

Summary
Phosphorylation has been generally thought to activate the SR family of splicing factors for efficient splice-site recognition, but this idea is incompatible with an early observation that overexpression of an SR protein kinase, such as the CDC2-like kinase 1 (CLK1), weakens splice-site selection. Here we report that CLK1 binds SR proteins, but lacks the mechanism to release phosphorylated SR proteins, thus functionally inactivating the splicing factors. Interestingly, CLK1 overcomes this dilemma through a symbiotic relationship with the serinearginine protein kinase 1 (SRPK1). We show that SRPK1 interacts with an RS-like domain in the N-terminus of CLK1 to facilitate the release of phosphorylated SR proteins, which then promotes efficient splice-site recognition and subsequent spliceosome assembly. These findings reveal an unprecedented signaling mechanism by which two protein kinases fulfill separate catalytic features that are normally encoded in single kinases to institute phosphorylation control of pre-mRNA splicing in the nucleus.
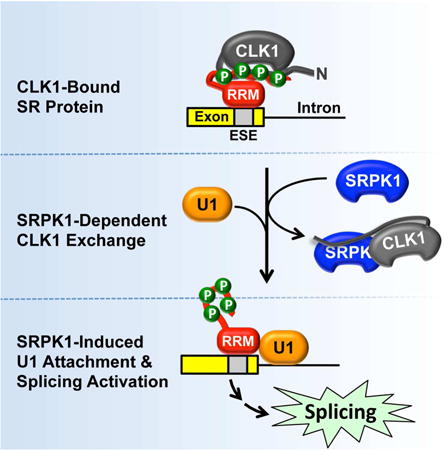
Graphical abstract
Introduction
Alternative mRNA splicing, prevalent in higher eukaryotic genomes, has the potential to singularly magnify the proteome size from a rather limited set of genes in development and disease (Nilsen and Graveley, 2010; Wang and Cooper, 2007). Splicing is tightly coupled with transcription, nuclear export and mRNA stability, thereby profoundly influencing the expression of gene products (Maniatis and Reed, 2002; Moore and Proudfoot, 2009; Pandit et al., 2008). Despite significant progress in the past two decades, we still have very limited knowledge of the molecular mechanisms underlying regulated splicing (Shin and Manley, 2004). Since up to 60% of disease-causing mutations are linked to defects in alternative splicing (Wang and Cooper, 2007), understanding of splicing mechanisms and their regulation continues to represent a major challenge in the post-genome era.
Recent studies have elucidated the role of cellular signaling in the regulation of alternative splicing (Fu and Ares, 2014; Zhou et al., 2012). Most important among different regulatory strategies are the protein kinases and phosphatases that control the activities of various splicing factors through reversible phosphorylation (Stamm, 2008). Protein kinases are key regulators of various cellular processes and have uniquely evolved mechanisms to carry out selective phosphorylation of their substrates. Their substrate selectivity has been attributed, in part, to the catalytic domain that recognizes preferred amino acids (Endicott et al., 2012; Taylor and Kornev, 2011). Protein kinases also encode modular domains in their gene structure that impart secondary binding surfaces for substrate selection as well as serve as signals for their sub-cellular localization and guidance toward physiological substrates (Parsons and Parsons, 2004; Taylor et al., 2012). These modular domains can bind to scaffold proteins that serve as platforms for both the protein kinases and their cognate substrates (Parsons and Parsons, 2004). Additionally, protein kinases have also adopted dimerization strategies for activation and substrate phosphorylation. Some protein kinases form homodimers (e.g. receptor tyrosine kinases) whereas a few form heterodimers (Lavoie et al., 2014; Lemmon and Schlessinger, 2010). The heterodimer assembly between inactivated B-RAF and active C-RAF activates the latter during MEK-ERK signaling (Poulikakos et al., 2010). RAF heterodimerization suggests a critical disease mechanism because such elegant arrangement was found in melanoma cells that develop resistance in the background of the most common B-Raf (V600E) mutation (Lavoie et al., 2014; Poulikakos et al., 2010).
Alternative splicing is facilitated by the reversible phosphorylation of the SR protein family of splicing regulators (Pandit et al., 2008). Two families of protein kinases specifically phosphorylate these essential splicing factors. The serine-arginine protein kinases (SRPK1-3) strictly phosphorylate Arg-Ser dipeptides whereas the CDC2-like kinases (CLK1-4) can phosphorylate both Arg-Ser and Ser-Pro dipeptides, common in all SR proteins (Ghosh and Adams, 2011) (Fig. 1A). Although SRPKs use a docking groove in the C-terminal lobe of the kinase domain for substrate recognition and phosphorylation, CLKs lack such a groove and instead use a disordered N-terminal domain to bind SR proteins with very high affinity (Fig. 1B).
Figure 1. SRPK1 and CLK1 form a nuclear complex.
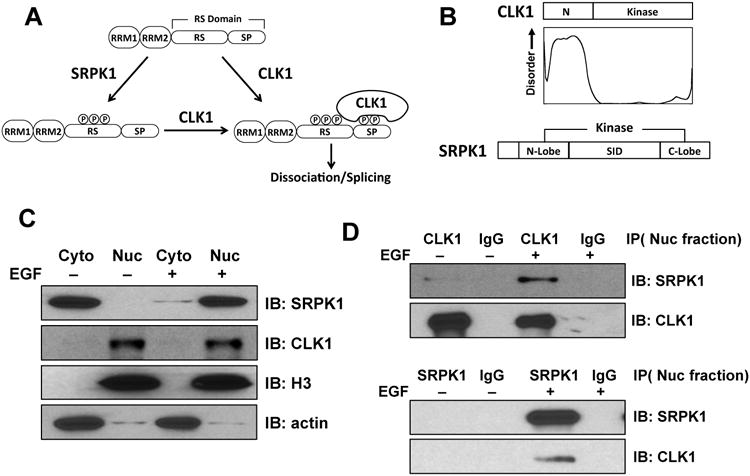
A) SRPK1 phosphorylates Arg-Ser repeats (RS) and CLK1 phosphorylates these and Ser-Pro dipeptides (SP) in the SRSF1 RS domain. B) SRPK1 and CLK1 domain structures. CLK1 N-Terminus is predicted to be disordered using DISOPRED. C) Cytoplasmic and nuclear fractionations of SRPK1 and CLK1 in HeLa cells with and without EGF stimulation. D) Co-immunoprecipitation of SRPK1 and CLK1 in nuclear lysates of HeLa cells.
Although SRPKs are located in both the cytoplasm and nucleus, their function in the cytoplasm is best understood. SRPKs support phosphorylation-dependent transport of SR proteins from the cytoplasm to the nucleus via an SR-specific transportin protein, TRN-SR2 (Kataoka et al., 1999; Lai et al., 2001; Yun et al., 2003). Unlike SRPKs, CLKs possess a nuclear localization signal in their N-termini, and thus, are localized strictly to the nucleus where they play a vital role in mobilizing SR proteins from speckles to sites of active gene splicing via Ser-Pro phosphorylation (Keshwani et al., 2015a). This has led to a simple model in which SRPKs generate basal SR protein phosphorylation levels in the cytoplasm and CLKs enhance phosphorylation in the nucleus for splicing regulation (Ding et al., 2006; Keshwani et al., 2015a). However, this simplistic relay mechanism fails to explain two long-standing issues regarding the role of phosphorylation in alternative splicing. First, while phosphorylation is thought to enhance the activity of SR proteins in splice-site recognition, high levels of CLK paradoxically inhibit this function (Prasad et al., 1999). Second, as both SRPKs and CLKs are also present in the nucleus, this sets up a potential competitive rather than a cooperative relationship because we recently found that although SRPK has built-in sequences that assist in the release of SR proteins after phosphorylation (Aubol et al., 2014; Koizumi et al., 1999), CLK does not release fully phosphorylated SR proteins (Aubol et al., 2014). These findings also suggest that CLKs may require a release factor to generate functional SR proteins for splicing function.
