRNA from 40% to 4%, knockdown of 4E-BP1 by shRNA effectively antagonized FGF-induced growth inhibition (Fig. S5A). Similar results were obtained when total protein synthesis was analyzed. In the cells overexpressing 4E-BP1 shRNA the level of protein synthesis was barely affected by FGF treatment (Fig. 5D), whereas control shRNA cells exhibited a 75% decline. These data demonstrate that 4E-BP1 plays an important role in inhibiting both protein synthesis and cell-cycle progression in response to FGF treatment.
Fig. 5.

The 4E-BP1 activity is crucial for mediating growth arrest in chondrocytes. (A) Relative levels of 4E-BP1-3 mRNAs in chondrocytes were determined by qRT-PCR. The 18S was used as a normalization control. (B) FGF signaling stimulates 4E-BP1/eIF4E and 4E-BP2/eIF4E complex formation. RCS cells were treated with FGF1, and protein extracts were incubated with agarose-conjugated eIF4E antibody. Five times more (5X) of the IPs were loaded to visualize 4E-BP2 in eIF4E IPs compared with the 4E-BP1 detection. The asterisk denotes an unspecific band caused by a protein marker. 5X and 1X eIF4A and eIF4E are the same exposure. (C–E) RCS cells were infected with either 4E-BP1 or nonsilencing (ns) shRNAs. (C) Relative levels of 4E-BP1-3 mRNAs in sh4E-BP1 cells compared with the control cells. Five micrograms of total protein were analyzed by WB. (D) Protein synthesis was measured by S35-Met/Cys incorporation. The data are representative of three independent experiments, and error bars represent mean ± SD. (E) Overexpression of wt4E-BP1 but not a 4E-BP1 mutant deficient in eIF4E binding (ΔE4E-BP) restores FGF response in the cells with low levels of 4E-BP1 (4E-BP1 shRNA cells). Five micrograms of total protein were analyzed by WB to validate protein expression. Protein synthesis was measured by S35-Met/Cys incorporation. The data are representative of three independent experiments.
Table S1.
qPCR primers used in the study
| Gene | Accession no. | Forward | Reverse |
| Eif4ebp1 | NM_053857.2 | 5-TCCTGATGGAGTGTCGGAAC-3 | 5-AAACTGTGACTCTTCACCACCT-3 |
| Eif4ebp2 | NM_001033069.1 | 5-CAAGAATCGTCCTGCCCTATTA-3 | 5-GAACAGCAATGGGCACTAAAC-3 |
| Eif4ebp3 | NM_001202552.1 | 5-GCCTTCCTGCTGCTCACTAT-3 | 5-AGATGATCCTGGTGCCTCCC-3 |
| Fabp4 | NM_024406.2 | 5-GATGCCTTTGTGGGAACCT-3 | 5-CTGTCGTCTGCGGTGATTT-3 |
| Col2a1 | NM_001113515.2 | 5-GGCAACAGCAGGTTCACATA-3 | 5-CCACACCAAATTCCTGTTCA-3 |
| Col10a1 | NM_009925.4 | 5-AAGGAGTGCCTGGACACAAT-3 | 5-GTCGTAATGCTGCTGCCTAT-3 |
| Alpl | NM_007431.3 | 5-AACCCAGACACAAGCATTCC-3 | 5-CCAGCAAGAAGAAGCCTTTG-3 |
Fig. S4.

Activation of 4E-BP2 by FGF in chondrocytes. (A) RCS cells were transfected with FLAG-tagged 4E-BP1 and 4E-BP2 constructs and analyzed using FLAG, 4E-BP1, and 4E-BP2 antibodies. Note that tagged 4E-BP1 migrates much slower than endogenous 4E-BP1, whereas 4E-BP2-FLAG has only eight extra amino acids and migrates similarly to the endogenous protein. (B) RCS cells were treated with FGF1 for the times indicated. Equal amounts of protein extracts were immunoprecipitated with 4E-BP1(S65) antibody. The presence of 4E-BP2 was assayed by using specific antibodies. As a reference, 5% of the immunoprecipitated whole-cell lysate (“Input”) was loaded. Equal amount of protein loading was confirmed by α-tubulin immunodetection.
Fig. S5.

