

Abstract
From both the structural and functional points of view, β-turns play important biological roles in proteins. In the present study, a novel two-stage hybrid procedure has been developed to identify β-turns in proteins. Binary logistic regression was initially used for the first time to select significant sequence parameters in identification of β-turns due to a re-substitution test procedure. Sequence parameters were consisted of 80 amino acid positional occurrences and 20 amino acid percentages in sequence. Among these parameters, the most significant ones which were selected by binary logistic regression model, were percentages of Gly, Ser and the occurrence of Asn in position i+2, respectively, in sequence. These significant parameters have the highest effect on the constitution of a β-turn sequence. A neural network model was then constructed and fed by the parameters selected by binary logistic regression to build a hybrid predictor. The networks have been trained and tested on a non-homologous dataset of 565 protein chains. With applying a nine fold cross-validation test on the dataset, the network reached an overall accuracy (Qtotal) of 74, which is comparable with results of the other β-turn prediction methods. In conclusion, this study proves that the parameter selection ability of binary logistic regression together with the prediction capability of neural networks lead to the development of more precise models for identifying β-turns in proteins.
Keywords: beta-turns, binary logistic regression, neural networks, secondary structure prediction, sequence parameters
Introduction
Protein secondary structure prediction is a preceding step to the more complicated tertiary structure prediction (Richardson, 1981[27]). Among many structural elements, tight turns play an important role in protein folding and stability. They are classified as δ-turns, γ-turns, β-turns, α-turns and π-turns, depending on the number of residues forming the turn (Chou, 2000[3]). β-turns are the most existing type of tight turns in proteins and include almost 25 % of all residues in globular proteins (Kabsch and Sander, 1983[11]). They consist of four consecutive residues defined by positions i, i+1, i+2 and i+3. The distance between Cα (i) and Cα (i+3) is less than 7 Ǻ (Chou, 2000[3]). According to the Ø, ψ angles of the residues i+1 and i+2, β-turns can be classified into 9 different types: I, I΄, II, II΄, IV, Via1, Via2, VIb and VIII.
Both from structural and functional points of view, β-turns play important biological roles in proteins. They tend to be found at solvent-exposed surfaces and therefore involve in molecular recognition processes between proteins, as well as in interactions between peptide substrates and receptors (Rose et al., 1985[28]). β-turn formation is a determining stage during the process of protein folding. Also, β-turns are responsible for the compact globular shape of proteins since they have the ability for reserving the alignment of protein chain (Takano et al., 2000[31]). Hence, the development of a precise method for analysis and prediction of β-turns (according to the amino acid sequences) would be useful for protein folding studies as well as for predicting the overall three-dimensional structure of proteins.
Many efforts have been made for analysis and prediction of β-turns in proteins. They can be divided into two categories: statistics-based and machine learning-based methods. The majority of statistics-based methods used positional preferences of amino acids in β-turns (Lewis et al., 1973[17]; Chou and Fasman, 1974[4]; Wilmot and Thornton, 1988[33]; Hutchinson and Thornton, 1994[9]; Zhang and Chou, 1997[34]; Fuchs and Alix, 2005[5]) .The second category includes neural network (NN) (McGregor et al., 1989[21]; Shepherd et al., 1999[29]; Kaur and Raghava, 2003[14], 2004[12]; Kirschner and Freshman, 2008[15]; Petersen et al., 2010[24]) as well as support vector machine (SVM) (Cai et al., 2003[2]; Pham et al., 2003[25], 2005[26]; Zhang et al., 2005[35]; Zheng and Kurgan, 2008[36]; Hu and Li, 2008[8]; Liu et al., 2009[19]; Meissner et al., 2009[22]; Kountouris and Hirst, 2010[16]; Shi et al., 2011[30]; Tang et al., 2011[32]) approaches. To compare main methods of β-turn prediction, Kaur and Raghava (2002[13]) have made an evaluation on the benchmark data set. They showed that neural network approach by Shepherd et al. (1999[29]) presented the best prediction performance among other evaluated methods. In a previous study based on a hybrid approach, we employed the multinomial logistic regression as well as neural networks for analysis and identification of β-turn types (Asgary et al., 2007[1]). More recently, Zheng and Kurgan (2008[36]) used SVM for β-turn prediction getting the performance of their method is the highest among all. As a result, machine learning methods (especially SVM approach) can be considered as the most accurate ones for prediction of β-turns.