EGF signaling increases nuclear levels of SRPKs and enhances bulk SR protein phosphorylation (Zhou et al., 2012). This mechanism involves the Akt-dependent release of several chaperones (Hsp70/90) from the spacer insert domain (SID) in SRPK1 that pins the kinase in the cytoplasm (Zhong et al., 2009) (Fig. 1B). Such signaling affects the alternative splicing of numerous genes and hints to a role for SRPKs in the nucleus. Furthermore, osmotic stress increases nuclear SRPK levels by also shedding several chaperones suggesting that multiple signals converge on this cytoplasmic kinase complex (Zhong et al., 2009; Zhou et al., 2012). Given the presence of SRPKs in the same compartment as the CLKs, we wish to further delineate the unique function of SRPKs in the nucleus. We here report that endogenous SRPK1 and CLK1 interact in the nucleus forming a complex, and surprisingly, rather than acting in a competitive manner, the dual kinase complex acts symbiotically to associate with and phosphorylate their substrates with SRPK1 functioning as a release factor for CLK1 unleashing phosphorylated SR proteins for the promotion of spliceosome assembly. These findings solve some major puzzles from previous studies, and more importantly, reveal a signaling paradigm where two protein kinases are required to catalyze a full phosphorylation cycle (from binding to release) of their substrates.
Results
SRPK1 Forms a Complex With CLK1 in Cells
SRPKs and CLKs have separate functions in the cell with regard to SR proteins. Whereas cytoplasmic SRPKs facilitate phosphorylation-dependent transport of SR proteins to the nucleus, CLKs perform additional phosphorylation steps that mobilize SR proteins for splicing activity in the nucleus (Keshwani et al., 2015a). What has gone unappreciated is that SRPKs are present not only in the cytoplasm but also in the nucleus (Colwill et al., 1996; Nayler et al., 1997; Wang et al., 1998). We showed in a previous study that, upon EGF stimulation, SRPK1 levels can be greatly increased in the nucleus where CLKs are localized (Zhou et al., 2012), an observation that we confirmed through fractionation studies (Fig. 1C). This raises the question of how the two kinases function in the same compartment. To begin to address this, we determined whether these kinases can interact with one another in the nucleus by co-immunoprecipitation using specific monoclonal antibodies. We found that anti-CLK1 efficiently co-immunoprecipitates with SRPK1 (Fig. 1D, top panel) and similarly, anti-SRPK1 can capture CLK1 (Fig. 1D, bottom panel) in EGF-treated HeLa cells. These findings indicate that CLK1 and SRPK1 have the capacity to form a stable complex in the nucleus.
CLK1 N-Terminus Interacts with the SRPK1 Kinase Domain
We showed previously that the CLK1 N-terminus binds with high affinity to the RS domain of its substrate SRSF1 (Aubol et al., 2014). Furthermore, we showed that the N-terminus also interacts with its kinase domain but is not a substrate, suggesting that it can bind different proteins in intra- and intermolecular fashions (Aubol et al., 2014; Colwill et al., 1996). To determine whether the CLK N-terminus also interacts with SRPK, we performed in vitro pull-down experiments using GST-tagged SRPK1 with either full-length His-tagged CLK1 or a form lacking its N-terminus (CLK1(ΔN)). We found that GST-SRPK1 pulled down the full-length CLK1 but not CLK1(ΔN) (Fig. 2A,B). As controls, we showed that CLK1 and CLK1(ΔN) did not interact with the glutathione-agarose resin. These findings suggest that the N-terminus is the principle domain that stabilizes the CLKSRPK complex.
Figure 2. CLK1 N-terminus interacts with the SRPK1 kinase domain.
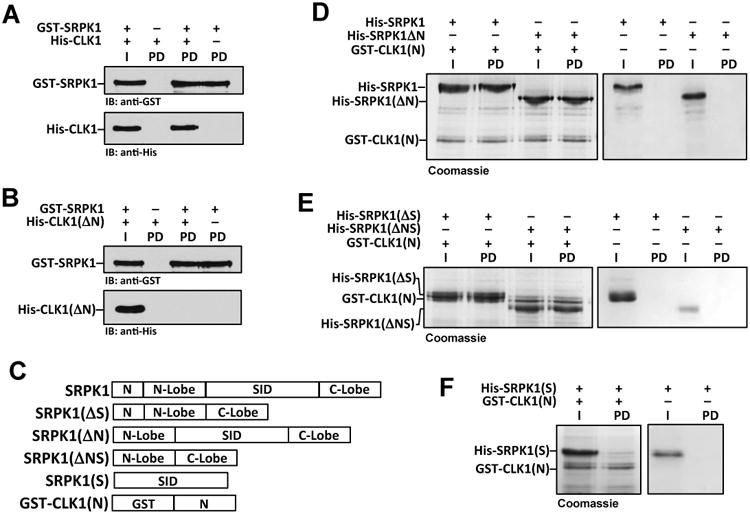
A,B) Pull downs of GST-SRPK1 and His-CLK1 (A) or His-CLK1(ΔN) (B). I = Input and PD = pull down. C) Deletion constructs of His-SRPK1 and GST-CLK1(N). D-F) Pull downs of His-SRPK1 deletions using GST-CLK1(N). I = Input and PD = pull down.
To determine how the N-terminus interacts with SRPK1, we performed a series of pull-down experiments using a GST-tagged form of the CLK1 N-terminus (GST-CLK1(N)) and several SRPK1 truncation mutants (Fig. 2C). We found that GST-CLK1(N) interacts strongly with SRPK1 and all truncations that include the kinase domain (Fig. 2D,E). In comparison, GST-CLK1(N) did not pull down SRPK1(S), suggesting that the N-terminus does not form a stable complex with the spacer insert domain (SID) (Fig. 2F). As controls, we showed that all SRPK1 forms did not interact with glutathione-agarose resin without GST-CLK1(N) (Fig. 2D-F). Together, these results suggest that the CLK1 N-terminus interacts with the SRPK1 kinase domain.
CLK1 Regulates Nuclear Levels of SRPK1
Although prior studies showed that SRPKs are maintained in the cytoplasm through chaperone binding (Hsp70/90) (Zhong et al., 2009), it is unclear whether there are nuclear constituents that help maintain nuclear pools of the kinase. We addressed whether complex formation with CLK anchors SRPK1 in the nucleus. By monitoring endogenous SRPK1 in HeLa cells using confocal microscopy, we found SRPK1 predominantly in the cytoplasm with a small fraction in the nucleus (Fig. 3A), consistent with our fractionation studies and previous findings (Gui et al., 1994). We then transfected an RFP-tagged CLK1 (CLK1-RFP) that expressed to an observed level 6 times that of the endogenous kinase and found that it increased the nuclear SRPK1 levels relative to the cytoplasmic pools (Fig. 3B,E). This result suggests that the SRPK1 cytoplasmic/nuclear levels are controlled by an equilibrium set by chaperones in the cytoplasm and CLK1 in the nucleus. Expressed CLK1 shifts this equilibrium toward greater levels of nuclear SRPK1 in accordance with mass action. To determine whether this phenomenon is dependent on catalytic activity, we expressed a kinase inactive CLK1 (kdCLK1-RFP) and found that it also induced nuclear SRPK1 (Fig. 3C,E). To determine whether the N-terminal RS-like domain of CLK1 plays a role in nuclear retention of SRPK1, we expressed a version of CLK1 that replaces the N-terminus with a nuclear localization sequence from nucleoplasmin 2 (KRLVPQKQASVAKKKK) and found that this new construct, nCLK1ΔN-RFP, did not increase nuclear SRPK1 levels (Fig. 3D,E). CLK1-RFP, kdCLK1-RFP and nCLK1ΔN-RFP are all exclusively present in the nucleus, indicating that the increased nuclear SRPK1 is due to increased nuclear CLK1 (Fig. 3F).
Figure 3. CLK1 expression and SRPK1 subcellular localization.
