

Abstract
Structural differences between conformers sustain protein biological function. Here, we studied in a large dataset of 745 intrinsically disordered proteins, how ordered‐disordered transitions modulate structural differences between conformers as derived from crystallographic data. We found that almost 50% of the proteins studied show no transitions and have low conformational diversity while the rest show transitions and a higher conformational diversity. In this last subset, 60% of the proteins become more ordered after ligand binding, while 40% more disordered. As protein conformational diversity is inherently connected with protein function our analysis suggests differences in structure‐function relationships related to order‐disorder transitions.
Keywords: disorder, conformational diversity, protein function, transitions
Abbreviations
- DHPP
6‐hydroxymethyl‐7,8‐dihydropterin monophosphate
- ID
intrinsically disordered
- RMSD
root mean squared deviation
- SAXS
small‐angle X‐ray scattering
- TTR
transthyretin
Introduction
The relationship between protein structure and function is well‐established in structural biology.1 In light of the overwhelming amount of experiments relating biological function with structural changes, that relationship can be rephrased in terms of dynamic properties.2, 3, 4, 5 The notion that the protein native state could contain different conformers with similar energies in dynamic equilibrium was early proposed to explain the binding capacity of seroalbumin by Karush in 1950.6 The structural differences between conformers define differential binding capacities towards ligands, an essential feature underneath protein function. During the sixties, Koshland and Monod respectively proposed the induced‐fit7 and the pre‐equilibrium8 models to explain how structural changes are required to understand cooperative and allosteric behaviors, key properties related with biological function (for a review see Ref. 9). According to Koshland, ligands induce conformational transitions while Monod's model considered that a dynamic equilibrium between conformers pre‐exists ligands. In this last model, the role of the ligands is to select the best conformation in terms of affinity, promoting an equilibrium shift towards this highest affinity conformer according to the mass action law. During the last years, several studies support the pre‐equilibrium model against induced‐fit,10, 11, 12 although a joined model for both theories has been recently proposed.13 Whatever the mechanisms underlying conformational change, it is clear that protein function is related with the switching between different structures composing the native state. Structural differences among conformers could be as large as relative movements of subunits or complete domains, but could also involve rearrangements of loops or secondary structure elements. These differences are usually measured by structure based scores (i.e., carbon‐alpha root mean squared deviation, RMSD) that are used to characterize protein conformational diversity. Recently, protein databases with different experimental based conformational diversity degrees have been developed (e.g., MolMovDB14 and CoDNaS15).
However, this outlined description of the native state is incomplete when intrinsically disordered (ID) proteins are considered.16, 17 ID proteins lack a well‐defined structure under physiological conditions as a whole or in different sequence regions.18, 19 In fact, disorder is commonly defined as a region with missing density in protein X‐ray crystallography, evidencing the presence of high mobility or disorder.20 Derived from proteomic analysis and showing a broad phylogenetic distribution,21 IDPs have been associated with different biological functions such as regulation of cell division and signaling, signal transduction, chaperon action, transcription and regulation of self‐assembling complexes such as ribosomes, cytoskeleton and chromatin.22 The complete conformational description of IDPs (both folded and natively unfolded regions) is still difficult, despite important advances in the last years using NMR23 and RX techniques such as small‐angle X‐ray scattering (SAXS) combined with coarse‐grain molecular dynamics simulations17 to study the large conformational space involved in ID regions. Interestingly these efforts have recently produced collections of structural ensembles of IDPs and unfolded proteins.24 However, a fraction of the disordered behavior may be studied with transitions involving ID regions between conformers and several biological factors associated with them. These factors could be classified as active, for example, ligands, post‐translational modifications, and protein–protein interactions, or passive, for example, changes in pH, temperature or light.20 Transitions could either cause ID regions to become structured or a structured region to become ID.20, 25, 26 Some ID proteins may also remain disordered after exposure to the active factors mentioned above.27 Disorder‐to‐order transitions are associated with the so‐called folding upon binding process28 and more recently have received the name of conditional disorder.29 Several ID proteins become more ordered after interaction with ligands or other factors under physiological conditions and it was suggested that the majority of ID proteins are cases of conditional disorder.27 On the other hand, order‐to‐disorder transitions have been recently reviewed under the name of cryptic disorder,20 referring to those ID regions where one conformation is ordered but turns disordered under different physiological conditions.