In the year 2002, Kaur and Raghava suggested that combining a statistics method with a machine learning method may provide substantially better results than either one alone (Kaur and Raghava, 2002[13]). In the present study, we followed their recommendation by combining the binary logistic regression as statistical method with the neural network as machine learning one for identification of β-turns.
The binary logistic regression method, which has not been applied for β-turn analysis so far, is useful when the presence or absence of a characteristic or an outcome based on a set of predictor variables is needed to be predicted. It is similar to a linear regression model but is suited to models with dichotomous dependent variable (Hosmer and Lemeshow, 2000[7]). We used binary logistic regression to select the most effective set of parameters which then were fed into a well-established neural network. In this way, we increased the accuracy and reliability of neural networks, in β-turns identification.
Materials and Methods
The dataset
Our dataset consisted of 565 non-homologous protein chains (Table 1(Tab. 1)). These protein chains were selected using the PDB-REPRDB server (Noguchi et al., 2001[23]). In this dataset, no two proteins have more than 25 % sequence uniformity. All proteins have reported X-ray structures with 2.0 Å resolution or better. The program PROMOTIF (Hutchinson and Thornton, 1996[10]) was employed to identify β-turns in the proteins. Sequence parameters including 80 amino acid positional occurrences as well as 20 amino acid percentages (of existence) in β-turn sequences were generated using IF and COUNTIF functions of EXCEL software (2003), respectively.
Table 1. The PDB (Protein Data Bank) codes of 565 protein chains.

Model design
Initially, binary logistic regression serves as a non-linear model on the dataset to select significant parameters due to the ''Re-substitution Test''. This test is absolutely necessary because it reflects the self-consistency of an identification method, especially for its algorithm part. Certainly, a prediction algorithm cannot be deemed as a good one if its self-consistency is poor. In other words, the re-substitution test is necessary but not sufficient for evaluating an identification method. When this test was implemented, each tetra peptide in the dataset concerned is in turn identified using the rule parameters derived from the same dataset, the so-called training dataset. After using the binary logistic regression in this manner, the NNs (which act non-linearly in the last stage of this hybrid procedure) were fed by the outputs of binary logistic regression to predict β-turns. The NN method has been trained and tested using 9-fold cross-validation techniques, whereby the dataset is divided into nine subsets (i.e. 8 subsets containing 62 protein chains, 1 subset containing 61 protein chains). The method has been trained on eight subsets and the performance was calculated on the remaining ninth subset. This procedure was repeated nine times, once for each subset. Actually, the ''Cross-validation Test'' can reflect the effectiveness of an identification method in practical application.
Binary logistic regression model
This model is used only when the dependent variable is dichotomous, that is, there are only two possible answers for the dependent variable. Let the dependent variable be Y. Since it is dichotomous, it takes on 0 or 1 for failure and success, respectively. The logistic regression model can be expressed as follows (Hosmer and Lemeshow, 2000[7]):
Log [p/(1-p)] = β0 + β1x1 + β2x2 + … +βnxn,
where p is the probability of Y=1, β0 is a constant, and β1 - βn are unknown logistic regression coefficients of independent variables x1-xn (amino acid occurrences or percentages in β-turn sequence). The ratio p/1-p takes on values between 0 and plus infinity. Therefore, the logarithm of this ratio (logit) is a continuous variable that takes on values between minus infinity and plus infinity. Using this equation, the value of logit is determined. Then a cutoff should be taken to recode logit values into two possible states of dependent variable (i.e. non-β-turn sequence and β-turn sequence) (Hosmer and Lemeshow, 2000[7]). The optimized cutoff value in this research was 0.5.
Several different options are available during the creation of logistic regression model. Independent variables can be entered into the model in the order specified by the researcher and logistic regression can test the fit of the model after each coefficient is added or deleted, called ''stepwise regression''. Stepwise regression is used in the exploratory phase of research. We used the Backward Wald (stepwise) binary logistic regression routine in SPSS program to develop our model. This routine appears to be the preferred method of exploratory analysis, where the analysis begins with a full or saturated model and independent variables are eliminated from the model in an iterative process. The fit of the model is tested after the elimination of each independent variable to ensure that the model still adequately fits the data. When no more independent variables can be eliminated from the model, the analysis has been completed. The measure for model fitness in each step is an index called -2 Log Likelihood. In general, as the model becomes better, this index (-2LL) will decrease in magnitude. In fact, in backward Wald routine, the first step has the minimum value of -2 log likelihood and hence its reported result (i.e. Parameter Estimates Table) is the main output of binary logistic regression model (Hosmer and Lemeshow, 2000[7]).
