

Abstract
Regulation of protein abundance is crucial to virtually every cellular process. Protein abundance reflects the integration of the rates of protein synthesis and protein degradation. Many assays reporting on protein abundance (e.g., single-time point western blotting, flow cytometry, fluorescence microscopy, or growth-based reporter assays) do not allow discrimination of the relative effects of translation and proteolysis on protein levels. This article describes the use of cycloheximide chase followed by western blotting to specifically analyze protein degradation in the model unicellular eukaryote, Saccharomyces cerevisiae (budding yeast). In this procedure, yeast cells are incubated in the presence of the translational inhibitor cycloheximide. Aliquots of cells are collected immediately after and at specific time points following addition of cycloheximide. Cells are lysed, and the lysates are separated by polyacrylamide gel electrophoresis for western blot analysis of protein abundance at each time point. The cycloheximide chase procedure permits visualization of the degradation kinetics of the steady state population of a variety of cellular proteins. The procedure may be used to investigate the genetic requirements for and environmental influences on protein degradation.
Keywords: Biochemistry, Issue 110, Saccharomyces cerevisiae, budding yeast, mutants, endoplasmic reticulum-associated degradation, protein degradation, cycloheximide chase, translocon, ubiquitin-proteasome system
Introduction
Proteins perform crucial functions in virtually every cellular process. Many physiological processes require the presence of a specific protein (or proteins) for a defined period of time or under particular circumstances. Organisms therefore monitor and regulate protein abundance to meet cellular needs 1. For example, cyclins (proteins that control cell division) are present at specific phases of the cell cycle, and the loss of regulated cyclin levels has been associated with malignant tumor formation 2. In addition to regulating protein levels to meet cellular needs, cells employ degradative quality control mechanisms to eliminate misfolded, unassembled, or otherwise aberrant protein molecules 3. Control of protein abundance involves regulation of both macromolecular synthesis (transcription and translation) and degradation (RNA decay and proteolysis). Impaired or excessive protein degradation contributes to multiple pathologies, including cancer, cystic fibrosis, neurodegenerative conditions, and cardiovascular disorders 4-8. Proteolytic mechanisms therefore represent promising therapeutic targets for a range of illnesses 9-12.
Analysis of proteins at a single time point (e.g., by western blot 13, flow cytometry 14, or fluorescence microscopy 15) provides a snapshot of steady state protein abundance without revealing the relative contributions of synthesis or degradation. Similarly, growth-based reporter assays reflect steady state protein levels over an extended time period without discriminating between the influences of synthesis and degradation 15-20. It is possible to infer the contribution of degradative processes to steady state protein levels by comparing abundance before and after inhibiting specific components of the degradative mechanism (e.g., by pharmacologically inactivating the proteasome 21 or knocking out a gene hypothesized to be required for degradation 13). A change in steady state protein levels after inhibiting degradative pathways provides strong evidence for the contribution of proteolysis to the control of protein abundance 13. However, such an analysis still does not provide information regarding the kinetics of protein turnover. Cycloheximide chase followed by western blotting overcomes these weaknesses by allowing researchers to visualize protein degradation over time 22-24. Further, because protein detection following cycloheximide chase is typically performed by western blotting, radioactive isotopes and lengthy immunoprecipitation steps are not required for cycloheximide chase, unlike many commonly used pulse chase techniques, which are also performed to visualize protein degradation25.
Cycloheximide was first identified as a compound with anti-fungal properties produced by the gram-positive bacterium Streptomyces griseus 26,27. It is a cell-permeable molecule that specifically inhibits eukaryotic cytosolic (but not organellar) translation by impairing ribosomal translocation 28-31. In a cycloheximide chase experiment, cycloheximide is added to cells, and aliquots of cells are collected immediately and at specific time points following addition of the compound 22. Cells are lysed, and protein abundance at each time point is analyzed, typically by western blot. Decreases in protein abundance following the addition of cycloheximide can be confidently attributed to protein degradation. An unstable protein will decrease in abundance over time, while a relatively stable protein will exhibit little change in abundance.
Mechanisms of selective protein degradation have been highly conserved throughout Eukarya. Much of what is known about protein degradation was first learned in the model unicellular eukaryote, Saccharomyces cerevisiae (budding yeast) 25,32-36. Studies with yeast are likely to continue providing novel and important insights into protein degradation. A method for cycloheximide chase in yeast cells followed by western blot analysis of protein abundance is presented here.
Protocol
1. Growth and Harvest of Yeast Cells
If not analyzing degradation kinetics of an endogenous yeast protein, transform desired yeast strain(s) with a plasmid encoding the protein of interest. Reliable methods for yeast transformation have been previously described 37.