In this work we performed a large‐scale study to explore the extension and contribution of conformational changes in the folded part of IDPs as a function of the presence of order‐disorder transitions and their interplay to sustain protein function. To this end, a large collection of IDPs with experimentally based conformational diversity was considered. Using pairs of conformers differing in a property associated with protein function (presence of biological ligand), two main measures were studied: conformational changes in folded regions were derived from structural based scores and order‐disorder transitions were quantified comparing transitions between regions in the conformers. These changes were then associated with different biological properties to understand their possible role in protein function.
Results
Analysis of order‐disorder transitions between conformers
The CoDNas15 and MobiDB30 database were used to retrieve a large dataset of proteins with experimentally based conformational diversity and annotated ID content. Our initial dataset contained at least two different conformers (with 100% sequence identity) obtained from alternative crystallization conditions of the same protein. ID changes were characterized in each pair of conformers for each protein. As it is explained in Methods, to study these transitions we considered any change between conformers in any ID segment of at least three contiguous residues. As each structure in CoDNaS is associated with the corresponding crystallization conditions (presence of ligands, pH, and so on) the change in order‐disorder could be linked with the change in crystallization conditions between the compared conformers. Following conformational selection theory, these factors could shift the conformational equilibrium towards different conformers producing structural changes and transitions in ordered or disordered regions. After removal of structures with mutations and with a resolution below 2.5 Å, we obtained 745 proteins with a total of 3444 conformers (Table 1 and Supporting Information). All possible pairs of conformers for each protein were considered to compare the extension of order‐disorder transitions. In this first set almost 50% of the proteins (365) show changes in order‐disorder proportion between conformers. The remaining 380 proteins show no differences in segments with a minimum of three continuous ID residues. Only five proteins show compensatory changes, that is, the protein becoming ordered in one region and disordered in another with no net ID change. We further filtered that set to obtain pairs of conformers without differences in pH and temperature with the aim to obtain a single parameter or change associated with the corresponding conformational change. We then obtained 398 proteins for which the presence of ligands was the only difference and just 24 proteins showing only differences in oligomeric state as derived from author annotation. As shown in Table 1, the effect of ligands or oligomeric change and their combination produce, on average, an ID increase in 17.07% of the proteins while for 27.88% order is increased. These ratios are very close to those found in the set of 416 proteins containing in addition changes in pH and temperature. In this later dataset, proteins could have a pH variation between conformers as high as 8.5 and 52 cases also show a difference in temperature. Apparently, these changes do not affect the global tendency observed in the cases where pH and temperature are unchanged.
Table 1.
Datasets Used in This Study
| Dataset | Number of proteins | Number of conformers | Factors describing transitions between conformers | Proteins gaining disorder | Proteins gaining order | Proteins with no‐change |
|---|---|---|---|---|---|---|
| 1 | 745 | 3444 | ΔpH, ΔT, ligands, change in oligomeric state | 153(20.54) | 212(28.46) | 380(51.01) |
| 2 | 416 | 1378 | ligands, change in oligomeric state | 71(17.07) | 116(27.88) | 229(55.05) |
| 3 | 398 | 1328 | ligands | 69(17.34) | 111(27.89) | 218(54.77) |
| 4 | 24 | 59 | Oligomeric change | 5(20.83) | 7(29.17) | 12(50.00) |
Figure 1 shows the fraction of ID characterizing the transition between two conformers for a given factor affecting the conformational equilibrium, with colors corresponding to the longest section changing ID. As shown in the figure for the 745 proteins used, the sequence length responsible for ID change has a median of 2, with the 75% quartile in a length of 5 contiguous residues changing. If only proteins with a change of three or more consecutive ID residues are considered, and crystallizations with changes in temperature and pH are removed, the length of the change has a median of 6 and a 75% quartile of 10 residues. It is important to note that for a given protein, a factor could increase disorder while another could increase order considering different conformers. There are 18 proteins with this behavior considering only the presence of ligands, as shown in both panels in Figure 1 (Supporting Information Table II). This is an expected result from pre‐equilibrium theory, mainly because different ligands could shift the conformational equilibrium towards different conformers. In this set of 18 proteins, we found that 10 have the same apo form but two different ligands promote the change to more ordered or disordered conformers. In the eight remaining, instead we have two different apo forms, mainly due to changes in pH. For these, conformational diversity in the apo form is as high as 1.67 Å RMSD and binding of different ligands again promotes ID content changes.
Figure 1.