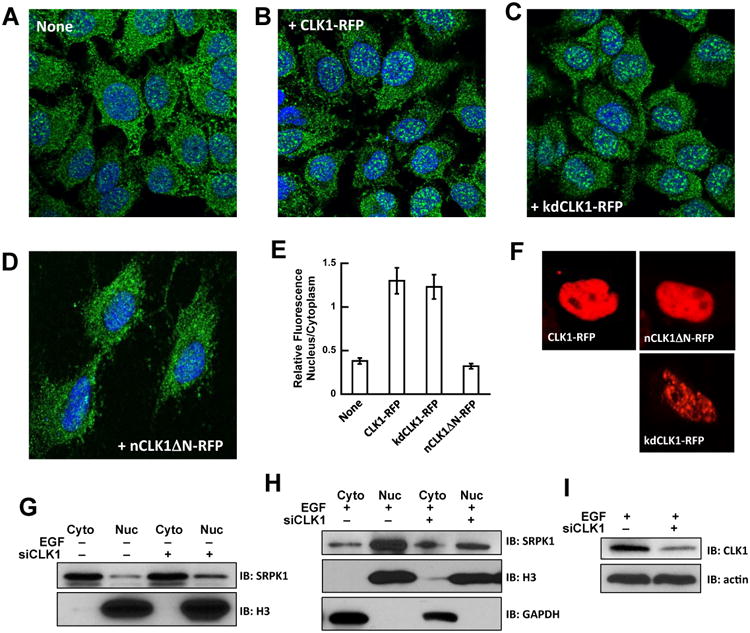
A-D) Confocal imaging of endogenous SRPK1 (green) in HeLa cells transfected with mock (A), CLK1-RFP (B), kdCLK1-RFP (C) and nCLK1ΔN-RFP (D). Nuclei are stained with DAPI (blue). E) Relative amounts of nuclear and cytoplasmic SRPK1 with and without CLK1 expression. Fractional amounts are calculated using ImageJ. The data represent an average of n=3 and error bars indicate ± SD. F) Confocal imaging of CLK1-RFP, kdCLK1-RFP and nCLK1ΔN-RFP showing nuclear localization. G,H) Nuclear and cytoplasmic SRPK1 levels in HeLa cells treated with CLK1 siRNA in the absence (G) and presence (H) of EGF stimulation. I) CLK1 levels in HeLa cells treated with CLK1 siRNA.
We next addressed whether CLK1 is required for SRPK1 nuclear localization by disrupting CLK1 using siRNA methods. CLK1 depletion did not affect SRPK1 levels in unstimulated cells where most of the kinase is present in the cytoplasm (Fig. 3G). However, in EGF-stimulated cells when SRPK1 is mostly in the nucleus, CLK1 depletion had little effect on the levels of SRPK1 in the cytoplasm but prevented the accumulation of SRPK1 in the nucleus (Fig. 3H), likely due to unstable SRPK1 in the nucleus in the absence of CLK1. In control experiments we showed that siRNA-treated cells displayed a significant decrease in CLK1 (Fig. 3I). These observations suggest that CLK1 acts as a nuclear anchor for SRPK1 through complex formation and that the CLK1 N-terminus is necessary for this function.
SRPK1 Releases Phospho-SRSF1 from CLK1
Although SRPK1 forms a highly stable complex with its substrate SRSF1, multi-site RS domain phosphorylation induces dissociation (Aubol and Adams, 2011; Ma et al., 2010). We verified these results in pull-down assays by incubating GST-SRSF1 with SRPK1 in the absence and presence of ATP. GST-SRSF1, bound to glutathione beads, strongly pulls down SRPK1 in the absence but not in the presence of ATP (Fig. 4A). In comparison, although CLK1 also forms a very stable complex with SRSF1 similar to that observed with SRPK1, phosphorylation of the RS domain does not reduce the stability of the CLK1-SRSF1 complex (Fig. 4B). This suggests that, when acting alone, CLK1 would behave as an inhibitor by titrating free SR proteins in the nucleus.
Figure 4. SRPK1 releases CLK1 from phosphorylated SRSF1.
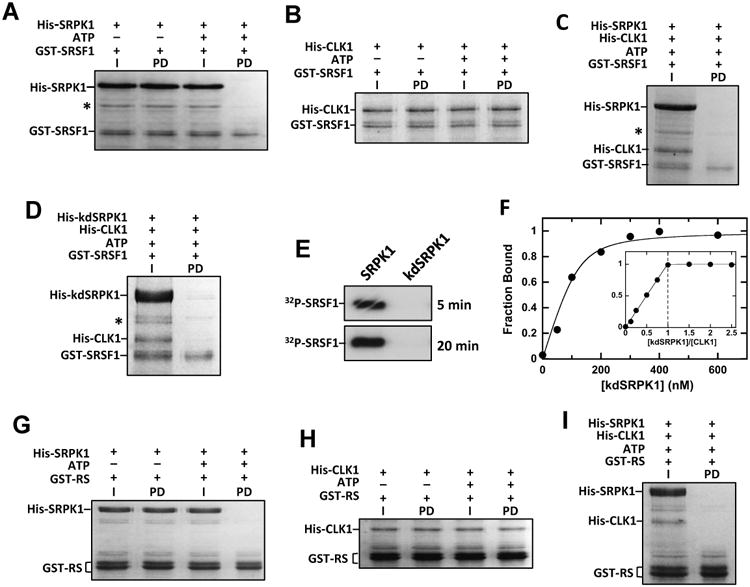
A,B) Phosphorylation disrupts SRSF1 interaction with SRPK1 but not CLK1. Immobilized GST-SRSF1 is incubated with His-SRPK1 (A) or His-CLK1 (B) with ATP, washed and run on SDS-PAGE. I = Input and PD = pull down. C,D) Binding of phosphorylated SRSF1 to His-CLK1 is disrupted by His-SRPK1 (C) and His-kdSRPK1 (D). Immobilized GST-SRSF1 is incubated with His-CLK1 and ATP and treated with His-SRPK1 or His-kdSRPK1. I = Input and PD = pull down. E) SRSF1 (0.2 μM) is phosphorylated by SRPK1 (1 μM) but not kdSRPK1 (1 μM). F) Complex affinity and stoichiometry. His-CLK1 fraction bound versus His-kdSRPK1 using 100 nM His-CLK1 is fit to equation (1) to obtain a Kd of 16 ± 6 nM and Ro of 120 ± 17 nM. Fraction bound using 400 nM His-CLK1 is plotted as a ratio of total His-kdSRPK1 to His-CLK1 establishing a complex stoichiometry of 1:1. G,H) Phosphorylation disrupts RS domain interactions with His-SRPK1 but not His-CLK1. GST-RS is incubated with His-SRPK1 (G) or His-CLK1 (H) in the presence of ATP, bound to glutathione-agarose resin, washed and run on SDS-PAGE. I = Input and PD = pull down. I) Binding of phosphorylated RS domain to His-CLK1 is disrupted by His-SRPK1. GST-RS is incubated with His-CLK1 and ATP, treated with His-SRPK1, bound to glutathioneagarose resin, washed and run on SDS-PAGE. I = Input and PD = pull down.
Given the ability of SRPK1 to interact with CLK1, we were intrigued by the possibility that SRPK1 might serve as a release factor for phospho-SRSF1 from CLK1. To this possibility, we performed pull-down experiments using phosphorylated GST-SRSF1 in the presence of SRPK1. We showed that CLK1-phosphorylated GST-SRSF1 readily dissociates CLK1 in the presence of SRPK1 (Fig. 4C). This function does not depend on catalytic activity since the kinase inactive SRPK1 (kdSRPK1) also leads to dissociation (Fig. 4D). In control experiments, we showed that kdSRPK1 is inactive and does not phosphorylate SRSF1 in the time frame of the exchange reaction (Fig. 4E). Thus, while CLK1 forms a complex with phosphorylated SRSF1, SRPK1 facilitates the release of the splicing factor from CLK1. This process is not a simple competitive event with regard to the SR protein since SRPK1 does not bind phosphorylated SRSF1 under these conditions (Fig. 4A).
To measure the affinity of the SRPK1-CLK1 complex, we used the ability of SRPK1 to displace phospho-SRSF1 from CLK1 as a reporter for the kinase-kinase complex. We phosphorylated GST-SRSF1 (500 nM) with His-tagged CLK1 (100 nM) and 32P-ATP, bound the kinase to the Ni-resin and then added varying kdSRPK1 to displace the phospho-SR protein (Fig. 4F). We used kdSRPK1 to avoid any potential additional phosphorylation of the substrate. The fraction bound of CLK1 is plotted against total kdSRPK1 to obtain an observed Kd of 16 nM using equation (1), a quadratic function for tight-binding ligands (Taira and Benkovic, 1988) (Fig. 4F). We next repeated this experiment using higher CLK1 concentrations (400 nM) and was able to titrate CLK1 with kdSRPK1 establishing a kdSRPK1-CLK1 stoichiometry of 1:1 (Fig. 4F, inset). To address whether the RRMs are necessary for SRSF1 displacement by SRPK1, we performed pull-down assays using a substrate lacking these domains (GST-RS). We found that although GST-RS binds SRPK1 and CLK1 tightly, phosphorylation leads to dissociation of SRPK1 but not CLK1, similar to that for the full-length substrate (Fig. 4 G,H). Similar to the full-length SR protein, SRPK1 dissociates the CLK1-pRS complex, suggesting that the RRMs are not involved in the release mechanism (Fig. 4I). Overall, the data show that, in addition to phosphorylating Arg-Ser dipeptides in SRSF1, SRPK1 has a secondary function in releasing the tightly bound phospho-SRSF1 from CLK1.