Neural network model
The neural network (NN) was utilized as a robust non-linear predictor in hybrids with the binary logistic regression. In this way, the selected variables from binary logistic regression model were used as input nodes of neural network. This is supposed to decrease the number of input nodes, simplify the network architecture and shorten the time needed for model building. We used feed-forward back propagation networks with a single hidden layer. Using such algorithm, the parameters related to the training cases were fed into the networks. The final outputs estimated by the networks were compared with the actual type of cases; generating a mean of the sum of square error (MSE). This quantity was propagated back into the networks to adjust the randomly chosen weights. The training cases were then tested with new weights and the procedure repeated. Through such process, the MSE was minimized.
Three layer networks were used in this study. Each unit in the input layer was fed by one independent variable which has been selected by binary logistic regression model. The output layer included two units which represented 1 0 and 0 1 for β-turn and non-β-turn sequence, respectively.
The MSE was utilized as an index of network efficiency in determining the optimized number of hidden units (Hayatshahi et al., 2005[6]). To do so, the number of hidden units was changed in every network in order to develop networks producing the minimal MSE. At last, after such optimizing process, the number of units for hidden layer reached 8.
The final neural network structure was consisted of 33 units in input layer, 8 units in hidden layer and 2 units in output layer. The activation function of hidden layer units was logsig. We used the Quasi-Newton training function in this research. This training function is prior to simple 'batch' gradient-descent and lead to significantly better solutions requiring fewer training steps. Besides, this method does not suffer from the specification problem of the learning rate parameter which is crucial for the performance of the gradient-descent method (Likas and Stafylopatis, 2000[18]).
Training has been done for 1000 epochs for nine networks. The value of the learning rate parameter has been set to 0.2. The software employed to build neural networks was in-house written in the MATLAB programming language.
Performance measures
Four different parameters have been used to measure the performance of prediction methods. These four parameters can be derived from the four scalar indices: TP (true positives: number of correctly classified β-turns), TN (true negatives: number of correctly classified non-β-turns), FP (false positives: number of non-β-turns incorrectly classified as β-turns) and FN (false negatives: number of β-turns incorrectly classified as non-β-turns). Using the following formulas which have been previously reported in the published material, we calculated these parameters for the output of binary Logistic Regression and NN models.

which is the fraction of correctly predicted β-turns and non-β-turns among all predictions.

which is the percentage of correctly predicted β-turns.
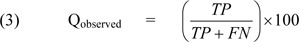
which is the percentage of observed β-turns that are correctly predicted.
(4) Matthews correlation coefficient (MCC): We used MCC as a more robust measure to evaluate the reliability of the established method (Matthews, 1975[20]). The MCC is defined by

The MCC is a limited number between -1 and 1. If there is no relationship between the predicted values and the actual values, the MCC should be 0 or very low (the predicted values are not better than random numbers). In contrast, the MCC value would increase as the strength of the relationship between the predicted values and actual values increases. It is obvious that a perfect fit gives a coefficient of 1.0. Furthermore, the higher MCC indicates the better performance of the prediction for the model.
Statistical analysis was performed using SPSS 13 for Windows (SPSS Inc., Chicago, USA).
Results
Binary logistic regression analysis
Binary logistic regression model was runned on the dataset using the re-substitution test. One of output tables of binary logistic regression model was ''Omnibus Tests of Model Coefficients'' (Table 2(Tab. 2)). This table reports significance levels by the traditional chi-square method and is an alternative to the ''Hosmer-Lemeshow Test'' (Hosmer and Lemeshow, 2000[7]). Among 24 steps, the only positive chi-square value (i.e. significant) can be seen in step 1 which is 6429.548 (P-value= 0.000) (Table 2(Tab. 2)). Since the probability of the first step chi-square was less than the significance level (0.05), the existence of a relationship between independent variables (100 structural parameters) and the dependent variable (β-turn and non-β-turn sequences) was supported.
Table 2. Omnibus tests of model coefficients.

Also, according to ''Model summary'' table in the output of binary logistic regression model (Table 3(Tab. 3)), we see that -2 log likelihood index has its minimum value in the first step of the model (47450.373). The lowest value of this index indicates the best step of the model (Hosmer and Lemeshow, 2000[7]). Therefore, the first step was recognized as the reference step.