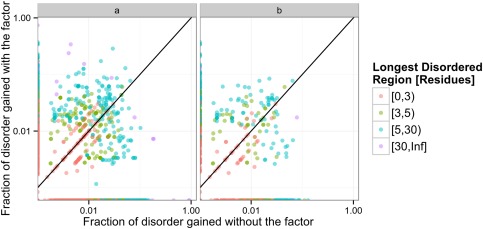
Fraction of disorder and order gain characterizing the transition between two conformers, colored as the longest involved region. (a) whole dataset (745 proteins) and (b) set containing conformers with differences in ligand presence.
To obtain a further understanding of ID transitions between conformers, we also studied the relationship between ligand size and transition extent. No correlation was found between ligand size (measured by molecular weight) and extent of disorder change between conformers (Spearman's rank correlation coefficient −0.078, P‐value 0.314). A similar behavior was observed using conformational diversity measured by RMSD (Spearman's rank correlation coefficient −0.055, P‐value 0.4867).
Conformational change and disorder transitions
Conformational changes measured by RMSD in folded regions were studied to explore if the disorder transitions were accompanied with major structural changes. In all cases, segments undergoing ID transitions were excluded to calculate the RMSD between the compared conformers. As can be seen in Figure 2, we found three dataset distributions corresponding to the proteins showing no change in disorder transition, proteins showing a gain in order and those gaining disorder. The three distributions are statistically different using a Wilcoxon test (P‐value < 0.05). Proteins gaining order or disorder are more flexible than proteins showing no change in ID segments. Considering the 75% quartile of each distribution, the RMSDs are 2.24, 1.86 and 0.87 Å. The same result is obtained using RMSD100,31 a RMSD measure normalized for every 100 residues that avoids RMSD length dependence. In addition, more flexible proteins also show a slight but statistically significant increase in disorder percentage (Wilcoxon test P‐value < 0.05) with 6.98, 10.22 and 12.12 of the 75% quartile for no‐change, gaining order and gaining disorder distributions. As expected, regions with ID transitions showed a higher than average B‐factors and this behavior is independent of crystallographic resolution (Fig. 3).
Figure 2.
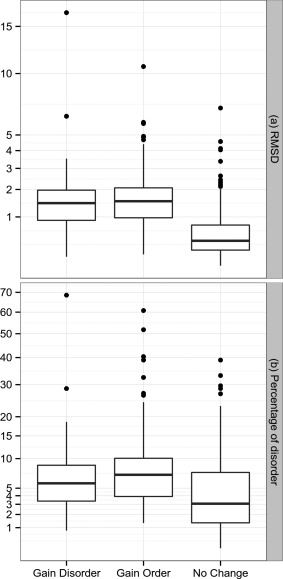
Box‐plot representing (a) RMSD distribution of proteins gaining order, disorder or with no‐detectable changes in order‐disorder content, (b) same distributions but as a function of percent of disorder. The box‐plot indicates 25, median and 75% quartile and in black dots the outliers. The y axis is in square root scale to gain a resolution at low values.
Figure 3.
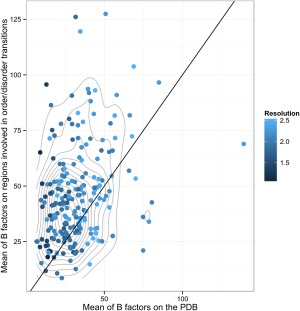
B‐factor comparison between regions under order‐disorder transitions and the average of the whole protein. The blue scale indicates different resolutions and the gray lines indicate the contour of the bi dimensional density: It can be seen that the mean B factor population of order‐disorder regions has a mean over the protein mean B factor since the center of the bi dimensional density is over the x = y line.
