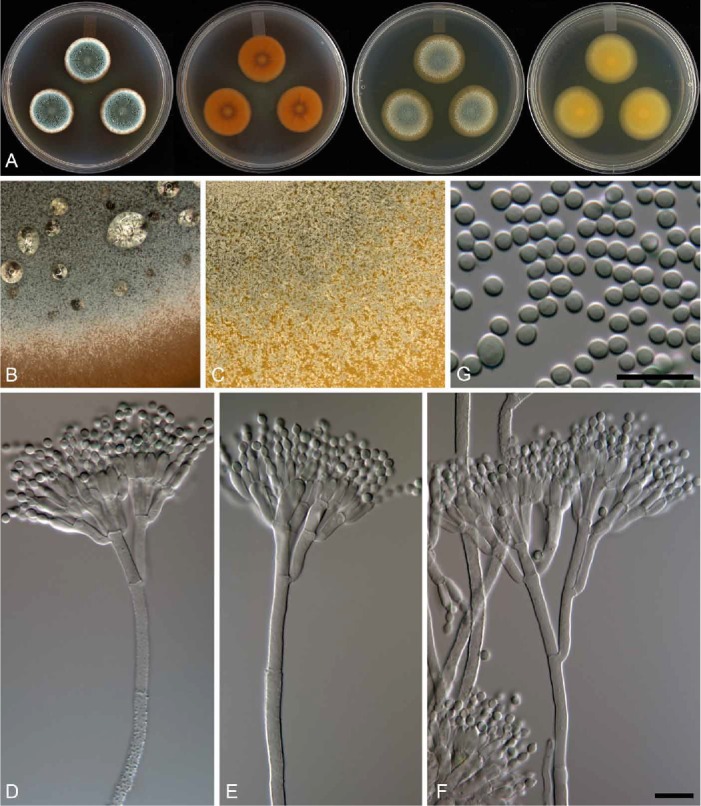
Abstract
The genomes of Armillaria fuscipes, Ceratocystiopsis minuta, Ceratocystis adiposa, Endoconidiophora laricicola, E. polonica, and Penicillium freii DAOMC 242723 are presented in this genome announcement. These six genomes are from plant pathogens and otherwise economically important fungal species. The genome sizes range from 21 Mb in the case of Ceratocystiopsis minuta to 58 Mb for the basidiomycete Armillaria fuscipes. These genomes include the first reports of genomes for the genus Endoconidiophora. The availability of these genome data will provide opportunities to resolve longstanding questions regarding the taxonomy of species in these genera. In addition these genome sequences through comparative studies with closely related organisms will increase our understanding of how these pathogens cause disease.
Keywords: Armillaria root rot, grain spoilage, insect vectored fungi, sap stain fungus, sugarcane root rot
Acknowledgments
Genome sequencing of Penicillium freii DAOMC 242723 was funded by Growing Forward 2 funding from Agriculture & Agri-Food Canada, with additional support from Canadian Safety and Security Programme grant CRTI 09-462RD/CSSP 30vv01.
The genome sequencing of the species Armillaria fuscipes, Ceratocystiopsis minuta, Ceratocystis adiposa, Endoconidiophora laricicola and E. polonica were financed by the University of Pretoria, the Department of Science and Technology (DST)/National Research Foundation (NRF) Centre of Excellence in Tree Health Biotechnology (CTHB), and the Genomics Research Institute (University of Pretoria). This work is based on the research supported in part by a number of grants from the National Research Foundation of South Africa (including Grant specific unique reference number (UID) 83924 and 87332). The Grant holders acknowledge that opinions, findings and conclusions or recommendations expressed in any publication generated by the NRF supported research are that of the author(s), and that the NRF accepts no liability whatsoever in this regard.