Inoculate yeast in 5 ml of appropriate medium (e.g., selective synthetic defined (SD) medium for plasmid maintenance of transformed cells or non-selective yeast extract-peptone-dextrose (YPD) medium for non-transformed cells). Incubate overnight at 30 °C, rotating. NOTE: 30 °C is the optimal growth temperature for typical wild-type laboratory yeast strains 38. However, because some mutant yeast strains do not grow optimally at 30 °C and some proteins undergo temperature-dependent degradation, the temperatures used for yeast cell growth and cycloheximide chase should be empirically determined 39.
Measure the optical density at 600 nm (OD600) of each overnight culture. NOTE: Cells may be in logarithmic or stationary growth phase but should have minimally reached a density that will allow dilution to an OD600 = 0.2 in 15 ml of fresh medium.
Dilute the overnight cultures to an OD600 value of 0.2 in 15 ml of fresh medium.
Incubate yeast at 30 °C, shaking until the cells reach mid-logarithmic growth phase (i.e., an OD600 between 0.8 and 1.2).
- During yeast cell growth, perform the following in preparation for the cycloheximide chase procedure:
- Set a heat block that can accommodate 15 ml conical tubes to 30 °C for incubation of cells in the presence of cycloheximide. Add water to the wells of the heat block to allow efficient heat distribution to cultures. Add water to each well such that a 15 ml conical tube will cause the water level to rise to, but not overflow, the lip of the well.
- Set a second heat block that can accommodate 1.5 ml microcentrifuge tubes to 95 °C for protein denaturation following cell lysis.
- Pre-warm fresh growth medium (1.1 ml per time point per culture to be assayed) to 30 °C.
- Add 50 µl 20x Stop Mix to pre-labeled microcentrifuge tubes (one tube per time point per culture to be assayed). Place tubes on ice. CAUTION: Sodium azide, an ingredient in 20x Stop Mix, is toxic via oral ingestion or dermal contact. Follow manufacturer recommendations when preparing, storing, and handling sodium azide. In the event of accidental exposure, consult material safety data sheet provided by manufacturer.
When cells have reached mid-logarithmic growth, collect 2.5 OD600 units of each culture per time point to be assayed (e.g., 7.5 OD600 units to analyze protein abundance at three time points). Centrifuge collected cells in 15 ml conical tubes at 3,000 x g at room temperature for 2 min. NOTE: One OD600 unit is equal to the amount of yeast present in 1 ml culture at an OD600 of 1.0. The volume of culture (in ml) required to harvest X OD600 units (V) may be determined by using the following equation: V = X OD600 units/Measured OD600. For example, to harvest 7.5 OD600 units of yeast cell culture at an OD600 of 1.0, collect 7.5 OD600 units/1.0 = 7.5 ml yeast culture.
Resuspend each cell pellet in 1 ml of 30 °C (pre-warmed) fresh growth medium per 2.5 OD600 units of cells (e.g., 3 ml for 7.5 OD600 units).
2. Cycloheximide Chase
Equilibrate yeast cell suspensions by incubation for 5 min in the 30 °C heat block.
Prepare a timer to count up from 0:00.
- To begin the cycloheximide chase, press "Start" on the timer. Swiftly, but carefully, perform the following steps:
- Add cycloheximide to a final concentration of 250 µg/ml to the first yeast cell suspension (e.g., add 37.5 µl of 20 mg/ml cycloheximide stock to 3 ml of cell suspension), and vortex briefly to mix. CAUTION: Cycloheximide is a dermal irritant and is toxic via oral ingestion. Follow manufacturer recommendations when preparing, storing, and handling cycloheximide. In the event of accidental exposure, consult material safety data sheet provided by manufacturer.
- Immediately after adding cycloheximide and vortexing, transfer 950 µl (~2.4 OD600 units) of the yeast cell suspension with added cycloheximide to a pre-labeled microcentrifuge tube containing 50 µl ice-cold 20x stop mix. Vortex the microcentrifuge tube, and place on ice until all samples have been collected.
- Return the yeast cell suspension to 30 °C.
Repeat steps 2.3.1 through 2.3.3 for each of the remaining yeast cell suspensions at regular time intervals (e.g., every 30 sec, such that cycloheximide is added to Sample #1 at 0:00, Sample #2 at 0:30, Sample #3 at 1:00, etc.).
- At each subsequent time point, vortex yeast cell suspensions and transfer 950 μl to labeled microcentrifuge tubes containing 50 μl pre-chilled 20x Stop Mix. Vortex and place collected cells on ice. Return 15 ml conical tubes to 30 °C heat block.
- For example, for collection of cells 30 min after cycloheximide addition (assuming 30-sec intervals between cycloheximide addition to yeast cell suspensions at the beginning of the time course), vortex and remove 950 μl of cell suspension from Sample #1 at 30:00. Repeat for Sample #2 at 30:30, and so on. NOTE: To prevent settling of yeast, vortex cell suspensions in 15 ml conical tubes approximately every 5 min throughout the course of the chase. Alternatively, yeast cell suspensions may be maintained in a continuously agitating water bath for the duration of the cycloheximide chase experiment.