As protein motions, or more precisely conformational diversity, are associated with biological function,14, 32 we explored how the presence of order‐disorder transitions were associated with specific functions. We used human proteins for this analysis because they are overrepresented in the three datasets. We were then unable to find a clear relationship between molecular function and the extension of ID content change using goProfile33 (P‐value 0.44). ID transitions are also neither particularly associated with cell compartment (P‐value 0.38) nor biological process (P‐value 0.71). We also explored the differential enrichment for several protein properties derived from UniProt annotations between the protein set with ID and without transitions. We found that the protein set showing ID transitions is differentially enrich in proteins participating in nucleotide binding where such transitions have already been observed and characterized,34 for example, the SecA helicase35 and G protein family.36 This set also confirms a previously reported enrichment for proteins undergoing post‐translational modifications (PTMs).13 Furthermore, it was early proposed that the occurrence of chemical modifications of residues in ID regions could be higher than in ordered regions due to the lack of strong physicochemical constraints. This propensity could also enhance protein–protein recognition due to the presence of ID transitions.37 The trend that some PTMs occur differentially associated with disordered regions has also been recently confirmed.38, 39
Biological examples of ID transitions
Browsing the top scoring proteins undergoing ID transitions (see Supporting Information Tables III and IV) we found some typical cases of important proteins associated with the presence of ID. Proteins associated with the cytoskeleton like actin and microtubule proteins, known to be associated with ID to achieve their functions,40 were found in the set of proteins becoming more ordered after a binding event. Another interesting case is human superoxide dismutase (SOD1), an enzyme associated with several genetic disorders such as familial amyotrophic lateral sclerosis (FALS or Lou Gehrig's disease) also undergoing ID transitions.41 Eight protein kinases in the dataset (see Supporting Information Table IV) share the same catalytic domain fold (CATH domain 3.20.200.20). Interestingly, two belong to the set becoming more ordered, two to the set becoming more disordered and four do not show significant changes. These proteins have 2.18, 3.45 and 0.67 Å of RMSD between their conformers respectively confirming the general trend (Fig. 2), but also demonstrating how the same function may be achieved with different flexibility by the same fold.
Finally, to illustrate some of the ID transitions in our dataset, some examples with experimental evidence about their biological meaning were analyzed. DivK is a response regulator in the Gram‐negative bacterium Caulobacter crescentus and functions in a complex phosphorelay system controling metabolic pathways related to both cell division and motility. DivK represents a good example of order to disorder transition upon ligand binding. Recently, DivK was crystallized in its metal‐free and metal‐bound forms.42 Mn++ binding has a destabilizing effect, increasing flexibility of the N‐terminal β4‐α4 loop involving residues 84 to 97 which are missing in the structures [Fig. 4(A)]. The main effect of this increased flexibility upon ligand binding is solvent exposure of the critical Cys99 that favors adoption of a dimeric form through disulfide formation. Another example is dihydropteroate synthase (DHPS), a key enzyme in the folate pathway of bacteria and some eukaryotes [Fig. 4(B)]. DHPS binds 6‐hydroxymethyl‐7,8‐dihydropterin monophosphate (DHPP) and p‐aminobenzoic acid in a specific pocket closed by two loops that are completely disordered in the unbound conformer. However, after binding the two substrates and in presence of Mg++, the two conserved and flexible loops become ordered, contributing to substrate stabilization during catalysis.43 We have also found proteins that undergo order to disorder, as well as disorder to order transitions when different conformers are studied under different conditions. One of these examples is transthyretin (TTR), a human plasma protein that transports thyroxine (T4), frequently associated with amyloidogenic processes. Several ligands have been developed to bind and stabilize the native state avoiding the amyloidogenic process.44 TTR has 26 crystallized conformers included in CoDNaS and we found that binding of different ligands produces a gain of order as well as ID depending on the selected conformers [see Fig. 4(C)].
Figure 4.
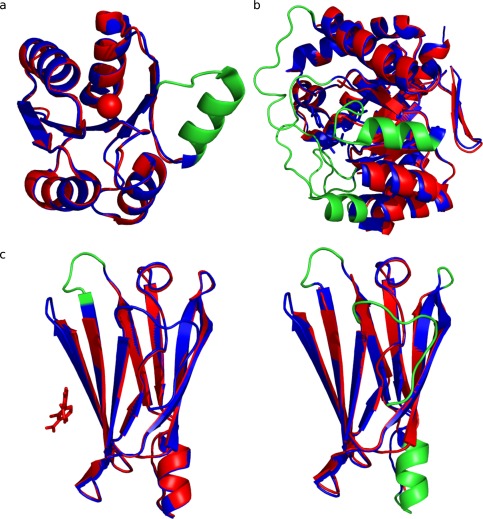
Biological examples of order‐disorder transitions between protein conformations. The region suffering order‐disorder transition is coloured in red. (a) Protein DivK conformations present an order‐disorder transition between apo (PDB code: 1M5T_A ‐ color blue) and holo (PDB code: 1MB0_A ‐ color cyan) forms. These structures present a C‐apha RMSD of 0.345 A. (b) Enzyme dihydropteroate synthase (DHPS) shows an order‐desorden transition in the conformers PDB code: 3TYZ_B (color blue) and 3TZN (color cyan). These structures present a C‐alpha RMSD of 0.548 A. (c) Conformers of human Transthyretin (TTR), (c1) Apo form (PDB code: 1TTA_B ‐ color blue) and holo form (PDB code: 3D2T_B ‐ color cyan) in complex with diflunisal. (c2) Apo conformation of TTR (PDB code: 1TTA_B ‐ color blue) and apo form (PDB code: 3CBR_B ‐ color cyan) at pH 3.5.