REFERENCES
- Agustian A, Mohammed C, Guillaumin J-J, Botton B. (1994) Discrimination of some African Armillaria species by isozyme electrophoretic analysis. New Phytologist 128: 135–143. [DOI] [PubMed] [Google Scholar]
- Aljanabi SM, Martinez I. (1997) Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques. Nucleic Acids Research 25: 4692–4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, et al. (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology 19: 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgartner K, Coetzee MPA, Hoffmeister D. (2011) Secrets of the subterranean pathosystem of Armillaria. Molecular Plant Pathology 12: 515–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belbahri L. (2015) Genome sequence of Ceratocystis platani, a major pathogen of plane trees. URL http://www.ncbi.nlm.nih.gov/nuccore/814603118. [Google Scholar]
- Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. (2011) Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27: 578–579. [DOI] [PubMed] [Google Scholar]
- Boetzer M, Pirovano W. (2012) Toward almost closed genomes with GapFiller. Genome Biology 13: R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottomley AM. (1937) Some of the more important diseases affecting timber plantations in the Transvaal. South African Journal of Science 33: 373–376. [Google Scholar]
- Butler EJ. (1906) Fungus diseases of sugar-cane in Bengal. Memoirs of the Department of Agriculture in India by Botanical Series 1: 1–53. [Google Scholar]
- Cantarel BL, Korf I, Robb SM, Parra G, Ross E, et al. (2008) MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Research 18: 188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikhi R, Medvedev P. (2014) Informed and automated k-mer size selection for genome assembly. Bioinformatics 30: 31–37. [DOI] [PubMed] [Google Scholar]
- Chillali M, Idder-Ighili H, Agustian A, Guillaumin J-J, Mohammed C, et al. (1997) Species delimitation in the African Armillaria complex by analysis of the ribosomal DNA spacers. Journal of General and Applied Microbiology 43: 23–29. [DOI] [PubMed] [Google Scholar]
- Christiansen E, Horntvedt R. (1983) Combined Ips/Ceratocystis attack on Norway spruce, and defensive mechanisms of the trees. Zeitschrift für angewandte Entomologie 96: 110–118. [Google Scholar]
- Coetzee MP, Wingfield BD, Bloomer P, Wingfield MJ. (2005) Phylogenetic analyses of DNA sequences reveal species partitions amongst isolates of Armillaria from Africa. Mycological Research 109: 1223–1234. [DOI] [PubMed] [Google Scholar]
- Coetzee MPA, Bloomer P, Wingfield MJ, Wingfield BD. (2011) Paleogene radiation of a plant pathogenic mushroom. PLoS ONE 6: e28545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee MPA, Wingfield BD, Coutinho TA, Wingfield MJ. (2000) Identification of the causal agent of Armillaria root rot of Pinus species in South Africa. Mycologia 92: 777–785. [Google Scholar]
- Collins C, Keane TM, Turner DJ, O’Keeffe G, Fitzpatrick DA, et al (2013) Genomic and proteomic dissection of the ubiquitous plant pathogen, Armillaria mellea: Toward a new infection model system. Journal of Proteome Research 12: 2552–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuomo CA, Rodriguez-Del Valle N, Perez-Sanchez L, et al (2014) Genome sequence of the pathogenic fungus Sporothrix schenckii (ATCC 58251). Genome Announcements 2: e00446–00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessandro E, Giosa D, Huang L, Zhang J, Gao W, et al (2016) Draft genome sequence of the dimorphic fungus Sporothrix pallida, a nonpathogenic species belonging to Sporothrix, a genus containing agents of human and feline sporotrichosis. Genome Announcements 4: e00184-00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson RW. (1942) Some additional species of Ceratostomella in the United States. Mycologia 34: 650–662. [Google Scholar]
