

Abstract
Experimental studies show that detrimental effects of ischemia-reperfusion (I/R) injury can be attenuated by hyperoxic preconditioning in normal hearts, however, there are few studies about hyperoxia effects in diseased myocardium. The present study was designed to assess the cardioprotective effects of hyperoxia pretreatment (≥ 95 % O2) in acute diabetic rat hearts. Normal and one week acute diabetic rats were either exposed to 60 (H60) and 180 (H180) min of hyperoxia or exposed to normal atmospheric air (21 % O2). Then hearts were isolated immediately and subjected to 30 min of regional ischemia followed by 120 min of reperfusion. Infarct size, cardiomyocyte apoptosis, enzymes release and ischemia induced arrhythmias were determined. Heart of diabetic control rats had less infarct size and decreased LDH and CK-MB release compared to normal hearts. 60 and 180 min of hyperoxia reduced myocardial infarct size and enzymes release in normal hearts. 180 min of hyperoxia also decreased cardiomyocytes apoptosis in normal state. On the other hand, protective values of hyperoxia were not significantly different in diabetic hearts. Moreover, hyperoxia reduced severity of ventricular arrhythmias in normal rat hearts whereas; it did not confer any additional antiarrhythmic protection in diabetic hearts. These findings suggest that diabetic hearts are less susceptible to ischemia-induced arrhythmias and infarction. Hyperoxia greatly protects rat hearts against I/R injury in normal hearts, however, it could not provide added cardioprotective effects in acute phase of diabetes.
Keywords: hyperoxia, diabetes, infarct size, apoptosis, arrhythmias
Introduction
Ischemic preconditioning (IPC) is an endogenous phenomenon in which brief periods of ischemia and reperfusion (I/R) render a tissue more resistant to subsequent severe ischemic injury (Murry et al., 1986[13]). Beneficial effects of IPC have been demonstrated in a variety of organs including the heart (Rajesh et al., 2004[15]). Preconditioning (PC) can also be induced by several other stimuli, such as hypoxia (Cohen et al., 1995[4]; Neckar et al., 2002[14]), oxidative stress (Baines et al., 1997[1]), heat stress (Yamashita et al., 1997[28]) and lipopolysaccharides (Rowland et al., 1997[20]). Most of these stimuli, however, lack potential for clinical application due to associated complication.
Pre-exposure to hyperoxia was shown to induce protective effects against ischemia-reperfusion (I/R) injury in different tissues, including the heart (Bigdeli et al., 2007[2]; Esmaili Dehaj et al., 2009[6]; Rasoulian et al., 2008[16]), perhaps through the generation of oxygen free radicals and induction of low graded oxidative stress (Tiravanti et al., 2007[24]; Zhang et al., 2004[29]). These beneficial effects of hyperoxia have been proved clearly in normal myocardium (Tahepold et al., 2003[23]; Tiravanti et al., 2007[24]), however little is known whether mechanisms of hyperoxic preconditioning are operating in pathologically altered myocardium. The easy clinical application of hyperoxia draws the attention with the development of its basic research especially in pathologic status of complicating disease such as diabetes.
Diabetes mellitus is a major risk factor for myocardial infarction and death and leads to an increased incidence of myocardial ischemia as well as mortality following ischemia (Wang and Chatham, 2004[27]). Accordingly, reducing the consequences of coronary artery disease using strategies that target I/R injury would be particularly valuable in diabetic population. Therefore, the current study was designed to examine the beneficial effects of in vivo hyperoxia pre-exposure against I/R injuries in streptozotocin-induced acute diabetic rat heart.
Materials and Methods
Animals and study design
Male Wistar rats (250-350 g) fed a standard diet and tap water ad libitum and housed at 12:12-h light-dark cycle in a stress-free environment. The experimental protocols were approved by TarbiatModares University Ethics Committee for animal research.
The present study was performed in two experiments, normal (N) and diabetic (D) condition. Three groups of animals (7-11 rats in every group) were assessed in each experiment including control group (C), 60 min hyperoxia pretreatment group (H60) and 180 min hyperoxia pretreatment group (H180). Animals in hyperoxia groups were kept in a hyperoxic chamber (≥ 95 % O2), while non-diabetic and diabetic control animals were kept in the same chamber breathing normal atmospheric air (21 % O2). Oxygen was continuously delivered at a rate of 0.5-1 l/min into the chamber. The percentage of oxygen was continuously monitored with an oxygen meter (Lutron-DO 5510, Taiwan). Immediately after pretreatment with normoxia or hyperoxia, the hearts were excised for Langendorff perfusion. After a stabilization time of 20 min, the hearts were exposed to 30 min of regional ischemia, followed by 120 min of reperfusion in all experiments.