Table 3. Model summary.

Table 4(Tab. 4) shows parameter estimates (β), standard errors, Wald statistic, p-values and corresponding odds ratios for selected parameters among 100 ones, for β-turns in contrast to non-β-turns as reference group of the backward-Wald binary logistic regression procedure. This information is related to the first step of the model. Among sequence parameters, 13 amino acid percentages and 35 amino acid positional occurrences were found to be significant in determining β-turns and non-β-turns.
Table 4. Parameter estimates, indicating statistical results for the significant parameters in the output of binary logistic regression procedure (β-turns in contrast to non-β-turns (as reference group)).

Percentages of Gly, Ser, Asp, Asn, Pro, Thr, His, Cys, Lys, Arg and Gln, respectively, have the most positive parameter estimate values among percentages of other amino acids. Therefore the probability of the sequence to be β-turn increases as increasing their values. On the other hand, percentages of Ile and Leu, respectively, have the most negative parameter estimate values among percentages of other amino acids and hence the probability of the sequence to be non-β-turn will increase as increasing values.
Occurrences of Asp, Pro, Asn, Ser, Cys, Phe, Leu and Thr respectively in position i of sequence have the most positive logit coefficients (or parameter estimates) among occurrences of other amino acids in the same position and support of β-turn sequence. Vice versa, occurrences of Gln and Gly respectively in position i of β-turn sequence have the most negative logit coefficients among occurrences of other amino acids in that position and support of non-β-turn sequence.
In analysis of position i+1 of sequence, occurrences of Pro, Glu, Lys, Asp, Ala, Ser, Arg, Leu and Asn, respectively, have the highest logit coefficients and occurrences of Cys and Gly have the most negative logit coefficients among others. Obtained results can be interpreted like above-mentioned sentences.
Ultimately, the final position of sequence which is highlighted in the table is i+2. Occurrences of Asn, Asp, Gly, His, Glu, Phe, Tyr, Thr, Ser, Trp, Arg, Lys, Leu and Gln, respectively, have the highest parameter estimates among others and hence they support the sequence to be β-turn.
The result of re-substitution {Self-Consistency} test was evaluated by the performance measures. The results shown in Table 5(Tab. 5) are obtained according to the output of the model.
Table 5. Prediction results of our two-stage hybrid procedure.

Neural network
We fed our neural networks with 33 significant parameters selected in the re-substitution binary logistic regression procedure to a build two-stage hybrid model. The number of units in the hidden layer was optimized in networks regarding the least MSE rate (refer you to Materials and Methods section). The final MSE rate was 0.13 (with the eight units in the hidden layer) which was the lowest among different examined numbers of units in the hidden layer. Ultimately, we ended to an optimal neural network architecture with 33 input units and a single hidden layer with 8 units for our binary prediction (i.e. be β-turn or non-β-turn). A nine fold cross-validation procedure was used for prediction of β-turns. The performance of the model was evaluated by averaging the mentioned measures over nine sets.
The prediction results using neural networks are presented in Table 5(Tab. 5). With applying a nine fold cross-validation test on the dataset, it would be found that the network reached an overall accuracy (Qtotal) of 74. Also, the network yielded Qpredicted value of 45 and Qobserved value of 30. Ultimately, the value of MCC for the network was 0.21.
Discussion
An important advantage for using logistic regression model is its capability of determining weights of each selected significant parameter, which highlights its priority of importance in clarifying the sequence-structure relationship. This study shows that this parameter selection ability of binary logistic regression in combination to the prediction ability of neural networks leads to the development of more precise models.
Binary logistic regression analysis showed that only 48 structural parameters among 100 ones were significant in identification of β-turns. Percentage of Gly in sequence was the most important parameter with the parameter estimate (β) value of 3.779 and the odds ratio value of 43.762. The second major parameter was the percentage of Ser with the parameter estimate and odds ratio values of 1.509 and 4.522, respectively. The third important parameter was the occurrence of Asn in positions i+2 of the sequence. Consequently, these three parameters have the highest effect on the constitution of β-turn sequence, among others. On the other hand, the percentage of Leu in sequence had the most negative parameter estimate value (i.e. -0.889). Thus this parameter has the highest effect on the constitution of non-β-turn sequence, among others.