- de Beer ZW, Duong TA, Barnes I, Wingfield BD, Wingfield MJ. (2014) Redefining Ceratocystis and allied genera. Studies in Mycology 79: 187–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Beer ZW, Wingfield MJ. (2013) Emerging lineages in the Ophiostomatales, In: The Ophiostomatoid Fungi: expanding frontiers (Seifert KA, de Beer ZW, Wingfield MJ. eds): 21–46. [CBS Biodiversity Series no. 12.] Utrecht: CBS-KNAW Fungal Biodiversity Centre. [Google Scholar]
- DiGuistini S, Wang Y, Liao NY, Taylor G, Tanguay P, et al (2011) Genome and transcriptome analyses of the mountain pine beetle-fungal symbiont Grosmannia clavigera, a lodgepole pine pathogen. Proceedings of the National Academy of Sciences, USA 108: 2504–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong TA, de Beer ZW, Wingfield BD, Wingfield MJ. (2013) Characterization of the mating-type genes in Leptographium procerum and Leptographium profanum. Fungal Biology 117: 411–421. [DOI] [PubMed] [Google Scholar]
- Floudas D, Held BW, Riley R, Nagy LG, Koehler G, et al. (2015) Evolution of novel wood decay mechanisms in Agaricales revealed by the genome sequences of Fistulina hepatica and Cylindrobasidium torrendii. Fungal Genetics and Biology 76: 78–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forgetta V, Leveque G, Dias J, Grove D, Lyons R, et al (2013) Sequencing of the Dutch Elm Disease fungus genome using the Roche/454 GS-FLX Titanium system in a comparison of multiple genomics core facilities. Journal of Biomolecular Techniques 24: 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisvad JC, Samson RA. (2004) Polyphasic taxonomy of Penicillium subgenus Penicillium - a guide to identification of food and air-borne terverticillate Penicillia and their mycotoxins. Studies In Mycology 49: 1–174. [Google Scholar]
- Frisvad JC, Smedsgaard J, Larsen TO, Samson RA. (2004) Mycotoxins, drugs and other extrolites produced by species in Penicillium subgenus Penicillium. Studies In Mycology 49: 201–241. [Google Scholar]
- Garcia-Alcalde F, Okonechnikov K, Carbonell J, Cruz LM, Gotz S, et al. (2012) Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics 28: 2678–2679. [DOI] [PubMed] [Google Scholar]
- Gezahgne A, Coetzee MPA, Wingfield BD, Wingfield MJ, Roux J. (2004) Identification of the Armillaria root rot pathogen in Ethiopian plantations. Forest Pathology 34: 133–145. [Google Scholar]
- Gregory SC, Rishbeth J, Shaw CG. (1991) Pathogenicity and virulence. In: Armillaria Root Disease (Shaw CG, Kile GA, eds): 76–87 [Agriculture Handbook no. 691.] Washington D.: Forest Service, United States Department of Agriculture. [Google Scholar]
- Grigoriev IV, Nikitin R, Haridas S, Kuo A, Ohm R, et al. (2014) MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Research 42: D699–D704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, Tesler G. (2013) QUAST: quality assessment tool for genome assemblies. Bioinformatics 29: 1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haridas S, Wang Y, Lim L, Alamouti SM, Jackman S, et al (2013) The genome and transcriptome of the pine saprophyte Ophiostoma piceae, and a comparison with the bark beetle-associated pine pathogen Grosmannia clavigera. BMC Genomics 14: 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff KJ, Stanke M. (2013) WebAUGUSTUS - a web service for training AUGUSTUS and predicting genes in eukaryotes. Nucleic Acids Research 41: W123–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houbraken J, Samson RA. (2011) Phylogeny of Penicillium and the segregation of Trichocomaceae into three families. Studies In Mycology 70: 1–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller O, Kollmar M, Stanke M, Waack S. (2011) A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics 27: 757–763. [DOI] [PubMed] [Google Scholar]