Discussion
We have performed a study of disorder transitions and extension of conformational diversity in a large protein dataset (745 IDPs). We found that almost half the proteins undergoing variations in crystallization conditions (presence of ligands, pH, temperature and oligomeric state) do not show a significant change in ID ratio among conformers. Interestingly, these proteins also show the lowest change in RMSD when the ordered region of the conformers is considered [75% quartile < 0.87 Å, Fig. 2(A)]. Furthermore, their RMSD distribution is similar to previously reported differences between conformers with and without ligands (75% quartile < 1 Å RMSD).45 Considering that the estimated crystallographic error could be as large as 0.4 Å,46 this set has a rather low conformational diversity between conformers, as well as a low content of ID regions [Fig. 2(B)]. In the other half of proteins, almost 60% became more ordered after a given change in crystallization conditions while the remaining 40% became more disordered. Both protein sets with changes in ID between conformers also show more flexible ordered regions, that is, higher RMSD between conformers with a 75% quartile 2.24 and 1.86 Å for the set gaining order and disorder respectively (Fig. 2). As these proteins also show a higher ID percentage [Fig. 2(B)], it is tempting to suggest that the increase in protein flexibility is associated with the presence of ID regions.
Of note, our analysis divided the ID dataset into two main groups, with and without disorder transitions, possibly suggesting different structure‐function relationships. As mentioned in Methods, the set of proteins differing in the presence of ligands involves proteins with ligands bound in their ordered regions and is not expected that the ID regions participate in their binding. It is rather expected that ligand affinity variation in the group of proteins without transitions and with lower conformational diversity could possibly include cases of “allosteric entropy”, where variation of binding affinities is almost independent of conformational change.3, 13 This concept was formerly proposed by Dryden and Cooper47 suggesting that a change in the distribution around the average structure of a protein was enough to generate a change in binding affinities, without the requirement of a change in the average structure of the protein. This property is also known as “dynamic allostery”. Fast internal protein motions detected with NMR relaxation measurements have shown side‐chain rotations participating in local as well as long‐range perturbations.48, 49, 50 These probably involve side‐chain movements promoting variation in inner cavity sizes or producing the opening and closing of tunnels almost without a significant change in protein backbone but promoting changes in ligand affinity.51, 52, 53
On the contrary, the group showing transitions and higher conformational diversity could indicate the necessity of important structural changes related with protein function. It was recently suggested that disorder transitions are an essential feature of key properties such as cooperativism and allosterism.54 Both properties are related with change in ligand binding affinities as a function of the same ligand and as a function of a different ligand (allosteric modulator) respectively. As we mentioned before, ligand binding location in this work has been mapped in folded regions of proteins composed our dataset. It is then interesting to note that proteins with disorder transition show higher RMSD in average (compared to those in the dataset without transitions) and then we can suggest that these transitions itself could modify ligand binding affinities favoring allosteric and cooperativity properties.
Materials and Methods
Dataset generation
The CoDNaS database15 (version 2.0), containing a redundant collection of three‐dimensional structures for the same protein, was used to recruit proteins exhibiting conformational diversity. Structures for the same protein obtained under different crystallographic conditions (ligands, mutations, changes in oligomeric state and others) have been associated with snapshots of protein dynamism and could, consequently, characterize protein conformers. The retrieved protein chains were linked with MobiDB,30, 55 to provide data on ID. Residues whose Cα atoms are missing in X‐ray structures from the PDB were considered as disordered.
An initial set of 754,203 conformer pairs with ID data representing 8,921 proteins from CoDNaS were filtered using different parameters. First, proteins containing only conformers with a resolution lower than 2.5 Å and containing mutations were removed. This set was further filtered using a segment of at least 3 continuous ID residues to assess order‐disorder transitions between conformers (see below). For each protein, only the conformers obtained at the same temperature were included as the crystallization temperature could affect protein conformation.56 The remaining proteins were further filtered to obtain single factors associated to the transition between a given pair of conformers for each protein (see Table 1, all datasets are included as Supporting Information). In the last dataset we finally found two main factors possibly affecting the transition between conformer pairs: presence of ligands and changes in oligomeric state. Other factors associated with protein function like the presence of post‐translational modifications are not represented in our dataset due to the stringency of applied filters.