In conclusion, our research highlighted the efficiency of using the statistical model of binary logistic regression as a preprocessor in determining effective parameters. Besides, the optimal structure of neural network can be simplified by a preprocessor in the first stage of hybrid approach, which in turn causes decreasing the time needed for neural network training procedure in the second stage.
Acknowledgements
We thank Professor Anoshirvan Kazemnejad and Mr Samad Jahandideh for their helpful comments and discussions. We also thank the Department of Biophysics (from the Tarbiat Modares University, Iran) for financial support.
References
- 1.Asgary MP, Jahandideh S, Abdolmaleki P, Kazemnejad A. Analysis and identification of β-turn types using multinomial logistic regression and artificial neural network. Biinformatics. 2007;23:3125–3130. doi: 10.1093/bioinformatics/btm324. [DOI] [PubMed] [Google Scholar]
- 2.Cai YD, Liu XJ, Li YX, Xu XB, Chou KC. Prediction of β-turns with learning machines. Peptides. 2003;24:665–669. doi: 10.1016/s0196-9781(03)00133-5. [DOI] [PubMed] [Google Scholar]
- 3.Chou KC. Prediction of tight turns and their types in proteins. Anal Biochem. 2000;286:1–16. doi: 10.1006/abio.2000.4757. [DOI] [PubMed] [Google Scholar]
- 4.Chou PY, Fasman GD. Conformational parameters for amino acids in helical, β-sheet and random coil regions calculated from proteins. Biochemistry. 1974;13:211–222. doi: 10.1021/bi00699a001. [DOI] [PubMed] [Google Scholar]
- 5.Fuchs PFJ, Alix AJP. High accuracy prediction of β-turns and their types using propensities and multiple alignments. Proteins. 2005;59:828–839. doi: 10.1002/prot.20461. [DOI] [PubMed] [Google Scholar]
- 6.Hayatshahi SHS, Abdolmaleki P, Safarian S, Khajeh K. Non-linear quantitative structure-activity relationship for adenine derivatives as competitive inhibitors of adenosine deaminase. Biochem Biophys Res Com. 2005;338:1137–1142. doi: 10.1016/j.bbrc.2005.10.049. [DOI] [PubMed] [Google Scholar]
- 7.Hosmer DW, Lemeshow S. In: Applied logistic regression. Groves RM, Kalton G, Rao JNK, Schwarz N, Skinner C, editors. New York: Wiley; 2000. [Google Scholar]
- 8.Hu X, Li Q. Using support vector machine to predict beta- and gamma-turns in proteins. J Comput Chem. 2008;29:1867–1875. doi: 10.1002/jcc.20929. [DOI] [PubMed] [Google Scholar]
- 9.Hutchinson EG, Thornton JM. A revised set of potentials for β-turn formation in proteins. Protein Sci. 1994;3:2207–2216. doi: 10.1002/pro.5560031206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hutchinson EG, Thornton JM. PROMOTIF: A program to identify and analyze structural motifs in proteins. Protein Sci. 1996;5:212–220. doi: 10.1002/pro.5560050204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kabsch W, Sander C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 12.Kaur H, Raghava GPS. A neural network method for prediction of β-turn types in proteins using evolutionary information. Bioinformatics. 2004;20:2751–2758. doi: 10.1093/bioinformatics/bth322. [DOI] [PubMed] [Google Scholar]
- 13.Kaur H, Raghava GPS. An evaluation of β-turn prediction methods. Bioinformatics. 2002;18:1508–1514. doi: 10.1093/bioinformatics/18.11.1508. [DOI] [PubMed] [Google Scholar]
- 14.Kaur H, Raghava GPS. Prediction of β-turns in proteins from multiple alignments using neural network. Protein Sci. 2003;12:627–634. doi: 10.1110/ps.0228903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirschner A, Freshman D. Prediction of beta-turns and beta-turn types by a novel bidirectional Elman-type recurrent neural network with multiple output layers (MOLEBRNN) Gene. 2008;422:22–29. doi: 10.1016/j.gene.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 16.Kountouris P, Hirst GD. Predicting β-turns and their types using predicted backbone dihedral angles and secondary structures. BMC Bioinformatics. 2010;11:407. doi: 10.1186/1471-2105-11-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lewis PN, Momany FA, Scheraga HA. Chain reversals in proteins. Biochem Biophys Acta. 1973;303:211–229. doi: 10.1016/0005-2795(73)90350-4. [DOI] [PubMed] [Google Scholar]