- Khoshraftar S, Hung S, Khan S, Gong Y, Tyagi V, et al (2013) Sequencing and annotation of the Ophiostoma ulmi genome. BMC Genomics 14: 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kile GA. (1993) Plant diseases caused by species of Ceratocystis sensu stricto and Chalara. In: Ceratocystis and Ophiostoma: taxonomy, ecology, and pathogenicity (Wingfield MJ, Seifert KA, Webber JF, eds): 173–183. St Paul, MN: American Phytopathological Society Press. [Google Scholar]
- Kim J-J, Allen EA, Humble LM, Breuil C. (2005) Ophiostomatoid and basidiomycetous fungi associated with green, red, and grey lodgepole pines after mountain pine beetle (Dendroctonus ponderosae) infestation. Canadian Journal of Forest Research 35: 274–284. [Google Scholar]
- Langmead B, Salzberg SL. (2012) Fast gapped-read alignment with Bowtie 2. Nature Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín M, Wingfield MJ. (2006) A review of Ceratocystis sensu stricto with special reference to the species complexes C. coerulescens and C. fimbriata. Revista Facultad Nacional de Agronomía, Medellín 59: 3045–3075. [Google Scholar]
- Mathiesen A. (1951) Einige neue Ophiostoma-arten in Schweden. Svensk Botanisk Tidskrift 45: 203–232. [Google Scholar]
- Mathiesen-Käärik A. (1960) Studies on the ecology, taxonomy and physiology of Swedish insect-associated blue stain fungi, especially the genus Ceratocystis. Oikos 11: 1–25. [Google Scholar]
- Miller JR, Delcher AL, Koren S, Venter E, Walenz BP, et al (2008) Aggressive assembly of pyrosequencing reads with mates. Bioinformatics 24: 2818–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed C, Guillaumin J-J. (1993) Armillaria in tropical Africa. In: Aspects of Tropical Mycology (Isaac S, Frankland JC, Watling R, Whalley AJS, eds): 207–217. Cambridgel: Cambridge University Press. [Google Scholar]
- Morrison DJ, Williams RE, Whitney RD. (1991) Infection, disease development, diagnosis, and detection. In: Armillaria Root Disease (Shaw CG, Kile GA, eds): 62–75 [Agriculture Handbook no. 691.] Washington DC: Forest Service, United States Department of Agriculture. [Google Scholar]
- Mwenje E, Ride JP. (1996) Morphological and biochemical characterization of Armillaria isolates from Zimbabwe. Plant Pathology 45: 1036–1051. [Google Scholar]
- Mwenje E, Ride JP. (1997) The use of pectic enzymes in the characterization of Armillaria isolates from Africa. Plant Pathology 46: 341–354. [Google Scholar]
- Mwenje E, Ride JP. (1999) Purification and characterization of an endo-polygalacturonase (PG1) from a Zimbabwean species of Armillaria. Physiological and Molecular Plant Pathology 55: 131–139. [Google Scholar]
- Mwenje E, Wingfield BD, Coetzee MP, Wingfield MJ. (2003) Molecular characterisation of Armillaria species from Zimbabwe. Mycological Research 107: 291–296. [DOI] [PubMed] [Google Scholar]
- Nikolenko SI, Korobeynikov AI, Alekseyev MA. (2013) BayesHammer: Bayesian clustering for error correction in single-cell sequencing. BMC Genomics 14 (Suppl. 1): S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paine TD, Raffa KF, Harrington TC. (1997) Interactions among scolytid bark beetles, their associated fungi, and live host conifers. Annual Review of Entomology 42: 179–206. [DOI] [PubMed] [Google Scholar]
- Park Y-J, Baek JH, Lee S, Kim C, Rhee H, et al. (2014) Whole genome and global gene expression analyses of the model mushroom Flammulina velutipes reveal a high capacity for lignocellulose degradation. PloS One 9: e93560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra G, Bradnam K, Korf I. (2007) CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23: 1061–1067. [DOI] [PubMed] [Google Scholar]