SRPK1-CLK1 Complex Phosphorylates the RS Domain of SRSF1
To determine whether the SRPK-CLK complex functions productively as a dual kinase system, we performed several kinetic experiments. We initially confirmed that both SRPK1 and CLK1 interact with high affinity to SRSF1 in kinetic assays. We showed that the Km for SRSF1 to CLK1 (70 nM) is lower than that for SRPK1 (110 nM), consistent with high-affinity binding observed previously (Aubol et al., 2013) (Fig. 5A). To address whether these kinases compete for the RS domain of SRSF1, we performed single turnover experiments in which large amounts of each kinase (3 μM) are used to phosphorylate a lower amount of SRSF1 (0.3 μM). In prior competition studies we showed that the Kd for SRSF1 is 60 nM to SRPK1 and 6 nM to CLK1 so that no free substrate is present and the reaction is performed under true single-turnover conditions (Aubol et al., 2013). Both kinases achieve high levels of SRSF1 phosphorylation with SRPK1 attaining multi-site phosphorylation at a faster rate than CLK1 (> 10-fold) in keeping with prior studies (Aubol et al., 2014) (Fig. 5B). SRPK1 adds 10 phosphates very rapidly (t1/2 = 0.7 min) and another 5 phosphates in a second, slower phase (t1/2 = 7 min). The phosphorylation of SRSF1 by CLK1 is monophasic and comparatively slow (t1/2 = 11 min).
Figure 5. SRPK1-CLK1 complex phosphorylates SRSF1.
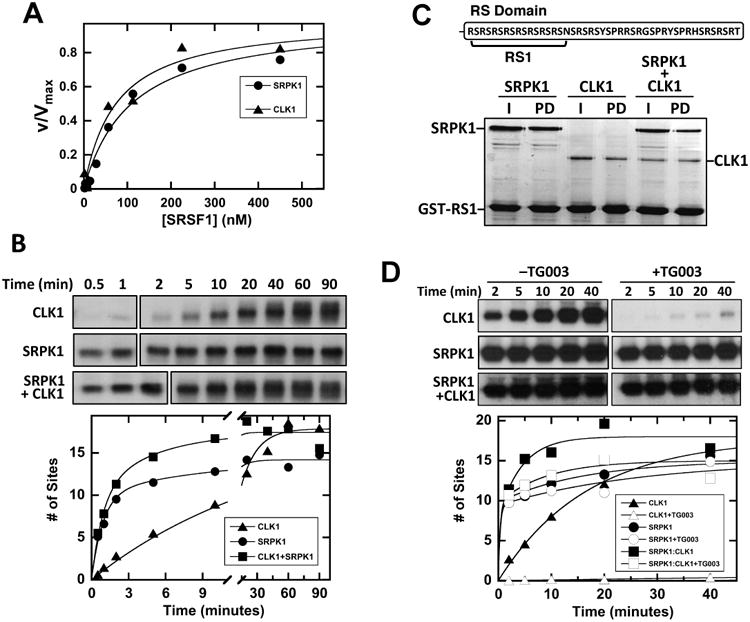
A) SRSF1 binds with similar, high affinity to CLK1 and SRPK1. Initial velocities with varying SRSF1 are fit to Km values of 70 ± 10 and 110 ± 10 nM for CLK1 and SRPK1. B) Single turnover curves for SRSF1 phosphorylation using SRPK1, CLK1 and the SRPK1-CLK1 complex. Full time courses were analyzed on separate gels as indicated by line breaks. SRSF1 phosphorylation with CLK1 is fit to a rate constant and amplitude of 0.065 ± 0.007 min-1 and 18 ± 1 sites. SRSF1 phosphorylation with SRPK1 or the SRPK1-CLK1 complex is fit to rate constants and amplitudes of 1.1 ± 0.1 and 0.11 ± 0.04 min -1 and 10 ± 1 and 4 ± 0.5 sites for SRPK1 or 1.1 ± 0.2 and 0.22 ± 0.10 min -1 and 10 ± 1 and 7.4 ± 0.6 sites for the SRPK1-CLK1 complex. C) SRPK1 and CLK1 bind the RS domain as a complex. Immobilized GST-RS1 is mixed with SRPK1, CLK1 and the SRPK1-CLK1 complex, washed and run on SDS-PAGE. D) Single turnover curves for SRSF1 phosphorylation using SRPK1, CLK1 and the SRPK1-CLK1 complex with and without TG003.
Although CLK1 binds better than SRPK1 to SRSF1 and is a much slower kinase, an equimolar amount of CLK1 did not slow down the SRPK1 reaction (Fig. 5B). These data suggest that SRPK1 does not simply compete with CLK1 for the substrate but rather takes on a dominant role in phosphorylating the RS domain when both kinases are present. Furthermore, since the concentrations of CLK1 and SRPK1 are equal and exceed the Kd for the complex by two orders of magnitude (Fig. 4F), the dominant catalytic species is the complex under our single-turnover conditions. Although it has no observable effect on the first kinetic phase, SRPK1 increases the net rate of RS domain phosphorylation as the second phase rate constant in the complex is about 3-fold larger than the rate constant for CLK1 by itself (0.22 vs. 0.065 min-1), suggesting kinase-kinase allosteric interactions. To rule out the possibility that CLK1 and SRPK1 may bind to distinct regions of the RS domain (50 aa), thus allowing two separate reactions on the same substrate, we expressed a short form of the RS domain (16 aa) that should bind only one kinase at a time (Fig. 5C). We showed previously that the RS1 segment of SRSF1 binds in both the docking groove and active site of SRPK1, and thus, cannot permit simultaneous association of both SRPK1 and CLK1 (Ngo et al., 2008). We showed that GST-RS1 binds well to both SRPK1 and CLK1 in separate pull-down experiments (Fig. 5C). However, SRPK1 and CLK1 binding was not affected when both were added, a result that is not consistent with a competitive relationship between the kinases. Instead, these findings suggest that both kinases can bind simultaneously to RS1. Given the limited binding surface of RS1, these observations suggest that CLK1 and SRPK1 form a stable complex that fully phosphorylates the RS domain of SRSF1.
SRPK1 Is the Primary Catalyst in the SRPK1-CLK1 Complex
Having shown that SRPK1 and CLK1 phosphorylate the SRSF1 RS domain as a complex, we next wished to determine which of the kinases take the principle role in phosphorylating the RS domain. To address this, we performed single-turnover experiments in which a CLK-specific inhibitor, TG003, was added to the SRPK1-CLK1 complex prior to reaction initiation. If SRPK1 is the principle kinase that binds the RS domain and rapidly phosphorylates the Arg-Ser dipeptides whereas CLK1 mostly modifies the Ser-Pro dipeptides, then we expect that TG003 addition will not affect the rapid, initial kinetic phase in the progress curve for the complex. Indeed, we observed that TG003 did not impact the fast phase for the complex, but instead slowed down later phosphorylation events suggesting that SRPK1 is the primary kinase that binds the RS domain and performs rapid Arg-Ser phosphorylation (Fig. 5D). To confirm inhibitor efficacy we showed that TG003 addition to CLK1 by itself significantly inhibited SRSF1 phosphorylation (Fig. 5D). These data suggest that SRPK1 takes on a dominant role in binding the RS domain in the SRPK1-CLK1 complex, rapidly phosphorylating Arg-Ser dipeptides. By itself, CLK1 can also phosphorylate Arg-Ser dipeptides, but in the complex, is likely to take on the specialized function of phosphorylating Ser-Pro dipeptides.
SRPK1-Induced Release Enhances Splicing of β-Globin pre-mRNA
Having demonstrated that SRPK1 is a release factor for CLK1 in vitro, we wished to determine whether SRPK1 could also serve a similar role in nuclear extracts to regulate splicing. To address this possibility, we studied splicing of β-Globin pre-mRNA in HeLa cell nuclear extracts. We found that the addition of SRPK1 to the extracts significantly inhibited β-Globin pre-mRNA splicing (Fig. 6A). This result was previously ascribed to a misbalance in the phosphorylation-dephosphorylation cycle necessary for spliceosome assembly and subsequent catalysis (Gui et al., 1994). As a control, we added kdSRPK1 to the nuclear extracts, and unexpectedly, found that it enhanced splicing of β-Globin pre-mRNA (Fig. 6A).