Induction of diabetes
Diabetes was induced by a single injection of streptozotocin (50 mg/kg, i.v.) diluted in buffer solution (0.1 M citrate buffer, pH 4.5). Development of the diabetes was confirmed by enhanced blood glucose levels (400-600 mg/dl). One week following induction of the diabetes, the animals were subjected to the experimental protocol.
Perfusion technique
Rats were anaesthetized (pentobarbital sodium 60 mg/kg, i.p.) and heparinized (300-400 IU, i.p.). Hearts were rapidly excised, placed in ice-cold Krebs-Henseleit buffer, cannulated via the aorta and perfused by the Langendorff method. Epical electrocardiogram (ECG) and left ventricular pressure were continuously recorded during the ischemia and reperfusion using a PowerLab analog to digital converter (AD Instruments, Australia). Coronary flow (CF) was measured by timed collections of the coronary effluent. Left ventricular systolic (LVSP) and end-diastolic (LVEDP) pressures were obtained by a latex water-filled balloon inserted into the left ventricle via the left atrium and connected to a pressure transducer (MLT 844). At the end of stabilization period, the volume of the balloon was adjusted to obtain end-diastolic pressure of 5-7 mm Hg and was unchanged for the remainder of the experiment. Left ventricular developed pressure (LVDP) was calculated as "LVSP-LVEDP". Rate pressure product (RPP) as an index of cardiac function was calculated by multiplying LVDP with heart rate (HR).
Induction of ischemia and reperfusion
A 5-0 silk suture was loosely placed under the left anterior descending coronary artery (LAD) 2 to 3 mm from its origin by inserting the needle into the left ventricular wall. Two ends of the suture were threaded through a 10 mm segment of sampler tip. Tightening and loosening this snare allowed the coronary artery occlusion and reperfusion, respectively (Curtis, 1998[5]).
Determination of infarct size
At the end of 120 min reperfusion, the coronary artery was re-occluded, and the area at risk (AAR) was delineated by perfusing 1 ml of 2 % Evans blue solution into the aortic cannula. After freezing at -20 °C, hearts were cut into transverse slices of 2 mm thickness from apex to base and slices were stained in 1 % triphenyltetrazolium chloride (TTC, Sigma) at 37 °C for 20 min. The slices were then photographed by a digital camera (Olympus, FE-160). AAR and infarct size were determined by computerized planimetry using image analysis software (Image Tool). Infarct size was calculated as percentage of AAR.
In situ nick end labeling
To determine myocardial apoptosis in a quantitative manner, five rats in each group were studied in an additional experiment. At the end of reperfusion, the hearts were washed with 0.9 % saline for 5 min , followed by 4 % paraformaldehyde in 0.1 M phosphate buffer saline (PBS, pH 7.4) for 10 min. AAR was cut and further fixed in 4 % paraformaldehyde in PBS for 48 h at room temperature. Fixed tissues were then embedded in a paraffin block. TUNEL staining was performed according to the manufacturer's protocol (Roche, Germany). In brief, 4-μm thick sections of the heart blocks were deparaffinized by two xylene wash and rehydrated by a descending ethanol series and then washed with phosphate-buffered saline. The sections were subsequently incubated with 20 μg/ml proteinase K for 15 min at room temperature, and endogenous peroxidase was inactivated by the treatment with 3 % hydrogen peroxide in methanol for 10 min. After 3 times wash with PBS and drying, sections were incubated with 50 μl of TUNEL reaction mixture, containing terminal deoxynucleotidyltransferase(TdT) and fluorescein-dUTP for 1 h at 37 °C. After end labeling was completed, the sections were washed with PBS and incubated with anti-fluorescein antibody conjugated with POD for 30 min at 37 °C. For the color development, 100 μl DAB was applied to the sections for 15 min at room temperature. Finally, the sections were counterstained with hematoxylin. After the procedures, 1000 nuclei in different regions in the area at risk were examined by light microscopy at a magnification of 400, and the number of TUNEL-positive myocyte nuclei (dark brown color) were counted and expressed as a percentage of the total number of myocyte nuclei in the AAR (% Apoptosis).
Estimation of lactate dehydrogenase (LDH) and creatine kinase (CK-MB) release
LDH and CK-MB activities were determined spectrophotometrically (TECHNICON RA-1000) using standard kits (Pars Azmun, Iran) for assessment of myocardial damage. The enzyme activities were expressed as units per liter.
Quantification of arrhythmias
Arrhythmias were defined according to the Lambeth Conventions (Walker et al., 1988[26]). Accordingly, arrhythmias were categorized as premature ventricular beats (PVBs, defined as discrete premature ventricular depolarization), ventricular tachycardia (VT, a run of four or more consecutive ventricular premature beats) and ventricular fibrillation (VF, a signal where individual QRS deflections could not easily be distinguished from each other). More complex forms of premature ventricular beats (bigeminy and salvos) were included in the count of ventricular premature beats and were not analyzed separately.