Measure of disorder change
Per‐residue disorder data for each crystallographic structure can be represented as a binary vector, where 1 represents a missing residue (disorder) and 0 an ordered residue as derived from MobiDB. Using this data structure, the disorder change between conformers was compared. Both vectors were subtracted in one sense and the other. The results could be 0, if both residues have the same state and −1 or 1 for the disorder or order gain respectively. The fraction of the binary vector with value 1 was calculated and represents the fraction of disorder gain. The difference of this value between conformers was used throughout this study. Also, we use this variable to define three groups: proteins that don't change disorder (that difference is 0), the proteins that gain the disorder in the presence of a factor and proteins that gain order. Only proteins that have a change of three or more contiguous residues are included in these groups. This discretization was useful to perform UniProt enrichment test and functional profile comparisons.
Other analyzed factors
Most of the analyzed data, for example, protein length and taxonomy annotations, are already included in the CoDNaS database. The remaining information was extracted from PDB files using BioPython scripts and linking with the UniProt database. For ligand binding, we extract the information from the PDB file. Any interaction between chain and ligand was only annotated when any atom of the chain is less than 4 Å away from any ligand atom. Ligand molecular weight was obtained parsing mmCIF and PDB files. Only the 367 H. sapiens proteins on the dataset with Bioconductor packages for the links between ID proteins were used for GO term analysis. Differences between GO terms of the proteins which gain or lose disorder with the external factor were analyzed with the R package goProfile. The flexibility of regions showing order/disorder transitions was measured as the average of the experimental B‐factors over regions with more than 6 continuous order‐disorder residues between conformers. This average was compared with the average B‐factors for the rest of the protein.
Supporting information
Supporting Information Table 1.
Supporting Information Table 2.
Supporting Information Table 3.
Supporting Information Table 4.
Acknowledgments
The authors thank Tomás Di Domenico for his initial help with the dataset. G.P. and M.S.F. are CONICET researchers and D.J.Z. and A.M. CONICET fellows.
References
- 1. Karplus M, Kuriyan J (2005) Molecular dynamics and protein function. Proc Natl Acad Sci USA 102:6679–6685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cui Q, Karplus M (2008) Allostery and cooperativity revisited. Protein Sci 17:1295–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Motlagh HN, Wrabl JO, Li J, Hilser VJ (2014) The ensemble nature of allostery. Nature 508:331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wrabl JO, Gu J, Liu T, Schrank TP, Whitten ST, Hilser VJ (2011) The role of protein conformational fluctuations in allostery, function, and evolution. Biophys Chem 159:129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Parisi G, Zea DJ, Monzon AM, Marino‐Buslje C (2015) Conformational diversity and the emergence of sequence signatures during evolution. Curr Opin Struct Biol 32:58–65. [DOI] [PubMed] [Google Scholar]
- 6. Karush F (1950) Heterogeneity of the binding sites of bovine serum albumin. J Am Chem Soc 72:2705–2713. [Google Scholar]
- 7. Koshland JDE, Ray JWJ, Erwin MJ (1958) Protein structure and enzyme action. Fed Proc 17:1145–1150. [PubMed] [Google Scholar]
- 8. Monod J, Wyman J, Changeux JP (1965) On the nature of allosteric transitions: a plausible model. J Mol Biol 12:88–118. [DOI] [PubMed] [Google Scholar]
- 9. Changeux J‐P (2012) Allostery and the Monod‐Wyman‐Changeux model after 50 years. Annu Rev Biophys 41:103–133. [DOI] [PubMed] [Google Scholar]
- 10. James LC, Roversi P, Tawfik DS (2003) Antibody multispecificity mediated by conformational diversity. Science 299:1362–1367. [DOI] [PubMed] [Google Scholar]