Figure 6. SRPK1-induced release affects splicing of β-Globin pre-mRNA.
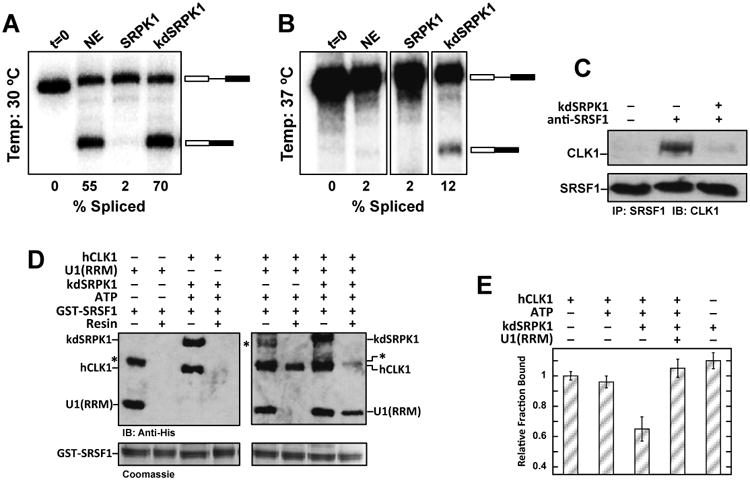
A,B) Splicing of the β-Globin gene in nuclear extracts in the absence and presence of exogenous SRPK1 and kdSRPK1 at 30 °C (A) and 37 °C (B). Several unrelated lanes were removed from the gel in panel B. C) SRPK1 induces release of SRSF1 from CLK1 in nuclear extracts. SRSF1 is immunoprecipitated and probed for CLK1 with and without exogneous kdSRPK1. Upper panel (first lane) is nuclear lysate with only protein G beads and no SRSF1 antibody. D) CLK1 release promotes binding of U1(RRM) to SRSF1. Interaction of U1(RRM) with GST-SRSF1 bound to glutathione-agarose beads is probed using an anti-His antibody. An asterisk represents an impurity in the U1(RRM) preparation. E) Binding of the Ron ESE to SRSF1 with and without CLK1 and kdSRPK1. The data are an average of n=3. Error bars indicate ± SD.
In vitro splicing assays are typically performed at 30°C, a temperature optimum for nuclear extracts (Movassat et al., 2014; Xiao and Manley, 1998). Given that SRPKs are part of the spliceosome (Mathew et al., 2008), we wished to determine whether the addition of exogenous SRPK1 could stabilize the spliceosome, thus enabling splicing at non-permissable temperatures. We showed that while nuclear extracts are not capable of facilitating splicing at 37°C, kdSRPK1 addition, indeed, stimulated splicing function (Fig. 6B). These findings indicate that SRPK1 can facilitate splicing in a phosphorylation-independent manner. To determine whether the general splicing enhancement at either temperature could be linked to SRPK1-dependent substrate release, we monitored the interaction of CLK1 and SRSF1 in nuclear extracts. We found that although endogenous CLK1 interacts with endogenous SRSF1 in nuclear extracts, the addition of recombinant kdSRPK1 dissociates this complex (Fig. 6C). These findings strongly suggest that SRPK1-induced release of SRSF1 from CLK1 may account for enhanced splicing.
CLK1 Dissociation Induces U1 RRM binding to SRSF1
Previous studies from the Ghosh lab showed that a phosphomimetic form (poly-serine-to-glutamate mutant) representing the CLK-phosphorylated state of SRSF1 associated favorably with the RRM from the 70K subunit of U1 snRNP (U1(RRM)) whereas the unphosphorylated, wild-type SRSF1 did not (Cho et al., 2011). These studies suggest that CLK-phosphorylated SRSF1 supports establishment of the 5′ splice-site through interactions between the RRMs from SRSF1 and U1 snRNP. Given the pivotal role for CLK1 in establishing this interaction, we wished to determine whether SRPK1-induced release of SRSF1 could play a role in this key step of splicing initiation. We performed pull-down assays and found that U1(RRM) did not interact with unphosphorylated GST-SRSF1, consistent with the previous study (Cho et al., 2011) (Fig. 6D, second lane in left panel). Surprisingly, we found that CLK1-phosphorylated GST-SRSF1 also did not interact with U1(RRM), suggesting that phosphorylation alone is not sufficient to initiate binding (Fig. 6D, second lane in right panel). Strikingly, kdSRPK1 addition dissociates CLK1 and strongly promotes U1(RRM) binding to SRSF1 (Fig. 6D, last lane in right panel). These findings indicate that SRPK1-induced release of CLK1-phosphorylated SRSF1 is needed to support the binding of U1(RRM) to SRSF1.
U1 RRM Binding and SRPK1-Induced Release alters RNA Binding to SRSF1
SR proteins promote pre-mRNA splicing by binding to ESEs in pre-mRNA. We wished to define the potential role of CLK1 phosphorylation and SRPK1-induced release on this recognition mechanism. To accomplish this, we monitored SRSF1 binding to the ESE from the Ron proto-oncogene (AGGCGGAGGAAGC) using a filter-binding assay (Aubol et al., 2014). We found that SRSF1 binds the ESE similarly in the presence of CLK1 whether or not the RS domain was phosphorylated (Fig. 6E, first two columns), indicating that CLK1 does not interfere with the RNA binding property of the SR protein. In contrast, kdSRPK1 addition to the CLK1-pSRSF1-RNA complex reduced RNA binding by about 35%, suggesting that CLK1 release causes the splicing factor to adopt a less productive conformation for RNA binding (Fig. 6E, third column). As control, we showed that kdSRPK1 alone does not reduce ESE affinity to SRSF1 in the absence of phosphorylation (Fig. 6E, last column). Interestingly, U1(RRM) addition to CLK1-phosphorylated SRSF1 lacking bound CLK1 led to restored high-affinity binding of the SR protein to the ESE (Fig. 6E, fourth column). Together, these findings suggest that, while SRPK1-induced release generates a form of SRSF1 that binds poorly to RNA in isolation, the released SR protein favors the formation of the ternary complex with U1 and ESE, which likely reflects the active process in 5′ splice site recognition in the cell.
Discussion
Enzymes have highly evolved active sites containing residues that specifically bind substrates with high affinity and position select functional groups for the ensuing catalytic steps. Indeed, the front end and internal portion of the enzymatic reaction is typically so fine-tuned that the product is oftentimes not easily released, thus leading to build up of an inhibitory complex. Enzymes have accordingly developed strategies to destabilize the product so that catalytic cycling can proceed unimpeded. The splicing kinase SRPK1 binds the RS domain of the SR protein SRSF1 with unusually high affinity and then initiates a series of fast phosphoryl transfer steps. During these modifications, significant negative charge develops that clashes with the negatively charged docking groove in the kinase domain. The result is a progressive destabilization of the phospho-RS domain and rapid release of the SR protein. Transient-state kinetic studies showed that the binding affinity of the product is reduced by about two orders of magnitude after 8 phosphate additions, the minimum number required for translocation into the nucleus (Aubol and Adams, 2011). In contrast, CLK1 incorporates an alternative strategy for SR protein recognition that uses a disordered N-terminal extension rather than a docking groove in the kinase domain (Aubol et al., 2014). This thematic variation is highly effective for binding SR proteins with very high affinity but does not offer a discriminatory mechanism between substrate and product. This observation raises the question of how CLKs release phospho-SR proteins for splicing.