Electrograms were analyzed for (1) the total number of premature ventricular beats (TPVBs) and (2) the incidence and duration of ventricular tachycardia and ventricular fibrillation during 30 min of ischemia. In addition, severity of arrhythmias was quantified by a scoring system. Each individual heart was evaluated by means of a 5-point arrhythmia score, where single PVBs were given a score of 1, bigeminy/salvos a score of 2, VT a score of 3, VF a score of 4, sustained VF (VF lasting more than two minutes) a score of 5 and an assigned number corresponded to the most severe type of arrhythmia observed in that heart. Scores were used for group analysis of severity of arrhythmias.
Statistics
Data were expressed as means ± SEM. The incidence of arrhythmias was presented as percentage of occurrence. Differences in the recovery of postischemic functional parameters, CK-MB and LDH were tested by two-way analysis of variance (ANOVA) with repeated measures on one factor, taking treatment as an independent factor and time as a dependent factor. Comparisons of infarct size, myocyte apoptosis and arrhythmias number were performed by one-way ANOVA with Duncanposthoc analysis. Differences in the arrhythmias score, VT and VF episodes number and duration between the groups were compared by the Mann-Whitney U test. P<0.05 was considered statistically significant.
Results
Cardiac function parameters
As shown in Table 1(Tab. 1), the baseline functional parameters including HR, LVDP, RPP and CF were not significantly different between the control and hyperoxia pretreatment groups both in normal and diabetic conditions. During reperfusion, 60 and 180 min of hyperoxia significantly improved coronary flow in normal hearts compared to respective control group (p<0.05 and p<0.001 respectively) whereas, there was no difference in values of diabetic hearts pretreated with hyperoxia versus diabetic control group. No important differences were found between normal and diabetic controls with respective hyperoxia treated groups regarding the values of HR, LVDP and RPP during reperfusion. The heart functional parameters were not changed in control diabetic hearts compared to control normal ones.
Table 1. Haemodynamic measurements in Langendorff-perfused rat hearts subjected to 30 min of regional ischemia and 120 min of reperfusion.

Myocardial infarct size
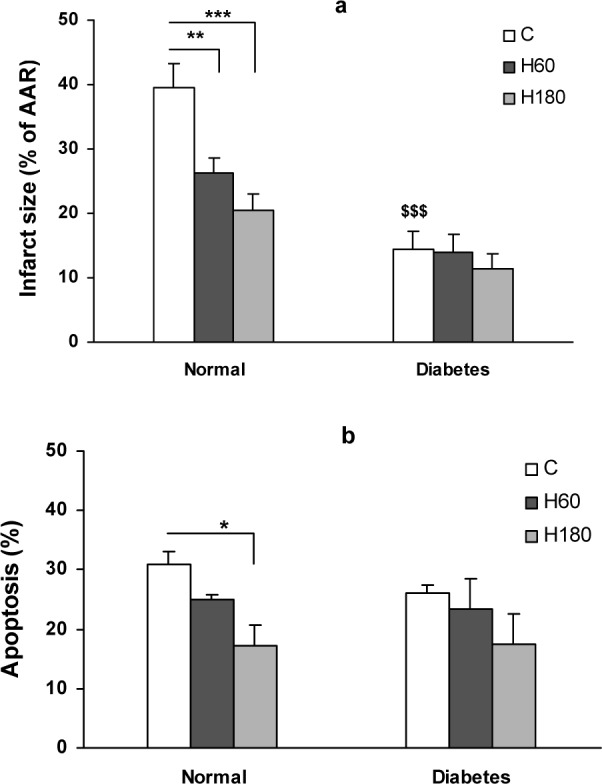
In the control normal group 39.5 ± 3.6 % of AAR was necrotic at the end of 2 h reperfusion. Pretreatment with 60 and 180 min hyperoxia reduced infarct size to 26.3 ± 2.3 (p<0.05) and 20.4 ± 2.6 % (p<0.01) respectively. There was no difference between two normal hyperoxia treated groups. Infarct size was also decreased in diabetic control hearts compared to normal control ones (14.5 ± 2.8 vs. 39.5 ± 3.6 %, p<0.001). Hyperoxia for 60 and 180 min did not cause further reduction in infarct size in the diabetic animals (Figure 1a(Fig. 1)).
Figure 1. Figure1: (a) Infarct size expressed as percentage of area at risk (AAR) in normal and diabetic rat hearts after 30 min ischemia and 120 min of reperfusion. (b) Percentage of TUNEL positive nuclei in AAR of left ventricle in non-diabetic and diabetic rat hearts. Data presented as mean ± SEM. *p<0.05, **p<0.01 and *** p<0.001 compared to respective control group. $$$ p<0.001, diabetic control vs. normal control group.