The translation repressor 4E-BP1 is crucial for mediating FGF response in chondrocytes. RCS cells were infected with either 4E-BP1 or nonsilencing (ns) shRNAs. (A) S-phase levels in the indicated cell lines treated with FGF1 were assayed by BrdU incorporation. (B) RCS cells overexpressing either ns or 4E-BP1 shRNAs were treated with FGF1, and equal amounts of protein extracts were incubated with agarose-conjugated eIF4E antibody. Two separate samples were used for the cells overexpressing 4E-BP1 shRNA (lanes 2a and 2b and lanes 4a and 4b). Two exposures are shown for 4E-BP1 WB to demonstrate that 4E-BP1 is dephosphorylated similarly in both cell lines.
We also evaluated eIF4F complex formation in 4E-BP1 shRNA cells treated with FGF. A significant amount of 4E-BP1 was bound to eIF4E in control cells, and eIF4G was not detectable (Fig. S5B). However, eIF4G was readily detected in eIF4E IPs from 4E-BP1 shRNA cells, and the amount of bound 4E-BP1 was significantly lower compared with control cells (Fig. S5B). These data demonstrate that proper remodeling of eIF4F complexes is crucial for mediating FGF-induced down-regulation of total protein synthesis. To support this hypothesis further we transiently overexpressed either wild-type 4E-BP1 or a Δ4E 4E-BP1 mutant [protein with a deleted binding site for eIF4E (15)]. Overexpression of wt4E-BP1 restored the original chondrocyte-specific inhibitory FGF response (Fig. 5E). In contrast, translation rate was not significantly affected when the mutant form was overexpressed (Fig. 5E). These experiments clearly indicate that binding of 4E-BP1 to eIF4E is vital for mediating the chondrocyte-specific inhibitory FGF response and, to our knowledge, provide the first evidence that this response is mediated by activation of translational repressor 4E-BP1, which leads to remodeling of eIF4F complexes.
Discussion
Down-Regulation of Protein Synthesis by FGF is Chondrocyte-Specific.
FGF-induced growth arrest in chondrocytes is a unique response, which has been well documented in the past two decades (5, 7, 18, 27); however, it was not known whether translational control plays any role in this unique case of FGF signaling. This is of particular interest, as it is well established that growth factors stimulate translation through the AKT/mTOR pathway. Here we show that FGF signaling can also inhibit protein synthesis, and this inhibition is an integral part of overall growth arrest. We demonstrated that this phenomenon is lineage-specific, as MSC acquired this ability only during differentiation into chondrocytes. Interestingly, we did not observe FGF-induced down-regulation of protein synthesis at later times, when the majority of chondrocytes become hypertrophic. This is consistent with known FGFR3 expression in vivo, as FGFR3 is mainly expressed in the proliferative zone and not in hypertrophic chondrocytes, making the FGF-inhibitory response a unique feature of proliferative chondrocytes. Suppression of protein synthesis by FGF observed in RCS cells likely reflects differences in the rate of protein synthesis between proliferative and hypertrophic chondrocytes in vivo, which one can expect to be substantial, as proliferative chondrocytes are a driving force of bone longitudinal growth.
A Novel Role of 4E-BP1 in Mediating FGF Inhibitory Growth Response.
Paradoxically, we found that upon FGF treatment, kinetics of mTOR and 4E-BP1 activities do not correlate, and despite high levels of mTOR activity detected in cells, 4E-BP1 phosphorylation status is not maintained. FGF-induced dephosphorylation of Ser65 was consistently detected in all experimental systems, including micromass and organ cultures, indicating that our in vitro data are relevant to the situation in vivo and likely reflect the effect of FGF signaling on the downstream targets during the process of endochondral ossification. mTORC1 complexes have been recently implicated in chondrogenesis (28). In this elegant work the authors used the Prx-Cre system to delete mTOR or Raptor at the mesenchymal progenitor stage and demonstrated that embryonic skeletal growth was significantly diminished with delays in chondrocyte hypertrophy. They concluded that mTORC1 mostly affects protein synthesis; however, extracellular signals responsible for dynamic regulation of mTORC1 activity at different stages of chondrogenesis are unknown. It is possible that FGF signaling might play an active role in the dynamics of mTOR activity during chondrogenesis. This would also be in line with reports demonstrating a role of the PI3K pathway in hypertrophic chondrocyte differentiation (29).