Our results describe a unique paradigm in which two splicing kinases (CLK and SRPK) develop a symbiotic relationship to functionally compensate for their limitations and cooperatively facilitate the release of phospho-SRSF1. In the nucleus, SRPK1 strips the N-terminus from the phospho-RS domain, thereby, destabilizing its interaction with CLK1 to generate free SR protein (Fig. 7A). This mechanism bears some resemblance to a classic exchange process akin to the guanine exchange factors (GEFs) that destabilize and promote GDP release from the Rho GTPases (Rossman et al., 2005). However, unlike a simple inert exchange factor, SRPK1 is also a catalyst and works symbiotically with CLK1 to yield phosphorylated SR proteins in the nucleus. This symbiosis generates an active SRPK1-CLK1 complex that sheds the limitations of each individual kinase by incorporating the favorable attributes of both kinases (Fig. 7B). Although SRPK1 is a very agile kinase that rapidly phosphorylates Arg-Ser dipeptide repeats in a C-to-N-terminal direction, it cannot modify Ser-Pro dipeptides that are common in SR proteins and whose phosphorylation mobilizes the splicing factors and alters splicing patterns (Keshwani et al., 2015a). CLK1 can phosphorylate Ser-Pro dipeptides along with Arg-Ser repeats but only at very reduced rates. In the hetero-kinase complex, SRPK1 takes on a superior position by binding and rapidly phosphorylating the lengthy Arg-Ser repeats in SRSF1. With its N-terminus re-purposed for interaction with SRPK1, the kinase domain of CLK1 can freely pivot within the complex and modify the individual Ser-Pro dipeptides in the RS domain. We believe that Ser-Pro dipeptide modification occurs later in the multisite reaction since a CLK1-specific inhibitor does not affect the rapid initial phase of the progress curves. There is likely to be some allosteric effects in the complex as the second reaction phase is much faster than the net rate of SR phosphorylation by CLK1 alone. In all, the heterocomplex avoids a competitive roadblock by melding the speed and dissociative mechanism of SRPK1 with the dipeptide specificity of CLK1 to achieve a fully phosphorylated SR protein that participates in spliceosome assembly.
Figure 7. Role of nuclear SRPK1 for SR protein phosphorylation and splicing.
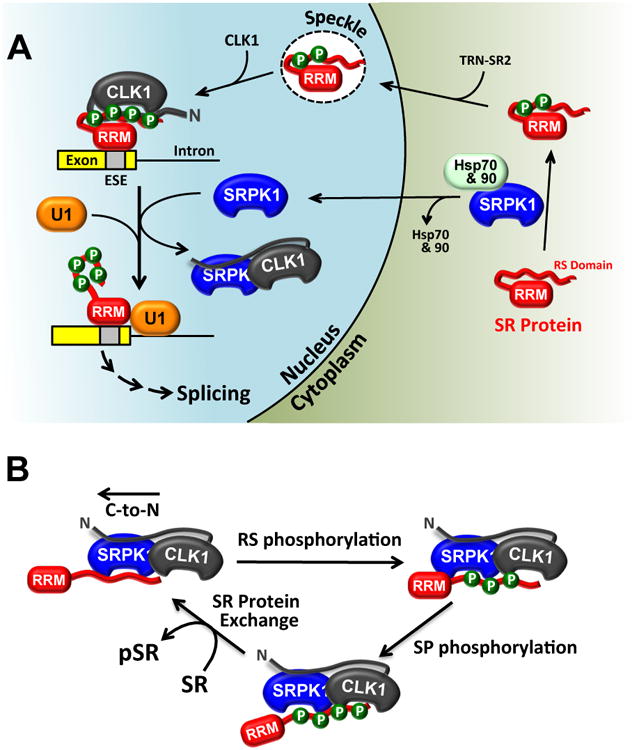
A) SRPK1 is drawn into the nucleus through contacts with CLK1. The SRPK1-CLK1 complex releases the SR protein from CLK1 and promotes splicing activity through enhanced contacts with U1. B) Catalytic cycle for SR protein phosphorylation by the SRPK1-CLK1 complex. The complex is held together by interactions between the N-terminus of CLK1 and the kinase domain of SRPK1. SRPK1 is the primary catalyst that phosphorylates Arg-Ser repeats in a C-to-N-terminal direction and CLK1 primarily phosphorylates Ser-Pro dipeptides. SRPK1 induces SR protein release by prohibiting contacts with the RS domain.
In addition to demonstrating how the dual kinase mechanism solves the biochemical release problem for CLK1, the SRPK1-CLK1 complex also provides a framework for understanding the biological role of these kinases in splicing regulation. Although we showed previously that SRPKs localize in the cytoplasm through interactions with molecular chaperones (Zhong et al., 2009), we could not explain why SRPKs are also found in the nucleus without a localization sequence. Furthermore, we could not explain why removal of the SID that interacts with the cytoplasmic chaperones leads to complete nuclear localization of SRPK1 (Ding et al., 2006). These phenomena can now be explained by the ability of CLK1 to retain SRPK1 in the nucleus, balancing the cytoplasmic tethering of the chaperones (Fig. 7A).
Once in the nucleus, SRPK1 serves a vital function in splicing. Prior studies using phospho-mimics of SRSF1 suggest that CLK1 phosphorylation promotes interaction of a component of the 70K subunit of U1 snRNP with the SR protein and subsequent formation of the 5′ splice site in pre-mRNA (Cho et al., 2011). We now show that CLK1-dependent phosphorylation is not sufficient for this process. Instead, CLK1 behaves as an inhibitor of splicing when bound to phosphorylated SRSF1. This explains the early observation that overexpressed CLK1 gave rise to similar functional consequences to SR protein depletion in transfected cells (Prasad et al., 1999). Moreover, formation of the SRPK1-CLK1 heterocomplex shifts internal binding contacts and promotes the association of the U1 RRM with phospho-SRSF1, a key step in establishing the 5′ splice site. Signals that re-attach SRPK1 to cytoplasmic chaperones (e.g., down-regulation of Akt) are then expected to increase cytoplasmic SRPK1 levels leading to dissociation of the SRPK1-CLK1 complex and resumption of CLK1-bound SR proteins.
Traditionally, SRPKs and CLKs have been viewed in light of their spatial orientation in eukaryotic cells. SRPKs phosphorylate Arg-Ser dipeptides in RS domains, releasing phospho-SR proteins for attachment to TRN-SR and nuclear import. Partially phosphorylated SR proteins are further phosphorylated at Ser-Pro dipeptides by nuclear CLKs, a modification that shunts the splicing factors toward the splicing machinery. We have now uncovered an arrangement of these two kinases in the nucleus that helps explain the mechanism of SR protein-dependent splicing that needs both kinases in the nucleus. The SRPK1-CLK1 complex brilliantly avoids the dilemma of establishing two kinases in a single compartment that, owing to their similar, high affinities, would compete for the SR protein pool. Such elegant intra-cellular symbiosis between kinases in the nucleus explains how the inhibitory effects of CLK1 are removed to facilitate spliceosome assembly and the splicing reaction upon signaling-induced nuclear translocation of SRPK1.
Experimental Procedures
Materials
ATP, Mops, Tris, MgCl2, NaCl, EDTA, glycerol, sucrose, acetic acid, lysozyme, DNAse, RNAse, Phenix imaging film, BSA, Protein G–agarose, Ni-resin and liquid scintillant were obtained from Fisher Scientific. 32P-ATP was obtained from NEN Products. RNA (AGGCGGAGGAAGC) was purchased from Integrated DNA Technologies. siRNA for CLK1 was obtained from Bioneer. Protease inhibitor cocktail was obtained from Roche and TG003 was obtained from Sigma. Anti-CLK1 monoclonal antibody was purchased from Aviva Systems Biology and anti-SRPK1 monoclonal antibody was purchased from BD Biosciences. InstantBlue was purchased from Expedeon, Hybond ECL nitrocellulose blotting membrane was purchased from Amersham and the KinaseMax™ Kit was purchased from Ambion.
Expression and Purification of Proteins
SRPK1, SRSF1, and CLK1(ΔN) (residues 148-484) were expressed and purified from pET19b vectors with an N-terminal His tag and GST-SRSF1, GST-RS1 and GST-CLK1(N) (residues 1–160) were expressed and purified from a pGEX vector as previously described (Aubol et al., 2014). kdSRPK1, inactive SRPK1 containing K109M, was expressed and purified from a pET19b vector. Deletion constructs SRPK1(ΔN), SRPK1(ΔS) and SRPK1(S) (residues 222-492) were expressed and purified as previously described (Aubol et al., 2014; Aubol et al., 2012). CLK1 virus was transfected and expressed in Hi5 insect cells and CLK1 was purified with a Nickel resin and a published procedure (Keshwani et al., 2015b).
Cell Fractionation Studies
HeLa cells were harvested and lysed in 10mM Hepes (pH 7.9), 1.5 mM MgCl2, 10 mM KCl, 1 mM DTT, 0.05% Triton and protease inhibitor cocktail. The lysates were centrifuged at 228g for 10 min to pellet nucleus and the supernatant was retained as the cytoplasmic fraction. The pellet was washed with lysis buffer and re-suspended in 200 μL of 0.25 mM sucrose and 10 mM MgCl2. The nuclear suspension was layered on 0.88 mM sucrose and 0.5 mM MgCl2 and spun at 2800g to obtain a nuclear pellet. The pellet was re-suspended in 1× RIPA buffer, spun at 2800g and the supernatant was retained as nuclear fraction.