Myocytes apoptosis
Total numbers of myocyte nuclei were counted in the AAR of histological sections obtained from each heart. The TUNEL positive nuclei were observed only in the AAR and the number of TUNEL positive nuclei in nonrisk region of the heart was sparse. The percentage of TUNEL positive nuclei was significantly reduced by 180 min of hyperoxia pretreatment in normal hearts (17.5 ± 3.5 % in H180 vs. 31 ± 2.3 % of control group, p<0.05), showing an attenuating effect of normobarichyperoxia pre-exposure against I/R induced apoptosis. In contrast, number of TUNEL positive nuclei in the area at risk region of diabetic hearts was not significantly changed by hyperoxia pretreatment (Figure 1b(Fig. 1)).
Lactate dehydrogenase (LDH) and creatine kinase (CK-MB) leakage
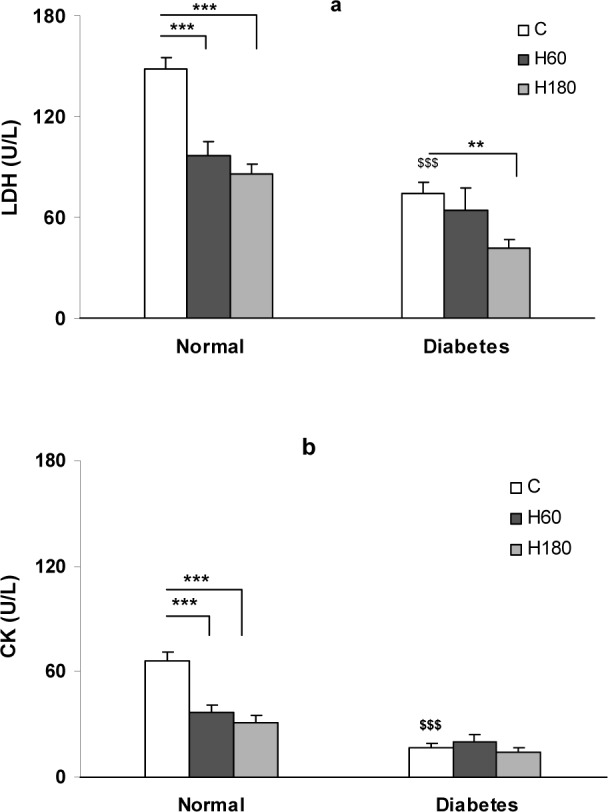
The extent of reperfusion induced injury was determined by measuring the extent of CK and LDH enzymes release into the coronary effluent (Figure 2a, b(Fig. 2)). In normal control group the values of LDH and CK release into the effluent at reperfusion were 148 ± 7 and 66 ± 5 U/L respectively. Pretreatment with hyperoxia, remarkably decreased the amount of LDH and CK release to 97 ± 8 and 37 ± 4 U/L in H60 and to 86 ± 6 and 31 ± 4 U/L in H180 groups respectively (p<0.001). Induction of diabetes, decreased the amount of LDH and CK release to 73.91 ± 6.71 and 16.96 ± 2.13 vs. 148 ± 7 and 66 ± 5 U/L in normal control group, p<0.001). In the diabetic hearts, 180 min hyperoxia reduced the extent of LDH release in the coronary effluent (41.62 ± 4.95 vs. 73.91 ± 6.71 U/L, p<0.01) whereas, hyperoxia could not significantly change the extent of CK-MB release in diabetic hearts.
Figure 2. LDH (a) and CK (b) release in coronary effluent of control and hyperoxia pretreatment groups in normal and diabetic hearts. Data presented as mean ± SEM. ** p<0.01 and *** p<0.001 compared to respective control group. $$$ p<0.001, diabetic control vs. normal control group.