Here, for the first time to our knowledge we determined that 4E-BP1 is the major 4E-BP isoform in chondrocytes. Importantly, FGF-induced down-regulation of protein synthesis requires its function, as it is sensitive to the levels of 4E-BP1 expression. This clearly indicates that ceasing the activity of the translational machinery in chondrocytes is not just a simple consequence of FGF-induced changes on a transcriptional level, but an important component of a well-orchestrated cellular response, which was attained during chondrogenesis. At this point it is not clear why the 4E-BP1/4E-BP2 double knockout mouse model does not exhibit any apparent bone phenotype (30). Although we did not observe any compensatory up-regulation of the other 4E-BP isoforms in the cells overexpressing 4E-BP1 shRNA after 2–3 wk selection, the situation in the whole organism is more complicated, and it would be interesting to use lineage-specific inducible knockout mouse models to validate the role of 4E-BP isoforms in vivo. This approach looks particularly promising, as adipogenesis—which, like chondrogenesis, is a property of the mesenchyme—is jeopardized in 4E-BP1/4E-BP2 double knockout mice (30).
Most importantly, we demonstrated that 4E-BP1 activation precedes suppression of total protein synthesis. It is possible that 4E-BP1 activation at earlier times of FGF treatment is directed to a subset of specific mRNAs whose requirement for eIF4F complexes is particularly high, whereas the effective protein synthesis remains indispensable for cells as they are about to enlarge and differentiate into hypertrophic chondrocytes. The subsets of mRNAs referred to as “eIF4E-sensitive” and characterized by long, highly structured 5′UTR have been described previously in other systems (31). This scenario implies that proliferative chondrocytes can use two different modes of protein synthesis repression: one for defined subsets of mRNAs and the other one on a global basis. It would be important to determine which mRNAs are enriched on polysomes at different times of FGF treatment to completely visualize the changes induced by FGF signaling to the chondrocyte translational landscape.
According to our data, activation of 4E-BP1 is a crucial step in mediating FGF-induced repression of protein synthesis and growth inhibition; however, it is currently unclear how 4E-BP1 is initially activated, especially as its dephosphorylation begins when the level of mTOR activity is comparable with that in untreated cells. It is possible that counteractive phosphatase activity is present in FGF-treated chondrocytes to activate 4E-BP1 in the presence of active mTOR. Most of the evidence points to PP2A as an activator of 4E-BP; however, a direct interaction between 4E-BP and PP2A failed to be detected experimentally (32). Similarly, we could not detect such an interaction in FGF-treated chondrocytes. Although this interaction might be transient and challenging to detect, there is a possibility that an alternate—perhaps PP2A family phosphatase—is responsible for 4E-BP1 dephosphorylation. It was reported that PPM1G (protein phosphatase, Mg2+/Mn2+ dependent, 1G) dephosphorylates 4E-BP under normal growth conditions (33); however, in our hands, 4E-BP1 phosphorylation status was sensitive to 5nM okadaic acid (OA) (Fig. S6) and overexpression of SV40ST, suggesting that a member of PP2A family phosphatases could be either directly or indirectly responsible for the 4E-BP1 dephosphorylation in chondrocytes. The well-documented role of PP2A in the modulation of activity of several key players of AKT/mTOR pathway, including AKT itself (34–36), complicates the interpretation of these data, and more experiments are needed to understand the precise mechanism of 4E-BP dephosphorylation.
Fig. S6.

Role of Ser/Thr phosphatase activity in 4E-BP1 dephosphorylation. RCS cells were pretreated with 5 nM of OA for 16 h followed by FGF1 treatment as indicated. Ten micrograms of total protein were used for immunodetection of 4E-BP, and equal amount of protein loading was confirmed by α-tubulin immunodetection.
Here, for the first time to our knowledge we demonstrated the capacity of FGF signaling to induce and to inhibit protein synthesis in a cell-type–specific manner. Furthermore, 4E-BP1 repressor activity is necessary to suppress protein synthesis and cell-cycle progression in FGF-treated chondrocytes. Importantly, FGF-induced changes in the 4E-BP1 activity observed in cell culture are also detected in vivo and reflect the action of FGF signaling on downstream targets during bone development.
Materials and Methods
Reagents and Antibodies.
All chemicals were from Sigma-Aldrich (unless otherwise stated). The following antibodies were used: 4E-BP1, phospho-4E-BP1 (S65), phospho-4E-BP1 (T70), ERK1/2, phospho-ERK1/2 (T202, Y204), eIF4G, eIF4E, eIF4AI, mTOR, phospho-mTOR (S2481), phospho-mTOR (S2448), rpS6, phospho-rpS6(S235/236), phospho-TSC2 (T1462), TSC2, and DEPTOR (Cell Signaling); α-tubulin (clone B-5-1-2), actin, GFP (Sigma); agarose-conjugated eIF4E, St SV40 (Santa Cruz Biotechnology).