- Paulin-Mahady AE, Harrington TC, McNew D. (2002) Phylogenetic and taxonomic evaluation of Chalara, Chalaropsis, and Thielaviopsis anamorphs associated with Ceratocystis. Mycologia 94: 62–72. [PubMed] [Google Scholar]
- Pérez-Sierra A, Guillaumin J-J, Spooner BM, Bridge PD. (2004) Characterization of Armillaria heimii from Africa. Plant Pathology 53: 220–230. [Google Scholar]
- Plattner A, Kim J-J, Reid J, Hausner G, Lim YW, et al (2009) Resolving taxonomic and phylogenetic incongruence within species Ceratocystiopsis minuta. Mycologia 101: 878–887. [DOI] [PubMed] [Google Scholar]
- Redfern DB, Stoakley JT, Steele H, Minter D. (1987) Dieback and death of larch caused by Ceratocystis laricicola sp. nov. following attack by Ips cembrae. Plant Pathology 36: 467–80. [Google Scholar]
- Ross-Davis AL, Stewart JE, Hanna JW, Kim MS, Knaus BJ, et al. (2013) Transcriptome of an Armillaria root disease pathogen reveals candidate genes involved in host substrate utilization at the host–pathogen interface. Forest Pathology 43: 468–477. [Google Scholar]
- Roux J, van Wyk M, Hatting H, Wingfield MJ. (2004) Ceratocystis species infecting stem wounds on Eucalyptus grandis in South Africa. Plant Pathology 53: 414–421. [Google Scholar]
- Sartoris GB. (1927) A cytological study of Ceratostomella adiposum (Butl.) comb. nov., the black-rot fungus of sugar cane. Journal of Agricultural Research 35: 577–583. [Google Scholar]
- Seifert KA, Wingfield MJ, Kendrick WB. (1993) A nomenclature for described species of Ceratocystis, Ophiostoma, Ceratocystiopsis, Ceratostomella and Sphaeronaemella. In: Ceratocystis and Ophiostoma: taxonomy, ecology, and pathogenicity (Winfield MJ, Seifert KA, Webber JF, eds): 269–287. St Paul, MN: American Phytopathological Society Press. [Google Scholar]
- Siemaszko W. (1939) Fungi associated with bark beetles in Poland. Planta Polonica 7: 1–54. [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31: 3210–3212. [DOI] [PubMed] [Google Scholar]
- Six DL, Wingfield MJ. (2011) The role of phytopathogenicity in bark beetle-fungus symbioses: A challenge to the classic paradigm. Annual Review of Entomology 56: 255–72. [DOI] [PubMed] [Google Scholar]
- Stanke M, Keller O, Gunduz I, Hayes A, Waack S, et al. (2006) AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Research 34: W435–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Steinkamp R, Waack S, Morgenstern B. (2004) AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Research 32: 309–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Tzvetkova A, Morgenstern B. (2006) AUGUSTUS at EGASP: using EST, protein and genomic alignments for improved gene prediction in the human genome. Genome Biology 7 (Suppl. 1): S11.11–S11.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot P. (1956) New and interesting records of South African fungi. Part II. Bothalia 6: 489–500. [Google Scholar]
- Tarailo-Graovac M, Chen N. (2009) Using RepeatMasker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics 25: 4.10.1–4.10.14. [DOI] [PubMed] [Google Scholar]
- Teixeira MM, de Almeida LG, Kubitschek-Barreira P, Alves FL, et al (2014) Comparative genomics of the major fungal agents of human and animal Sporotrichosis: Sporothrix schenckii and Sporothrix brasiliensis. BMC Genomics 15: 943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyay H. (1981) A monograph of Ceratocystis and Ceratocystiopsis. Athens, GA: University of Georgia Press. [Google Scholar]