Ischemia-induced arrhythmias
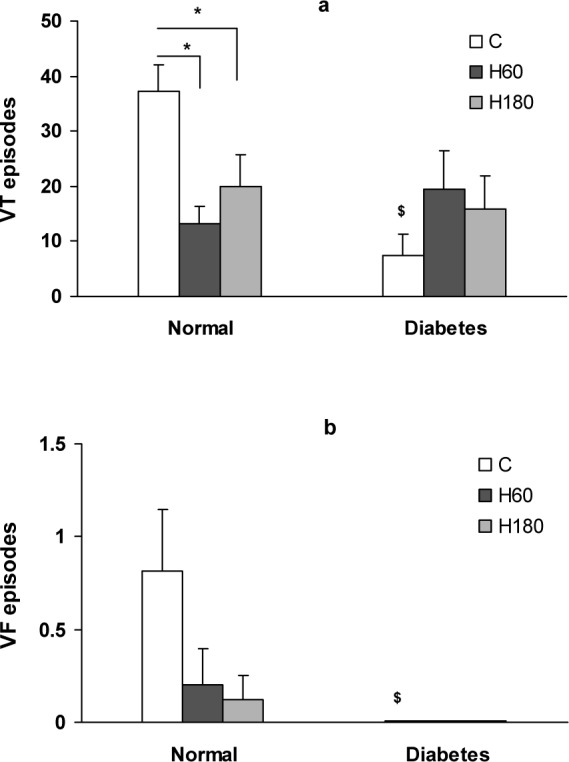
Occlusion of LAD coronary artery caused severe ventricular arrhythmias. The duration of arrhythmias was affected by hyperoxia. In normal hearts, the total duration of VT and VF was significantly reduced by 60 and 180 min of hyperoxia pretreatment (22.39 ± 5.96 and 24.65 ± 19.28 s respectively vs. 63.36 ± 7.56 s in control, p<0.05). Diabetic hearts showed an enhanced resistance to ischemia induced arrhythmias and the total duration of VT and VF was markedly suppressed (7.56 ± 3.78 vs. 63.36 ± 16.24 in normal group, p<0.001). The effect of hyperoxia was not evident in diabetic hearts (Figure 3(Fig. 3)). Number of VT and VF episodes was decreased by both 60 and 180 min of hyperoxia pretreatment in normal hearts (Figure 4a, b(Fig. 4)). Induction of diabetes was accompanied by a decrease in number of VT episodes (Figure 4a(Fig. 4)) and completely abolished VF incidence and episodes number (Figure 4b(Fig. 4)). Number of VT episodes did not significantly change by hyperoxia in diabetic hearts (Figure 4a(Fig. 4)) and VF episodes in diabetic hyperoxia groups remained zero (Figure 4b(Fig. 4)).
Figure 3. Total sum of VT and VF duration during 30 min ischemia. Data presented as mean ± SEM. *p<0.05 compared to normal control group. $$$ p<0.001, diabetic control vs. normal control group.
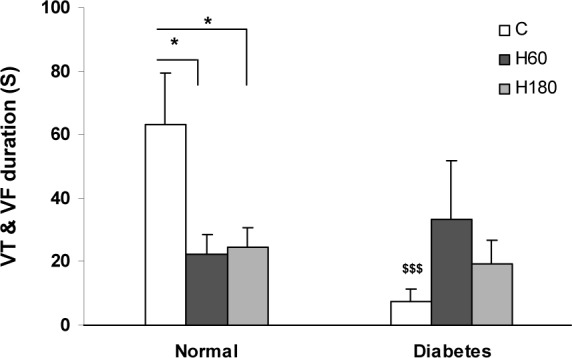
Figure 4. Number of VT (a) and VF (b) episodes during 30 min ischemia. Data presented as mean ± SEM. *p<0.05 compared to normal control group. $ p<0.05, diabetic control vs. normal control group.
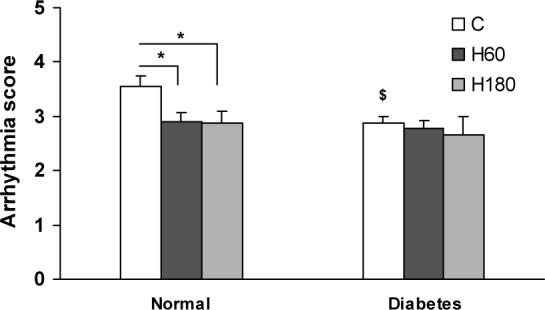
Severity of ischemia induced arrhythmias (evaluated by means of arrhythmia score) was also assessed. 60 and 180 min of hyperoxia pretreatment reduced arrhythmia score compared to normal group (2.9 ± 0.18 and 2.87 ± 0.22 vs. 3.54 ± 0.20 respectively, p<0.05). In diabetic control group severity was significantly lower compared to normal (2.87 ± 0.12 vs. 3.54 ± 0.20, p<0.05). Hyperoxia, however, failed to reduce the severity of arrhythmias in diabetic hearts (Figure 5(Fig. 5)).
Figure 5. Arrhythmias severity evaluated by means of arrhythmia score. Data presented as mean ± SEM. *p<0.05 compared to normal control group. $ p<0.05, diabetic control vs. normal control group.
Discussion
The first finding of the present study is that one week acute diabetes led to a great cardioprotection by reducing infarct size and severity of ischemic arrhythmias. However, hyperoxia pretreatment fails to confer additional protection in diabetic hearts. In additionhyperoxia induced significant protective effect in normal rats, in accordance with previous studies (Choi et al., 2006[3]; Esmaili Dehaj et al., 2009[6]).