- 18.Likas A, Stafylopatis A. Training the random neural network using quasi-Newton methods. Eur J Oper Res. 2000;126:331–339. [Google Scholar]
- 19.Liu L, Fang Y, Li M, Wang C. Prediction of beta-turn in protein using E-SSpred and support vector machine. Protein J. 2009;28:175–181. doi: 10.1007/s10930-009-9181-4. [DOI] [PubMed] [Google Scholar]
- 20.Matthews BW. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochem Biophys Acta. 1975;405:442–451. doi: 10.1016/0005-2795(75)90109-9. [DOI] [PubMed] [Google Scholar]
- 21.McGregor MJ, Flores TP, Sternberg MJE. Prediction of β-turns in proteins using neural network. Protein Eng. 1989;2:521–526. doi: 10.1093/protein/2.7.521. [DOI] [PubMed] [Google Scholar]
- 22.Meissner M, Koch O, Klebe G, Schneider G. Prediction of turn types in protein structure by machine-learning classifiers. Proteins. 2009;74:344–352. doi: 10.1002/prot.22164. [DOI] [PubMed] [Google Scholar]
- 23.Noguchi T, Matsuda TH, Akiyama Y. PDB_REPRDB: A database of representative protein chains from the Protein Data Bank (PDB) Nucleic Acids Res. 2001;29:219–220. doi: 10.1093/nar/29.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petersen B, Lundegaard C, Petersen TN. NetTurnP - Neural network prediction of Beta turns by use of evolutionary information and predicted protein sequence features. PloS One. 2010;5(11) doi: 10.1371/journal.pone.0015079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pham TH, Satou K, Ho TB. Prediction and analysis of β-turns in proteins by support vector machine. Genome Inform. 2003;14:196–205. [PubMed] [Google Scholar]
- 26.Pham TH, Satou K, Ho TB. Support vector machines for prediction and analysis of beta and gamma-turns in proteins. J Bioinform Comput Biol. 2005;3:343–358. doi: 10.1142/s0219720005001089. [DOI] [PubMed] [Google Scholar]
- 27.Richardson JS. The anatomy and taxonomy of protein structure. Adv Protein Chem. 1981;34:167–339. doi: 10.1016/s0065-3233(08)60520-3. [DOI] [PubMed] [Google Scholar]
- 28.Rose GD, Gierasch LM, Smith JA. Turns in peptides and proteins. Adv Protein Chem. 1985;37:100–109. doi: 10.1016/s0065-3233(08)60063-7. [DOI] [PubMed] [Google Scholar]
- 29.Shepherd AJ, Gorse D, Thornton JM. Prediction of the location and type of β-turns in proteins using neural networks. Protein Sci. 1999;8:1045–1055. doi: 10.1110/ps.8.5.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi X, Hu X, Li S, Liu X. Prediction of β-turn types in protein by using composite vector. J Theor Biol. 2011;286:24–30. doi: 10.1016/j.jtbi.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 31.Takano K, Yamagata Y, Yutani K. Role of amino acid residues at turns in the conformational stability and folding of human lysozyme. Biochemistry. 2000;39:8655–8665. doi: 10.1021/bi9928694. [DOI] [PubMed] [Google Scholar]
- 32.Tang Z, Li T, Liu R, Xiong W, Sun J, Zhu Y, et al. Improving the performance of β-turn prediction using predicted shape strings and a two-layer support vector machine model. BMC Bioinformatics. 2011;12:283. doi: 10.1186/1471-2105-12-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilmot CM, Thornton JM. Analysis and prediction of the different types of β-turns in proteins. J Mol Biol. 1988;203:221–232. doi: 10.1016/0022-2836(88)90103-9. [DOI] [PubMed] [Google Scholar]
- 34.Zhang CT, Chou KC. Prediction of β-turns in proteins by 1-4 & 2-3 correlation model. Biopolymers. 1997;41:673–702. [Google Scholar]
- 35.Zhang Q, Yoon S, Welsh WJ. Improved method for predicting β-turn using support vector machine. Bioinformatics. 2005;21:2370–2374. doi: 10.1093/bioinformatics/bti358. [DOI] [PubMed] [Google Scholar]
- 36.Zheng C, Kurgan L. Prediction of beta-turns at over 80 % accuracy based on an ensemble of predicted secondary structures and multiple alignments. BMC Bioinformatics. 2008;9:430. doi: 10.1186/1471-2105-9-430. [DOI] [PMC free article] [PubMed] [Google Scholar]