Immunoprecipitation Experiments
HeLa cell nuclear lysates (200μL) were pre-cleared with Protein A beads and incubated overnight in the cold room with 25μL of Protein A beads and either 3μL rabbit anti-CLK1 (Aviva systems) or mouse anti-SRPK1 antibody (BD Biosciences). The beads were spun at 2052g and washed with 200μL of lysis buffer followed by adding 2× SDS loading buffer. The heated slurry was run on a 12% SDS-PAGE followed by immunoblot analysis. SRSF1 was immunopreciptated from nuclear lysates (100 μl) using 3ul SRSF1 antibody with and without 5uM kdSRPK1 and probed using anti-CLK1 antibody.
Confocal Imaging
HeLa cells were plated on 2.5cm2 MatTek poly-d-lysine plates and transfected with mock, CLK1-RFP, kdCLK1-RFP and nCLK1ΔN-RFP DNA constructs (2μg) for 24 hrs. The cells were washed with PBS and fixed with 2% paraformaldehyde in PBS for 20 min followed by a 2× PBS wash. Cell permeabilization was done with 0.25% triton in PBS for 10 min followed by a 3× PBS wash. Cells were then blocked in 10% Donkey serum PBS for 1hr at room temperature followed by overnight incubation with SRPK antibody at 4°C. Cells were washed 3× with PBS followed by incubation with secondary fluorescent anti-mouse antibody conjugated with Alexafluor 488 (Life Technologies) for 1hr at room temperature followed by 3× PBS washes for 5 min. Cells were mounted with DAPI-containing mounting medium (Vectorlabs). For live-cell imaging, transfected HeLa cells were analyzed using an Olympus FV1000 as described previously (Keshwani et al., 2015a).
Pull-Down Assays
GST- and His-tagged proteins (4 μM) were incubated in 40 μL of binding buffer (0.1% NP40 (Nonidet P40), 20 mM Tris/HCl (pH 7.5), 75 mM NaCl) for 30 min before incubating with 25 μl of glutathione–agarose resin for 30 min at room temperature. Where phosphorylation was performed, GST- and His-tagged enzymes (4 μM) were mixed with and without 100 μM ATP in 10 mM Mg2+, 20 mM Tris/HCl (pH 7.5), and 75 mM NaCl at 37°C for 30 minutes, followed by incubation with 25 μl of glutathione–agarose resin for 30 min at room temperature. In some cases, 4 μM kdSRPK was added and incubated at room temperature for 10 minutes prior to wash steps. Resin was washed 4× with 200 μl of binding buffer, and the bound proteins were eluted with SDS quench buffer and boiled for 5 min. Retained protein was resolved by SDS-PAGE (12% gel) and visualized by Instant Blue Coomassie stain or Western blotting with a mouse anti-His antibody.
Phosphorylation Assays
Single-turnover assays were performed in assay buffer (100 mM Mops (pH 7.4), 10 mM Mg2+ and 5 mg/ml BSA), at 37°C using 3 μM enzyme, 300 nM SRSF1 and 100 μM 32P-ATP (4000–8000 cpm/pmol) with and without 50 μM TG003 and 5% DMSO. Steady-state assays using 5 nM SRPK1 or 25 nM CLK1 were performed in assay buffer with 100 μM 32P-ATP (4000–8000 cpm pmol−1) at 23 °C (SRPK1) and 37 °C (CLK1) and quenched with 10 μl SDS/PAGE loading buffer at 2 and 20 minutes, respectively. Phosphorylated SR proteins were cut from a dried 12% SDS-PAGE and counted in liquid scintillant. The amount of kdSRPK1 bound to CLK1 was measured by phosphorylating GST-SRSF1 (500 nM) with CLK1 (100 or 400 nM) and 100 μM 32P-ATP for 90 minutes at 37 °C and binding to the Ni-resin. Varying kdSRPK1 (0-1000 nM) was added to CLK1-bound resin and washed 2× with 400 μL of binding buffer. The CLK1 fraction bound (FB) was measured from the CPMs on the resin and fit to equation (1)
| (1) |
where Ro is the total CLK1, Lo is the total kdSRPK1 and Kd is the complex dissociation constant.
RNA Preparation
In vitro transcription was performed using 0.5 μg/μL linear β-Globin DNA in a reaction buffer containing 0.67 μM (α-32P) UTP, 0.4 mM ATP, 0.4 mM CTP, 0.1 mM UTP, 0.1 mM GTP, 2 mM m7G(5′)ppp(5′G) (cap analog), 2 mM DTT, 10 U/μL ribonuclease inhibitor, 7.5 U/μL T7 RNA polymerase (Promega) and 1× transcription buffer (Promega). Reactions were incubated at 37°C for 2 h, gel purified using denaturing PAGE, eluted from the gel (elution buffer: 0.5 mM NaOAc, pH 5.6, 0.1% SDS, 10 mM Tris, pH 7.5, 1 mM EDTA), ethanol precipitated, and resuspended in nuclease free water.
In Vitro Splicing
Splicing reactions were performed using α-32P labeled β-Globin RNA in 30% HeLa nuclear extract with 500 nM SRPK1 or kdSRPK1 in reaction buffer as previously described (Movassat et al., 2014) with final 70mM NaCl and incubated at 30 °C for 90 min. Reactions were digested using proteinase K, extracted with phenol chloroform, precipitated with ethanol and separated on denaturing PAGE. The gel was exposed to BaFBr:Eu screen and imaged on PhosphorImager (Bio-Rad). Appearance of final spliced product was determined by taking the percent of the sum of the adjusted intensity for the spliced band divided by the total signal for the spliced and unspliced product band (Movassat et al., 2014).
RNA Binding Assays
Pull-down experiments were performed with a 32P-labeled RNA oligomer based on the Ron ESE (AGGCGGAGGAAGC). Labeling was carried out using the KinaseMax™ kit from Ambion and confirmed by 12% urea PAGE. GST-tagged SRSF1 proteins (4 μM) were incubated with 3 pmol of 32P-labeled Ron ESE at 23°C for 30 min in 20 μl of binding buffer and mixed with 25 μl of glutathione–agarose resin for 30 min at room temperature. Where phosphorylation was required, GST- and His-tagged enzymes (4 μM) were incubated with and without 100 μM ATP and 10 mM Mg2+ in 20 mM Tris/HCl (pH 7.5) and 75 mM NaCl at 37°C for 30 minutes prior to the RNA addition step. In some cases, 4 μM kdSRPK or buffer blank was added and incubated at room temperature for 10 minutes prior to the RNA addition step. The resin was washed 3× with 200 μl of binding buffer and the retained RNA was counted in liquid scintillant.
Highlights.
SRPK1 and CLK1 form a stable complex in the nucleus of cells.
The kinase domain of SRPK1 interacts with the disordered N-terminus of CLK1.
SRPK1 acts as a release factor that strips CLK1 from tightly-bound SR proteins.
SRPK1-induced release promotes U1 binding and mRNA splicing.
Acknowledgments
This work was supported by NIH grants GM67969 to J.A.A., GM52872 to X.-D.F and GM62287 to K.J.H. We would like to thank Dr. Gourisankar Ghosh for providing U1(RRM).