- van der Nest MA, Beirn LA, Crouch JA, Demers JE, de Beer ZW, et al. (2014b) IMA Genome–F 3: Draft genomes of Amanita jacksonii, Ceratocystis albifundus, Fusarium circinatum, Huntiella omanensis, Leptographium procerum, Rutstroemia sydowiana, and Sclerotinia echinophila. IMA Fungus 5: 473–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Nest MA, Bihon W, De Vos L, Naidoo K, Roodt D, et al. (2014a) IMA Genome-F 2: Ceratocystis manginecans, Ceratocystis moniliformis, Diplodia sapinea: Draft genome sequences of Diplodia sapinea, Ceratocystis manginecans, and Ceratocystis moniliformis. IMA Fungus 5: 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Nest MA, Steenkamp ET, McTaggart AR, Trollip C, Godlonton T, et al. (2015) Saprophytic and pathogenic fungi in the Ceratocystidaceae differ in their ability to metabolize plant-derived sucrose. BMC Evolutionary Biology 15: 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victorian Bioinformatics Consortium (2012) VelvetOptimiser. bioinformatics.net.au/software.velvetoptimiser.shtml. [Google Scholar]
- Visagie CM, Houbraken J, Frisvad JC, Hong SB, Klaassen CHW, et al. (2014) Identification and nomenclature of the genus Penicillium. Studies In Mycology 78: 343–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilken PM, Steenkamp ET, Wingfield MJ, de Beer ZW, Wingfield BD. (2013) IMA Genome–F 1: Ceratocystis fimbriata: draft nuclear genome sequence for the plant pathogen, Ceratocystis fimbriata. IMA Fungus 4: 357–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilken PM, Steenkamp ET, Wingfield MJ, de Beer ZW, Wingfield BD. (2014) DNA loss at the Ceratocystis fimbriata mating locus results in self-sterility. PLoS ONE 9: e92180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AM, Godlonton T, van der Nest MA, Wilken PM, Wingfield MJ, et al. (2015) Unisexual reproduction in Huntiella moniliformis. Fungal Genetics and Biology 80: 1–9. [DOI] [PubMed] [Google Scholar]
- Wingfield BD, Ades PK, Al-Naemi FA, Beirn LA, Bihon W, et al. (2015a) IMA Genome-F 4: Draft genome sequences of Chrysoporthe austroafricana, Diplodia scrobiculata, Fusarium nygamai, Leptographium lundbergii, Limonomyces culmigenus, Stagonosporopsis tanaceti, and Thielaviopsis punctulata. IMA Fungus 6: 233–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingfield BD, Barnes I, de Beer ZW, De Vos L, Duong TA, et al. (2015b) IMA Genome-F 5: Draft genome sequences of Ceratocystis eucalypticola, Chrysoporthe cubensis, C. deuterocubensis, Davidsoniella virescens, Fusarium temperatum, Graphilbum fragrans, Penicillium nordicum, and Thielaviopsis musarum. IMA Fungus 6: 493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingfield MJ, Harrington TC, Solheim H. (1997) Two species in the Ceratocystis coerulescens complex from conifers in western North America. Canadian Journal of Botany 75: 827–834. [Google Scholar]
- Wingfield MJ, Seifert KA, Webber JF. (eds) (1993) Ceratocystis and Ophiostoma: taxonomy, ecology and pathogenicity. St Paul, MN: American Phytopathological Society Press. [Google Scholar]
- Yamaoka Y, Wingfield MJ, Ohsawa M, Kuroda Y. (1998) Ophiostomatoid fungi associated with Ips cembrae in Japan and their pathogenicity of Japanese larch. Mycoscience 39: 367–378. [Google Scholar]
- Yao G, Ye L, Gao H, Minx P, Warren WC, Weinstock GM. (2012) Graph accordance of next-generation sequence assemblies. Bioinformatics 28: 13–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XD, de Beer ZW, Wingfield BD, Wingfield MJ. (2001) Ophiostomatoid fungi associated with three pine-infesting bark beetles in South Africa. Sydowia 53: 290–300. [Google Scholar]
- Zhou XD, de Beer ZW, Ahumada R, Wingfield BD, Wingfield MJ. (2004a) Ophiostoma and Ceratocystiopsis spp. associated with two pine-infesting bark beetles in Chile. Fungal Diversity 15: 253–266. [Google Scholar]
- Zhou XD, de Beer ZW, Cibrian D, Wingfield BD, Wingfield MJ. (2004b) Characterisation of Ophiostoma species associated with pine bark beetles from Mexico, including O. pulvinisporum sp. nov. Mycological Research 108: 690–698. [DOI] [PubMed] [Google Scholar]