Cell Culture, ex Vivo Culture, S35-Methionine Incorporation, and FACS Analysis.
Rat chondrosarcoma (RCS), OB1, and C3H10T1/2 cells were maintained in culture in DMEM supplemented with 10% (vol/vol) FBS. Cells were treated with fibroblast growth factor as previously described (9). Cells were labeled with 110 μCi of [35S]Met/Cys (Perkin-Elmer) for 30 min, and lysates were prepared as previously described (9). The 35S incorporation was determined by trichloroacetic acid precipitation. Differentiation of C3H10T1/2 cells is described in SI Materials and Methods. Chondrocytes from the ribs of newborn rats were isolated as previously described (9) and treated with FGF after 48 h in culture. Flow cytometry was performed as previously described and analyzed using ModFit LT software (Verity Software House). Micromass and organ cultures are described in SI Materials and Methods. The study was approved by the Institutional Animal Use and Care Committees of New York University Langone Medical Center.
Immunoprecipitation and in Vitro Kinase Assay.
Protein lysates were prepared in modified RIPA buffer (50 mM Tris HCl pH 7.4, 150 mM NaCl, 10 mM KCl, 1% Nonidet P-40, 1mM EDTA) in the presence of phosphatase and protease inhibitor mixtures. Protein lysates (0.35 mg) were incubated with agarose-conjugated eIF4E antibody overnight at 4 °C. The immune complexes were washed three times with 1 mL of RIPA buffer, and 50% of immunoprecipitates was resolved on SDS/PAGE. Agarose-conjugated mouse IgG was used as negative control. mTOR kinase assay is described in SI Materials and Methods.
Immunohistochemistry and immunofluorescence analysis were performed according to the manufactures protocols with slight modifications that are described in SI Materials and Methods.
Expression Vectors, Cloning, Lentivirus Production, and siRNA Transfection.
To obtain 4E-BP1–depleted cells, primers targeting 4E-BP1 coding region (nt 382–404) were cloned into lentiviral vector pLKO.1 between AgeI and EcoRI sites according to the Addgene protocol. siRNAs were purchased from Sigma. The start sites of the targeted sequences were the following—DEPTOR1:1282, DEPTOR2:1421, TSC2-1:1090, and TSC2-2:2275. Transfections were performed using Lipofectamine RNAi Max according to the manufacturer's protocol. MISSION siRNA Universal Negative Control #1 (Sigma) was used as a negative control. 4E-BP2-FLAG vector was obtained by inserting PCR fragment from cDNA in pCMV6-Entry (Origen) into pUNO-mcs vector (InvivoGen) between BamHI and NheI.
SI Materials and Methods
Differentiation of C3H10T1/2 Cells.
Cells were plated in DMEM at 10ˆ5 cells per well in 12-well plates. When cells reached confluence, medium was switched to differentiation medium. (Osteogenesis: DMEM/10% FBS, 10 mM beta-Glycerol phosphate, 100 μg/mL abscorbic acid, and 50 ng/mL BMP2 (R&D Systems). Adipogenesis: DMEM/10% FBS, 10 μg/mL insulin, 100 nM dexamethasone, 250 μM IBMX, and 200 μM indomethacin). For chondrogenesis, cells were plated in Ham’s Nutrient Mixture F12 media in a 10 μL drop containing 105 cells in 24-well plates. After 3 h, chondrogenic media was added to the cells (F12/10% FBS, 100 ng/mL BMP2). Alkaline phosphatase assay and Oil Red O staining were carried out as described (37). For qPCR, cells were collected in 350 µL of RLT buffer in the presence of β-mercaptoethanol, and total cellular RNA was extracted simultaneously, after completion of the differentiation assay. Used primers are summarized in Table S1. The 18S rRNA was used as a normalization control.
Immunohistochemistry and Immunofluorescence Analysis.