- 11. del Sol A, Tsai C‐J, Ma B, Nussinov R (2009) The origin of allosteric functional modulation: multiple pre‐existing pathways. Structure 17:1042–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ma B, Nussinov R (2010) Enzyme dynamics point to stepwise conformational selection in catalysis. Curr Opin Chem Biol 14:652–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tsai C‐J, Nussinov R (2014) A unified view of “how allostery works”. PLoS Comput Biol 10:e1003394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gerstein M, Krebs W (1998) A database of macromolecular motions. Nucleic Acids Res 26:4280–4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Monzon AM, Juritz E, Fornasari S, Parisi G (2013) CoDNaS: a database of conformational diversity in the native state of proteins. Bioinformatics 29:2512–2514. [DOI] [PubMed] [Google Scholar]
- 16. Wright PE, Dyson HJ (1999) Intrinsically unstructured proteins: re‐assessing the protein structure‐function paradigm. J Mol Biol 293:321–331. [DOI] [PubMed] [Google Scholar]
- 17. Tompa P (2012) Intrinsically disordered proteins: a 10‐year recap. Trends Biochem Sci 37:509–516. [DOI] [PubMed] [Google Scholar]
- 18. Uversky VN, Gillespie JR, Fink AL (2000) Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 41:415–427. [DOI] [PubMed] [Google Scholar]
- 19. Dunker AK, Obradovic Z (2001) The protein trinity–linking function and disorder. Nat Biotechnol 19:805–806. [DOI] [PubMed] [Google Scholar]
- 20. Jakob U, Kriwacki R, Uversky VN (2014) Conditionally and transiently disordered proteins: awakening cryptic disorder to regulate protein function. Chem Rev 114:6779–6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT (2004) Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol 337:635–645. [DOI] [PubMed] [Google Scholar]
- 22. Tompa P (2002) Intrinsically unstructured proteins. Trends Biochem Sci 27:527–533. [DOI] [PubMed] [Google Scholar]
- 23. Jensen MR, Ruigrok RWH, Blackledge M (2013) Describing intrinsically disordered proteins at atomic resolution by NMR. Curr Opin Struct Biol 23:426–435. [DOI] [PubMed] [Google Scholar]
- 24. Varadi M, Kosol S, Lebrun P, Valentini E, Blackledge M, Dunker AK, Felli IC, Forman‐Kay JD, Kriwacki RW Pierattelli R, Sussman J, Svergun DI, Uversky VN, Vendruscolo M, Wishart D, Wright PE, Tompa P (2014) pE‐DB: a database of structural ensembles of intrinsically disordered and of unfolded proteins. Nucleic Acids Res 42:D326–D335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Babu MM, Kriwacki RW, Pappu RV (2012) Structural biology. Versatility from protein disorder. Science 337:1460–1461. [DOI] [PubMed] [Google Scholar]
- 26. Oldfield CJ, Dunker AK (2014) Intrinsically disordered proteins and intrinsically disordered protein regions. Annu Rev Biochem 83:553–583. [DOI] [PubMed] [Google Scholar]
- 27. Tompa P, Fuxreiter M (2008) Fuzzy complexes: polymorphism and structural disorder in protein‐protein interactions. Trends Biochem Sci 33:2–8. [DOI] [PubMed] [Google Scholar]
- 28. Wright PE, Dyson HJ (2010) Linking folding and binding. Curr Opin Microbiol 19:31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bardwell J, Jakob U (2012) Conditional disorder in chaperone action. Trends Biochem Sci 37:517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Di Domenico T, Walsh I, Martin AJM, Tosatto SCE (2012) MobiDB: a comprehensive database of intrinsic protein disorder annotations. Bioinformatics 28:2080–2081. [DOI] [PubMed] [Google Scholar]
- 31. Carugo O, Pongor S (2001) A normalized root‐mean‐square distance for comparing protein three‐dimensional structures. Protein Sci 10:1470–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bahar I, Lezon TR, Yang L‐W, Eyal E (2010) Global dynamics of proteins: bridging between structure and function. Annu Rev Biophys 39:23–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sánchez A, Salicrú M, Ocaña J (2007) Statistical methods for the analysis of high‐throughput data based on functional profiles derived from the gene ontology. J Stat Plan Inference 137:3975–3989. [Google Scholar]
- 34. Dyson HJ (2012) Roles of intrinsic disorder in protein‐nucleic acid interactions. Mol Biosyst 8:97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Keramisanou D, Biris N, Gelis I, Sianidis G, Karamanou S, Economou A, Kalodimos CG (2006) Disorder‐order folding transitions underlie catalysis in the helicase motor of SecA. Nat Struct Mol Biol 13:594–602. [DOI] [PubMed] [Google Scholar]
- 36. Hatley ME, Lockless SW, Gibson SK, Gilman AG, Ranganathan R (2003) Allosteric determinants in guanine nucleotide‐binding proteins. Proc Natl Acad Sci USA 100:14445–14450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z (2002) Intrinsic disorder and protein function. Proteins 41: 6573–6582. [DOI] [PubMed] [Google Scholar]