Hyperoxic preconditioning is thought to have useful cardioprotective effects against I/R injury. Simplicity and easy clinical applicability of O2 pretreatment cause it to become a potential clinical preconditioning tool in the near future (Tahepold et al., 2002[22]). However, it remains unknown whether hyperoxic preconditioning is still effective when certain co-morbidities accompany the ischemic heart disease. Thus, our study has investigated whether the beneficial effects of hyperoxic preconditioning can be preserved in diabetes which is a common clinical condition and predisposes to coronary artery disease (Monteiro et al., 2005[11]).
Although results obtained from clinical studies of diabetic cardiomyopathy suggests an increased risk of myocardial infarction and a rate of mortality, experimental results are not consistent to each other and point out to increased, unchanged or decreased susceptibility to ischemia-reperfusion injury in different experimental conditions (Ravingerova et al., 2000[18]).
The results of the present study show that one week of diabetes could not significantly alter the cardiac function parameters compared to non-diabetic control group, whereas infarct size and severity of ischemic arrhythmias were markedly reduced by diabetes. These results are consistent with the results obtained by other researchers. Ravingerova et al. (2003[17], 2000[19]) have shown that acute diabetes markedly suppresses ischemic arrhythmias and infarct size in rat heart models. In accord, Tosaki et al. (1996[25]) have demonstrated a reduction in the incidence of reperfusion-induced arrhythmias in the rat heart in the early phase of diabetes. Other studies of short term STZ-induced diabetes in rats have reported decreased apoptotic index as indicated by reduced cleaved caspase-3 as a marker of apoptosis (Ma et al., 2006[9]). Our data also showed that percent of apoptotic myocytes tends to decrease (non-significant) in area at risk of diabetic hearts compared normal ones. This result is consistent with the reduction in infarct size in the diabetic rats. Taken together, these findings indicate that acute diabetes activates some of cardioprotective mechanisms, which in turn reduce ischemia-reperfusion injuries.
One of the proposed mechanisms of increased heart tolerance to injury in diabetic condition is the concept of metabolic preconditioning. According to this, the metabolic and secondary molecular and cellular changes which occur in cardiomyocytes after diabetes might be responsible for enhanced myocardial resistance to ischemia (Hadour et al., 1998[8]). Diabetes decreases rate of glycolysis, and alleviates degree of intracellular acidosis in ischemia (Feuvray and Lopaschuk, 1997[7]). Moreover it has been shown that type 1 diabetes decreases activity of sarcolemmal Na+/Н+ exchanger which in turn, may lead to limitation of Ca2+ inward current through Na+/Ca2+ exchanger in cardiomyocytes, and therefore may retard the irreversible I/R injury (Feuvray and Lopaschuk, 1997[7]).
On the other hand, increased myocardial tolerance to I/R injury in diabetes may be due to difference in hemodynamic parameters between control and diabetic animals. It is known that lower values of blood pressure and heart rate are associated with smaller cardiac performance and, hence, smaller myocardial oxygen demand (Ma et al., 2006[9]) however, this factor was disregarded in present study, since there were no differences between non-diabetic control and diabetic animals in hemodynamic parameters.
The most important finding of the present study was the different response of normal and diabetic hearts to hyperoxic preconditioning. Until now studies related to the role of hyperoxia in heart preconditioning have only been performed in normal condition. In this study, 60 and 180 min of hyperoxia significantly reduced infarct size and ischemia-induced arrhythmias in the non-diabetic hearts, while it did not confer any additional protection in one-week diabetic rat hearts. Decreased protective value of hyperoxic preconditioning seen in diabetic hearts may be due to the great protective effect induced by diabetes itself and this cardioprotective effect which is already induced by diabetes, could potentially mask the additional protective value of hyperoxic preconditioning seen in normal hearts.
Studies demonstrate that in both conditions common protective mechanisms, such as activation of protein kinase C (Choi et al., 2006[3]), as well as opening of KATP channels (Tiravanti et al., 2007[24]) have been implicated. PKC activation of and KATP channels opening play major roles in ischemic preconditioning. It has been suggested that alteration in the activity of these protective mediators in acute phase of diabetes induce heart protection (Malhotra et al., 1997[10]; Moon et al., 1999[12]; Smith and Wahler, 1996[21]). Thus the protective effects, which were seen in the hyperoxia pretreated diabetic hearts, may be due to the effects of diabetes, hyperoxia or combination of both. Although possible beneficial effects of hyperoxia against I/R injury in diabetic hearts were not clear in the present study, we could not conclude that hyperoxic exposure had not any effects in diabetic rat hearts. It may be due to limitation of the current study including, the protocol or duration of hyperoxic gas exposure and/or duration of diabetes.
In conclusion, the results of the present study show that normobarichyperoxia pretreatment reduces infarct size, myocytes apoptosis and protects the heart against ischemic arrhythmias. Early period of diabetes is associated with an increased myocardial resistance to cell damage and decreased severity of ischemic arrhythmias. Hyperoxia does not exert obvious additional cardioprotective effect in acute diabetic hearts.