Immunohistochemistry was performed using antibodies against 4E-BP1, phospho-4E-BP1 (Ser65), p70S6K, and phospho-p70S6. Paraffin-embedded tissue sections were dewaxed in xylene, rehydrated, and washed in PBS (pH 7.4). For antigen retrieval, paraffin sections were heated in a microwave oven (900 W) in 10 mM citrate buffer followed by treatment with 3% H2O2 and blocked with 10% normal goat serum. Sections were then incubated with appropriate antibodies overnight at 4 °C, followed by incubation with a biotinylated rabbit secondary antibody (Vector Laboratories). An avidin–biotin complex was formed and developed using diaminobenzidine chromagen, followed by a counterstain with hematoxylin. Microscopy was performed on a Leica DM5000 microscope, and representative images were acquired using Leica Imaging Software. For immunofluorescence analysis, RCS cells were washed with PBS, fixed with 4% paraformaldehyde at 4 °C, permeabilized with 0.5% Triton-X 100 in PBS, blocked with 2% BSA-PBS, and then incubated with mouse anti-GFP antibody overnight. Cells were washed with 1X PBS five times and treated with secondary antibody [Alexa Fluor 488 goat anti-mouse (LifeTechnologies A-11001)] for 1 h. Cells were washed with 1X PBS four times. Nuclei were counterstained with DAPI, and cells were mounted in DAKO mounting medium.
Ex Vivo Culture.
Micromass cultures were established from E12.5 embryos. Briefly, forelimb and hindlimb buds were removed and pooled in an Eppendorf tube. After washing in PBS, the tissue was treated with 1 mg/mL collagenase B (Roche) in Ham’s F-12 medium at 37 °C for 1 h. Cells were washed and resuspended at 1 × 106 cells/mL in 1% FCS Ham’s F-12 medium. Cells were plated in 20 μL drops either in 6-well plates or onto chamber-slides. After 2 h at 37 °C, 1% FCS Ham’s F-12 medium was added to the culture. The medium was changed the next day before FGF1 treatment (10 ng/mL of FGF1 and 5 μg/mL heparin). Metatarsal long bone rudiments from E15.5 embryos were dissected under sterile conditions. Organ culture was carried out in α-MEM without nucleosides (GIBCO), supplemented with 50 μg/mL ascorbic acid, 1 mM b-glycerophosphate, and 0.2% FBS (complete medium). Each long bone was cultured individually in 24-well plates containing 400 μL of complete medium in the presence or absence of 10 ng/mL of FGF1 and 5 μg/mL heparin. The metatarsal cultures were maintained at 37 °C for up to 48 h, and the medium was changed every day. For the proliferation assay, the long bones were treated with FGF for 24 and 48 h, and BrdU (1 μg/mL) was added during the last 6 h of treatment. The long bones were fixed in 4% PFA overnight at 4 °C and embedded in paraffin.
In Vitro Kinase Assay.
To determine mTOR kinase activity, FGF-treated lysates were assayed for 4E-BP1 kinase activity. Ten micrograms of FGF-treated or untreated RCS lysates were used in each 30 μL reaction in 50 mM Hepes pH 7.5, 10 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 25 mM β-glycerophosphate. The reactions were supplemented with 200 μM ATP and 0.1 μg of recombinant 4E-BP1 (Prospec) either in the presence or absence of 0.2 μg of recombinant eIF4E (Prospec). Reaction mixtures were incubated for 30 min at 30 °C, stopped by adding 10 μL of 5XSDS loading buffer, and resolved on 12% SDS/PAGE following immunoblotting.
Gene Expression Analysis by RT-qPCR.
RNA was extracted from RCS or primary chondrocytes using the RNeasy mini kit (Life Technologies) according to the manufacturer’s instructions. Total RNA (500 ng) was used for cDNA synthesis using the iScript cDNA synthesis kit (Bio-Rad Laboratories). qPCR reactions were run on a MyiQ Single-Color Real-TimePCR Detection System (Bio-Rad) using iQ SYBR Green Supermix. Relative gene expression levels were calculated using the ΔΔCt method. Target mRNA levels were normalized to 18S rRNA. Expression of 4E-BP isoforms was presented as fold change compared with 4E-BP1 expression. Expression of the genes specific to different lineages was presented as fold change compared with untreated control. Primers used are listed in Table S1.
Acknowledgments
We thank Dr. Ian Mohr and Dr. Derek Walsh for the critical reviewing of the manuscript and Dr. Claudio Basilico, Dr. Upal Basu Roy, and Dr. Hannah Burgess for useful discussions. We are grateful to Dr. R. Schneider for the reagents. This work was supported by NIH Grant AR063128 (to V.K.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1605451113/-/DCSupplemental.