Footnotes
Author Contributions: B.E.A., G.W., M.M.K., L.F. and M.M. performed all experiments. K.J.H. and X.-D.F helped with data analysis and manuscript editing. J.A.A. planned the experiments and wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aubol BE, Adams JA. Applying the brakes to multisite SR protein phosphorylation: substrate-induced effects on the splicing kinase SRPK1. Biochemistry. 2011;50:6888–6900. doi: 10.1021/bi2007993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubol BE, Plocinik RM, Hagopian JC, Ma CT, McGlone ML, Bandyopadhyay R, Fu XD, Adams JA. Partitioning RS domain phosphorylation in an SR protein through the CLK and SRPK protein kinases. J Mol Biol. 2013;425:2894–2909. doi: 10.1016/j.jmb.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubol BE, Plocinik RM, Keshwani MM, McGlone ML, Hagopian JC, Ghosh G, Fu XD, Adams JA. N-terminus of the protein kinase CLK1 induces SR protein hyperphosphorylation. Biochem J. 2014;462:143–152. doi: 10.1042/BJ20140494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubol BE, Plocinik RM, McGlone ML, Adams JA. Nucleotide release sequences in the protein kinase SRPK1 accelerate substrate phosphorylation. Biochemistry. 2012;51:6584–6594. doi: 10.1021/bi300876h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S, Hoang A, Sinha R, Zhong XY, Fu XD, Krainer AR, Ghosh G. Interaction between the RNA binding domains of Ser-Arg splicing factor 1 and U1-70K snRNP protein determines early spliceosome assembly. Proc Natl Acad Sci U S A. 2011;108:8233–8238. doi: 10.1073/pnas.1017700108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwill K, Pawson T, Andrews B, Prasad J, Manley JL, Bell JC, Duncan PI. The Clk/Sty protein kinase phosphorylates SR splicing factors and regulates their intranuclear distribution. EMBO J. 1996;15:265–275. [PMC free article] [PubMed] [Google Scholar]
- Ding JH, Zhong XY, Hagopian JC, Cruz MM, Ghosh G, Feramisco J, Adams JA, Fu XD. Regulated cellular partitioning of SR protein-specific kinases in mammalian cells. Mol Biol Cell. 2006;17:876–885. doi: 10.1091/mbc.E05-10-0963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endicott JA, Noble ME, Johnson LN. The structural basis for control of eukaryotic protein kinases. Annu Rev Biochem. 2012;81:587–613. doi: 10.1146/annurev-biochem-052410-090317. [DOI] [PubMed] [Google Scholar]
- Fu XD, Ares M., Jr Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet. 2014;15:689–701. doi: 10.1038/nrg3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh G, Adams JA. Phosphorylation mechanism and structure of serine-arginine protein kinases. FEBS J. 2011;278:587–597. doi: 10.1111/j.1742-4658.2010.07992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui JF, Lane WS, Fu XD. A serine kinase regulates intracellular localization of splicing factors in the cell cycle. Nature. 1994;369:678–682. doi: 10.1038/369678a0. [DOI] [PubMed] [Google Scholar]
- Kataoka N, Bachorik JL, Dreyfuss G. Transportin-SR, a nuclear import receptor for SR proteins. J Cell Biol. 1999;145:1145–1152. doi: 10.1083/jcb.145.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshwani MM, Aubol BE, Fattet L, Ma CT, Qiu J, Jennings PA, Fu XD, Adams JA. Conserved proline-directed phosphorylation regulates SR protein conformation and splicing function. Biochem J. 2015a;466:311–322. doi: 10.1042/BJ20141373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshwani MM, Hailey KL, Aubol BE, Fattet L, McGlone ML, Jennings PA, Adams JA. Nuclear Protein Kinase CLK1 Uses A Nontraditional Docking Mechanism To Select Physiological Substrates. Biochem J. 2015b doi: 10.1042/BJ20150903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koizumi J, Okamoto Y, Onogi H, Mayeda A, Krainer AR, Hagiwara M. The subcellular localization of SF2/ASF is regulated by direct interaction with SR protein kinases (SRPKs) J Biol Chem. 1999;274:11125–11131. doi: 10.1074/jbc.274.16.11125. [DOI] [PubMed] [Google Scholar]
- Lai MC, Lin RI, Tarn WY. Transportin-SR2 mediates nuclear import of phosphorylated SR proteins. Proc Natl Acad Sci U S A. 2001;98:10154–10159. doi: 10.1073/pnas.181354098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie H, Li JJ, Thevakumaran N, Therrien M, Sicheri F. Dimerization-induced allostery in protein kinase regulation. Trends Biochem Sci. 2014;39:475–486. doi: 10.1016/j.tibs.2014.08.004. [DOI] [PubMed] [Google Scholar]
- Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma CT, Ghosh G, Fu XD, Adams JA. Mechanism of dephosphorylation of the SR protein ASF/SF2 by protein phosphatase 1. J Mol Biol. 2010;403:386–404. doi: 10.1016/j.jmb.2010.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis T, Reed R. An extensive network of coupling among gene expression machines. Nature. 2002;416:499–506. doi: 10.1038/416499a. [DOI] [PubMed] [Google Scholar]
- Mathew R, Hartmuth K, Mohlmann S, Urlaub H, Ficner R, Luhrmann R. Phosphorylation of human PRP28 by SRPK2 is required for integration of the U4/U6-U5 tri-snRNP into the spliceosome. Nat Struct Mol Biol. 2008;15:435–443. doi: 10.1038/nsmb.1415. [DOI] [PubMed] [Google Scholar]
- Moore MJ, Proudfoot NJ. Pre-mRNA processing reaches back to transcription and ahead to translation. Cell. 2009;136:688–700. doi: 10.1016/j.cell.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Movassat M, Mueller WF, Hertel KJ. In vitro assay of pre-mRNA splicing in mammalian nuclear extract. Methods Mol Biol. 2014;1126:151–160. doi: 10.1007/978-1-62703-980-2_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayler O, Stamm S, Ullrich A. Characterization and comparison of four serine- and arginine-rich (SR) protein kinases. Biochem J. 1997;326(Pt 3):693–700. doi: 10.1042/bj3260693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo JC, Giang K, Chakrabarti S, Ma CT, Huynh N, Hagopian JC, Dorrestein PC, Fu XD, Adams JA, Ghosh G. A sliding docking interaction is essential for sequential and processive phosphorylation of an SR protein by SRPK1. Mol Cell. 2008;29:563–576. doi: 10.1016/j.molcel.2007.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–463. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit S, Wang D, Fu XD. Functional integration of transcriptional and RNA processing machineries. Curr Opin Cell Biol. 2008;20:260–265. doi: 10.1016/j.ceb.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons SJ, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene. 2004;23:7906–7909. doi: 10.1038/sj.onc.1208160. [DOI] [PubMed] [Google Scholar]
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad J, Colwill K, Pawson T, Manley JL. The protein kinase Clk/Sty directly modulates SR protein activity: both hyper- and hypophosphorylation inhibit splicing. Mol Cell Biol. 1999;19:6991–7000. doi: 10.1128/mcb.19.10.6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–180. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- Shin C, Manley JL. Cell signalling and the control of pre-mRNA splicing. Nat Rev Mol Cell Biol. 2004;5:727–738. doi: 10.1038/nrm1467. [DOI] [PubMed] [Google Scholar]
- Stamm S. Regulation of alternative splicing by reversible protein phosphorylation. J Biol Chem. 2008;283:1223–1227. doi: 10.1074/jbc.R700034200. [DOI] [PubMed] [Google Scholar]
- Taira K, Benkovic SJ. Evaluation of the importance of hydrophobic interactions in drug binding to dihydrofolate reductase. J Med Chem. 1988;31:129–137. doi: 10.1021/jm00396a019. [DOI] [PubMed] [Google Scholar]
- Taylor SS, Ilouz R, Zhang P, Kornev AP. Assembly of allosteric macromolecular switches: lessons from PKA. Nat Rev Mol Cell Biol. 2012;13:646–658. doi: 10.1038/nrm3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, Kornev AP. Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem Sci. 2011;36:65–77. doi: 10.1016/j.tibs.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GS, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet. 2007;8:749–761. doi: 10.1038/nrg2164. [DOI] [PubMed] [Google Scholar]
- Wang HY, Lin W, Dyck JA, Yeakley JM, Songyang Z, Cantley LC, Fu XD. SRPK2: a differentially expressed SR protein-specific kinase involved in mediating the interaction and localization of pre-mRNA splicing factors in mammalian cells. J Cell Biol. 1998;140:737–750. doi: 10.1083/jcb.140.4.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao SH, Manley JL. Phosphorylation-dephosphorylation differentially affects activities of splicing factor ASF/SF2. EMBO J. 1998;17:6359–6367. doi: 10.1093/emboj/17.21.6359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun CY, Velazquez-Dones AL, Lyman SK, Fu XD. Phosphorylation-dependent and -independent nuclear import of RS domain-containing splicing factors and regulators. J Biol Chem. 2003;278:18050–18055. doi: 10.1074/jbc.M211714200. [DOI] [PubMed] [Google Scholar]
- Zhong XY, Ding JH, Adams JA, Ghosh G, Fu XD. Regulation of SR protein phosphorylation and alternative splicing by modulating kinetic interactions of SRPK1 with molecular chaperones. Genes Dev. 2009;23:482–495. doi: 10.1101/gad.1752109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Qiu J, Liu W, Zhou Y, Plocinik RM, Li H, Hu Q, Ghosh G, Adams JA, Rosenfeld MG, et al. The Akt-SRPK-SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus. Mol Cell. 2012;47:422–433. doi: 10.1016/j.molcel.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]