- 38. Gao J, Xu D (2012) Correlation between posttranslational modification and intrinsic disorder in protein. Pac Symp Biocomput 17: 94–103. [PMC free article] [PubMed] [Google Scholar]
- 39. Kurotani A, Tokmakov AA, Kuroda Y, Fukami Y, Shinozaki K, Sakurai T (2014) Correlations between predicted protein disorder and post‐translational modifications in plants. Bioinformatics 30:1095–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guharoy M, Szabo B, Contreras Martos S, Kosol S, Tompa P (2013) Intrinsic structural disorder in cytoskeletal proteins. Cytoskeleton 70:550–571. [DOI] [PubMed] [Google Scholar]
- 41. Uversky VN (2010) Targeting intrinsically disordered proteins in neurodegenerative and protein dysfunction diseases: another illustration of the D(2) concept. Expert Rev Proteomics 7:543–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guillet V, Ohta N, Cabantous S, Newton A, Samama J‐P (2002) Crystallographic and biochemical studies of DivK reveal novel features of an essential response regulator in Caulobacter crescentus. J Biol Chem 277:42003–42010. [DOI] [PubMed] [Google Scholar]
- 43. Yun M, Wu Y, Li Z, Zhao Y, Waddell MB, Antonio M, Lee RE, Bashford D, White SW (2012) Catalysis and Sulfa Drug Resistance in Dihydropteroate Synthase: crystal structures reveal the catalytic mechanism of DHPS and the structural basis of sulfa drug action and resistance. Science 335:1110–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kolstoe SE, Mangione PP, Bellotti V, Taylor GW, Tennent GA, Deroo S, Morrison AJ, Cobb AJA, Coyne A Mccammon MG, Warnerd TD, Mitchelld J, Gilla R, Smithe MD, Leyc SV, Robinsonc CV, Wooda SP, Pepysa MB (2010) Trapping of palindromic ligands within native transthyretin prevents amyloid formation. Proc Natl Acad Sci USA 107:20483–20488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gutteridge A, Thornton J (2005) Conformational changes observed in enzyme crystal structures upon substrate binding. J Mol Biol 346:21–28. [DOI] [PubMed] [Google Scholar]
- 46. Burra PV, Zhang Y, Godzik A, Stec B (2009) Global distribution of conformational states derived from redundant models in the PDB points to non‐uniqueness of the protein structure. Proc Natl Acad Sci USA 106:10505–10510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cooper A, Dryden DTF (1984) Allostery without conformational change. A plausible model. Eur Biophys J 11:103–109. [DOI] [PubMed] [Google Scholar]
- 48. Wand AJ (2013) The dark energy of proteins comes to light: conformational entropy and its role in protein function revealed by NMR relaxation. Curr Opin Struct Biol 23:75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Igumenova TI, Lee AL, Wand AJ (2005) Backbone and side chain dynamics of mutant calmodulin‐peptide complexes. Biochemistry 44:12627–12639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Igumenova TI, Frederick KK, Wand AJ (2006) Characterization of the fast dynamics of protein amino acid side chains using NMR relaxation in solution. Chem Rev 106:1672–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Laskowski RA, Luscombe NM, Swindells MB, Thornton JM, Thornton JM (1996) Protein clefts in molecular recognition and function Protein clefts in molecular recognition and function. Protein Sci 5:2438–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gora A, Brezovsky J, Damborsky J (2013) Gates of enzymes. Chem Rev 113:5871–5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Brylinski M, Skolnick J (2008) What is the relationship between the global structures of apo and holo proteins? Proteins 70:363–377. [DOI] [PubMed] [Google Scholar]
- 54. Tompa P (2014) Multisteric regulation by structural disorder in modular signaling proteins: an extension of the concept of allostery. Chem Rev 114:6715–6732. [DOI] [PubMed] [Google Scholar]
- 55. Potenza E, Domenico TD, Walsh I, Tosatto SCE (2014) MobiDB 2.0: an improved database of intrinsically disordered and mobile proteins. Nucleic Acids Res 43:D315–D320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fraser JS, van den Bedem H, Samelson AJ, Lang PT, Holton JM, Echols N, Alber T (2011) Accessing protein conformational ensembles using room‐temperature X‐ray crystallography. Proc Natl Acad Sci USA 108:16247–16252. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Table 1.
Supporting Information Table 2.
Supporting Information Table 3.
Supporting Information Table 4.