References
- 1.Bitterman PB, Polunovsky VA. Translational control of cell fate: From integration of environmental signals to breaching anticancer defense. Cell Cycle. 2012;11(6):1097–1107. doi: 10.4161/cc.11.6.19610. [DOI] [PubMed] [Google Scholar]
- 2.Gingras AC, et al. Regulation of 4E-BP1 phosphorylation: A novel two-step mechanism. Genes Dev. 1999;13(11):1422–1437. doi: 10.1101/gad.13.11.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10(5):307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 4.Lefebvre V, Bhattaram P. Vertebrate skeletogenesis. Curr Top Dev Biol. 2010;90:291–317. doi: 10.1016/S0070-2153(10)90008-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ornitz DM. FGF signaling in the developing endochondral skeleton. Cytokine Growth Factor Rev. 2005;16(2):205–213. doi: 10.1016/j.cytogfr.2005.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen L, Deng CX. Roles of FGF signaling in skeletal development and human genetic diseases. Front Biosci. 2005;10:1961–1976. doi: 10.2741/1671. [DOI] [PubMed] [Google Scholar]
- 7.Aikawa T, Segre GV, Lee K. Fibroblast growth factor inhibits chondrocytic growth through induction of p21 and subsequent inactivation of cyclin E-Cdk2. J Biol Chem. 2001;276(31):29347–29352. doi: 10.1074/jbc.M101859200. [DOI] [PubMed] [Google Scholar]
- 8.Sahni M, et al. FGF signaling inhibits chondrocyte proliferation and regulates bone development through the STAT-1 pathway. Genes Dev. 1999;13(11):1361–1366. doi: 10.1101/gad.13.11.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kolupaeva V, Laplantine E, Basilico C. PP2A-mediated dephosphorylation of p107 plays a critical role in chondrocyte cell cycle arrest by FGF. PLoS One. 2008;3(10):e3447. doi: 10.1371/journal.pone.0003447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mansukhani A, Bellosta P, Sahni M, Basilico C. Signaling by fibroblast growth factors (FGF) and fibroblast growth factor receptor 2 (FGFR2)-activating mutations blocks mineralization and induces apoptosis in osteoblasts. J Cell Biol. 2000;149(6):1297–1308. doi: 10.1083/jcb.149.6.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reznikoff CA, Brankow DW, Heidelberger C. Establishment and characterization of a cloned line of C3H mouse embryo cells sensitive to postconfluence inhibition of division. Cancer Res. 1973;33(12):3231–3238. [PubMed] [Google Scholar]
- 12.Ahrens PB, Solursh M, Reiter RS, Singley CT. Position-related capacity for differentiation of limb mesenchyme in cell culture. Dev Biol. 1979;69(2):436–450. doi: 10.1016/0012-1606(79)90303-8. [DOI] [PubMed] [Google Scholar]
- 13.Katagiri T, et al. The non-osteogenic mouse pluripotent cell line, C3H10T1/2, is induced to differentiate into osteoblastic cells by recombinant human bone morphogenetic protein-2. Biochem Biophys Res Commun. 1990;172(1):295–299. doi: 10.1016/s0006-291x(05)80208-6. [DOI] [PubMed] [Google Scholar]
- 14.Denker AE, Haas AR, Nicoll SB, Tuan RS. Chondrogenic differentiation of murine C3H10T1/2 multipotential mesenchymal cells: I. Stimulation by bone morphogenetic protein-2 in high-density micromass cultures. Differentiation. 1999;64(2):67–76. doi: 10.1046/j.1432-0436.1999.6420067.x. [DOI] [PubMed] [Google Scholar]
- 15.Gingras AC, et al. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 2001;15(21):2852–2864. doi: 10.1101/gad.912401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dailey L, Laplantine E, Priore R, Basilico C. A network of transcriptional and signaling events is activated by FGF to induce chondrocyte growth arrest and differentiation. J Cell Biol. 2003;161(6):1053–1066. doi: 10.1083/jcb.200302075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laederich MB, Horton WA. FGFR3 targeting strategies for achondroplasia. Expert Rev Mol Med. 2012;14:e11. doi: 10.1017/erm.2012.4. [DOI] [PubMed] [Google Scholar]
- 18.Laplantine E, Rossi F, Sahni M, Basilico C, Cobrinik D. FGF signaling targets the pRb-related p107 and p130 proteins to induce chondrocyte growth arrest. J Cell Biol. 2002;158(4):741–750. doi: 10.1083/jcb.200205025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heesom KJ, Gampel A, Mellor H, Denton RM. Cell cycle-dependent phosphorylation of the translational repressor eIF-4E binding protein-1 (4E-BP1) Curr Biol. 2001;11(17):1374–1379. doi: 10.1016/s0960-9822(01)00422-5. [DOI] [PubMed] [Google Scholar]