Acknowledgement
This work was supported by a research grant by Tarbiat Modares University.
References
- 1.Baines CP, Goto M, Downey JM. Oxygen radicals released during ischemic preconditioning contribute to cardioprotection in the rabbit myocardium. J Mol Cell Cardiol. 1997;29:207–216. doi: 10.1006/jmcc.1996.0265. [DOI] [PubMed] [Google Scholar]
- 2.Bigdeli MR, Hajizadeh S, Froozandeh M, Rasulian B, Heidarianpour A, Khoshbaten A. Prolonged and intermittent normobaric hyperoxia induce different degrees of ischemic tolerance in rat brain tissue. Brain Res. 2007;1152:228–233. doi: 10.1016/j.brainres.2007.03.068. [DOI] [PubMed] [Google Scholar]
- 3.Choi H, Kim SH, Chun YS, Cho YS, Park JW, Kim MS. In vivo hyperoxic preconditioning prevents myocardial infarction by expressing bcl-2. Exp Biol Med. 2006;231:463–472. doi: 10.1177/153537020623100412. [DOI] [PubMed] [Google Scholar]
- 4.Cohen MV, Walsh RS, Goto M, Downey JM. Hypoxia preconditions rabbit myocardium via adenosine and catecholamine release. J Mol Cell Cardiol. 1995;27:1527–1534. doi: 10.1016/s0022-2828(95)90293-7. [DOI] [PubMed] [Google Scholar]
- 5.Curtis MJ. Characterisation, utilisation and clinical relevance of isolated perfused heart models of ischaemia-induced ventricular fibrillation. Cardiovasc Res. 1998;39:194–215. doi: 10.1016/s0008-6363(98)00083-2. [DOI] [PubMed] [Google Scholar]
- 6.Esmaili Dehaj M, Baharvand B, Rasoulian B, Foadaddini M, Asgari A, Noroozzadeh A, et al. Delayed protective effects of hyperoxia against cardiac arrhythmias and infarction in anesthetized rats. J Surg Res. 2009;151:55–61. doi: 10.1016/j.jss.2007.12.802. [DOI] [PubMed] [Google Scholar]
- 7.Feuvray D, Lopaschuk GD. Controversies on the sensitivity of the diabetic heart to ischemic injury: the sensitivity of the diabetic heart to ischemic injury is decreased. Cardiovasc Res. 1997;34:113. doi: 10.1016/s0008-6363(97)00037-0. [DOI] [PubMed] [Google Scholar]
- 8.Hadour G, Ferrera R, Sebbag L, Forrat R, Delaye J, de Lorgeril M. Improved myocardial tolerance to ischaemia in the diabetic rabbit. J Mol Cell Cardiol. 1998;30:1869–1875. doi: 10.1006/jmcc.1998.0751. [DOI] [PubMed] [Google Scholar]
- 9.Ma G, Al-Shabrawey M, Johnson JA, Datar R, Tawfik HE, Guo D, et al. Protection against myocardial ischemia/reperfusion injury by short-term diabetes: enhancement of VEGF formation, capillary density, and activation of cell survival signaling. NSArchPharmacol. 2006;373:415–427. doi: 10.1007/s00210-006-0102-1. [DOI] [PubMed] [Google Scholar]
- 10.Malhotra A, Reich D, Reich D, Nakouzi A, Sanghi V, Geenen D, et al. Experimental diabetes is associated with functional activation of protein kinase Cε and phosphorylation of troponin I in the heart, which are prevented by angiotensin II receptor blockade. Circ Res. 1997;81:1027–1033. doi: 10.1161/01.res.81.6.1027. [DOI] [PubMed] [Google Scholar]
- 11.Monteiro P, Goncalves L, Providencia LA. Diabetes and cardiovascular disease: the road to cardioprotection. Heart. 2005;91:1621–1625. doi: 10.1136/hrt.2005.063008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moon CH, Jung YS, Lee SH, Baik EJ. Protein kinase C inhibitors abolish the increased resistance of diabetic rat heart to ischemia-reperfusion injury. Jpn J Physiol. 1999;49:409–415. doi: 10.2170/jjphysiol.49.409. [DOI] [PubMed] [Google Scholar]
- 13.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 14.Neckar J, Papousek F, Novakova O, Ost'adal B, Kolar F. Cardioprotective effects of chronic hypoxia and ischaemic preconditioning are not additive. Basic Res Cardiol. 2002;97:161–167. doi: 10.1007/s003950200007. [DOI] [PubMed] [Google Scholar]