- 20.Shuda M, et al. CDK1 substitutes for mTOR kinase to activate mitotic cap-dependent protein translation. Proc Natl Acad Sci USA. 2015;112(19):5875–5882. doi: 10.1073/pnas.1505787112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alayev A, Holz MK. mTOR signaling for biological control and cancer. J Cell Physiol. 2013;228(8):1658–1664. doi: 10.1002/jcp.24351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9(3):316–323. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- 23.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4(9):648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 24.Peterson RT, Desai BN, Hardwick JS, Schreiber SL. Protein phosphatase 2A interacts with the 70-kDa S6 kinase and is activated by inhibition of FKBP12-rapamycinassociated protein. Proc Natl Acad Sci USA. 1999;96(8):4438–4442. doi: 10.1073/pnas.96.8.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janssens V, Goris J. Protein phosphatase 2A: A highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353(Pt 3):417–439. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gingras AC, Kennedy SG, O’Leary MA, Sonenberg N, Hay N. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev. 1998;12(4):502–513. doi: 10.1101/gad.12.4.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krejci P, et al. FGF2 inhibits proliferation and alters the cartilage-like phenotype of RCS cells. Exp Cell Res. 2004;297(1):152–164. doi: 10.1016/j.yexcr.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 28.Chen J, Long F. mTORC1 signaling controls mammalian skeletal growth through stimulation of protein synthesis. Development. 2014;141(14):2848–2854. doi: 10.1242/dev.108811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ulici V, Hoenselaar KD, Gillespie JR, Beier F. The PI3K pathway regulates endochondral bone growth through control of hypertrophic chondrocyte differentiation. BMC Dev Biol. 2008;8:40. doi: 10.1186/1471-213X-8-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Le Bacquer O, et al. Elevated sensitivity to diet-induced obesity and insulin resistance in mice lacking 4E-BP1 and 4E-BP2. J Clin Invest. 2007;117(2):387–396. doi: 10.1172/JCI29528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Svitkin YV, et al. The requirement for eukaryotic initiation factor 4A (elF4A) in translation is in direct proportion to the degree of mRNA 5′ secondary structure. RNA. 2001;7(3):382–394. doi: 10.1017/s135583820100108x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nho RS, Peterson M. Eukaryotic translation initiation factor 4E binding protein 1 (4EBP-1) function is suppressed by Src and protein phosphatase 2A (PP2A) on extracellular matrix. J Biol Chem. 2011;286(37):31953–31965. doi: 10.1074/jbc.M111.222299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu J, Stevens PD, Eshleman NE, Gao T. Protein phosphatase PPM1G regulates protein translation and cell growth by dephosphorylating 4E binding protein 1 (4E-BP1) J Biol Chem. 2013;288(32):23225–23233. doi: 10.1074/jbc.M113.492371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Kanegan MJ, Adams DG, Wadzinski BE, Strack S. Distinct protein phosphatase 2A heterotrimers modulate growth factor signaling to extracellular signal-regulated kinases and Akt. J Biol Chem. 2005;280(43):36029–36036. doi: 10.1074/jbc.M506986200. [DOI] [PubMed] [Google Scholar]
- 35.Yan L, et al. PP2A T61 epsilon is an inhibitor of MAP4K3 in nutrient signaling to mTOR. Mol Cell. 2010;37(5):633–642. doi: 10.1016/j.molcel.2010.01.031. [DOI] [PubMed] [Google Scholar]
- 36.Hahn K, et al. PP2A regulatory subunit PP2A-B’ counteracts S6K phosphorylation. Cell Metab. 2010;11(5):438–444. doi: 10.1016/j.cmet.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 37.Caterson EJ, Nesti LJ, Danielson KG, Tuan RS. Human marrow-derived mesenchymal progenitor cells: Isolation, culture expansion, and analysis of differentiation. Mol Biotechnol. 2002;20(3):245–256. doi: 10.1385/MB:20:3:245. [DOI] [PubMed] [Google Scholar]