- 15.Rajesh KG, Sasaguri S, Suzuki R, Xing Y, Maeda H. Ischemic preconditioning prevents reperfusion heart injury in cardiac hypertrophy by activation of mitochondrial KATP channels. Int J Cardiol. 2004;96:41–49. doi: 10.1016/j.ijcard.2003.06.010. [DOI] [PubMed] [Google Scholar]
- 16.Rasoulian B, Mohammadhosseniakbari H, Kadkhodaee M, Mofid M, Baqeri G, Bigdeli MR, et al. Preconditioning with oxygen attenuates rat renal ischemia-reperfusion injury. J Surg Res. 2008;146:282–288. doi: 10.1016/j.jss.2007.04.034. [DOI] [PubMed] [Google Scholar]
- 17.Ravingerova T, Neckar J, Kolar F. Ischemic tolerance of rat hearts in acute and chronic phases of experimental diabetes. Mol Cell Biochem. 2003;249:167–174. doi: 10.1023/a:1024751109196. [DOI] [PubMed] [Google Scholar]
- 18.Ravingerova T, Stetka R, Pancza D, Ulicna O, Ziegelhoffer A, Styk J. Susceptibility to ischemia-induced arrhythmias and the effect of preconditioning in the diabetic rat heart. Physiol Res. 2000;49:607–616. [PubMed] [Google Scholar]
- 19.Ravingerova T, Stetka R, Volkovova K, Pancza D, Dzurba A, Ziegelhoffer A, et al. Acute diabetes modulates response to ischemia in isolated rat heart. Mol Cell Biochem. 2000;210:143–151. doi: 10.1023/a:1007129708262. [DOI] [PubMed] [Google Scholar]
- 20.Rowland RT, Meng X, Cleveland JC, Jr, Meldrum DR, Harken AH, Brown JM. LPS-induced delayed myocardial adaptation enhances acute preconditioning to optimize postischemic cardiac function. Am J Physiol. 1997;272:H2708–H2715. doi: 10.1152/ajpheart.1997.272.6.H2708. [DOI] [PubMed] [Google Scholar]
- 21.Smith JM, Wahler GM. ATP-sensitive potassium channels are altered in ventricular myocytes from diabetic rats. Mol Cell Biochem. 1996;158:43–51. doi: 10.1007/BF00225881. [DOI] [PubMed] [Google Scholar]
- 22.Tahepold P, Ruusalepp A, Li G, Vaage J, Starkopf J, Valen G. Cardioprotection by breathing hyperoxic gas-relation to oxygen concentration and exposure time in rats and mice. Eur J Cardiothorac Surg. 2002;21:987–994. doi: 10.1016/s1010-7940(02)00125-2. [DOI] [PubMed] [Google Scholar]
- 23.Tahepold P, Vaage J, Starkopf J, Valen G. Hyperoxia elicits myocardial protection through a nuclear factor kappaB-dependent mechanism in the rat heart. J ThoracCardiovascSurg. 2003;125:650–660. doi: 10.1067/mtc.2003.36. [DOI] [PubMed] [Google Scholar]
- 24.Tiravanti EA, Colantuono G, Di Venosa N, Cazzato A, D'Agostino D, Federici A, et al. Hyperoxia confers cardioprotection in rats through involvement of ROS and mitok-ATP channels opening. J Mol Cell Cardiol. 2007;42:S173. [Google Scholar]
- 25.Tosaki A, Engelman DT, Engelman RM, Das DK. The evolution of diabetic response to ischemia/reperfusion and preconditioning in isolated working rat hearts. Cardiovasc Res. 1996;31:526–536. [PubMed] [Google Scholar]
- 26.Walker MJ, Curtis MJ, Hearse DJ, Campbell RW, Janse MJ, Yellon DM, et al. The Lambeth Conventions: guidelines for the study of arrhythmias in ischaemia infarction, and reperfusion. Cardiovasc Res. 1988;22:447–455. doi: 10.1093/cvr/22.7.447. [DOI] [PubMed] [Google Scholar]
- 27.Wang P, Chatham JC. Onset of diabetes in Zucker diabetic fatty (ZDF) rats leads to improved recovery of function after ischemia in the isolated perfused heart. Am J Physiol. 2004;286:725–736. doi: 10.1152/ajpendo.00295.2003. [DOI] [PubMed] [Google Scholar]
- 28.Yamashita N, Hoshida S, Nishida M, Igarashi J, Aoki K, Hori M, et al. Time course of tolerance to ischemia-reperfusion injury and induction of heat shock protein 72 by heat stress in the rat heart. J Mol Cell Cardiol. 1997;29:1815–1821. doi: 10.1006/jmcc.1997.0416. [DOI] [PubMed] [Google Scholar]
- 29.Zhang X, Xiong L, Hu W, Zheng Y, Zhu Z, Liu Y, et al. Preconditioning with prolonged oxygen exposure induces ischemic tolerance in the brain via oxygen free radical formation. Can J Anaesth. 2004;51:258–263. doi: 10.1007/BF03019107. [DOI] [PubMed] [Google Scholar]