When all samples have been collected, pellet collected cells by centrifugation at 6,500 x g at room temperature for 30 sec. Remove the supernatant by pipetting or aspiration. Cells are now ready for alkaline lysis. Alternatively, snap-freeze pelleted cells in liquid nitrogen and store at -80 °C.
3. Post-alkaline Protein Extraction (Modified from 16,40)
Add 100 µl of distilled water to each cell pellet. Resuspend by pipetting up and down or vortexing.
Add 100 µl of 0.2 M NaOH to each sample. Mix by pipetting up and down or vortexing.
Incubate suspended cells at room temperature for 5 min. At this stage, yeast cells have not been lysed, and proteins have not been released 40.
Pellet cells by centrifugation at 18,000 x g at room temperature for 30 sec. Remove supernatant by pipetting or aspiration.
To lyse cells, add 100 µl of Laemmli sample buffer to each cell pellet. Resuspend by pipetting up and down or vortexing. NOTE: Sequential incubation of cells with NaOH and Laemmli sample buffer releases proteins in a form compatible with sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using a Tris-glycine running buffer system.
Incubate at 95 °C for 5 min to fully denature proteins. NOTE: Incubation at 95 °C may not be suitable for analysis of proteins that are prone to aggregation (e.g., proteins with several transmembrane segments). These proteins may become insoluble when incubated at elevated temperatures 41. Thus, for the analysis of such proteins, lysates should be incubated at lower temperatures (e.g., 37 °C - 70 °C) for 10 - 30 min, as empirically determined.
Centrifuge lysates at 18,000 x g at room temperature for 1 min to pellet insoluble material. The supernatant (solubilized extracted protein) is ready for separation by SDS-PAGE and subsequent western blot analysis. Alternatively, store lysates at -20 °C.
4. Representative SDS-PAGE and Western Blotting Procedure (Adapted from 16)
Load protein molecular weight standards and empirically determined volume of lysates onto an SDS-PAGE gel. NOTE: Choose the percentage of acrylamide and bis-acrylamide in the SDS-PAGE gel based on the molecular weight of the protein of interest. In general, lower percentage gels are better suited for resolving higher molecular weight proteins.
Run gel at 200 V until the dye front has reached the bottom edge of the gel.
Transfer proteins from the gel to polyvinylidene fluoride (PVDF) membrane by wet transfer at 20 V for 60 - 90 min at 4 °C.
Block membrane by incubating in Tris-Buffered Saline (TBS) containing 5% skim milk, rocking, at room temperature for 1 hr or at 4 °C overnight. NOTE: To prevent microbial growth in the presence of a membrane incubated overnight, it is recommended to include sodium azide in the blocking solution at a final concentration of 0.02%.
Incubate membrane with primary antibody specific for protein of interest in TBS with 0.1% Tween-20 (TBS/T) and 1% skim milk, rocking, at room temperature for 1 hr.
Wash membrane at room temperature 3 x 5 min with TBS/T, rocking.
Incubate membrane with appropriate fluorophore-conjugated secondary antibody in TBS/T with 1% skim milk at room temperature for 1 hr, rocking. NOTE: Fluorophores are light-sensitive. Therefore, dilutions of antibodies conjugated to fluorophores should be prepared in the dark, and incubation of membranes in the presence of antibodies conjugated to fluorophores and subsequent wash steps should occur in lightproof (e.g., foil-wrapped) containers.
Wash membrane at room temperature 3 x 5 min with TBS/T, rocking.
Image membrane using LI-COR Odyssey CLx and Image Studio software (or comparable imaging equipment and software), following manufacturer recommendations.
After acquiring membrane image, incubate the membrane with a primary antibody specific for a loading control protein in TBS/T with 1% skim milk at room temperature for 1 hr, rocking.
Wash membrane at room temperature 3 x 5 min with TBS/T, rocking.
Incubate membrane with appropriate fluorophore-conjugated secondary antibody in TBS/T with 1% skim milk at room temperature for 1 hr, rocking.
Wash membrane at room temperature 3 x 5 min with TBS/T, rocking.
Image membrane as in step 4.9.
Representative Results
To illustrate cycloheximide chase methodology, the stability of Deg1-Sec62 (Figure 1), a model yeast endoplasmic reticulum (ER)-associated degradation (ERAD) substrate, was analyzed 42-44. In ERAD, quality control ubiquitin ligase enzymes covalently attach chains of the small protein ubiquitin to aberrant proteins localized to the ER membrane. Such polyubiquitylated proteins are subsequently removed from the ER and degraded by the proteasome, a large, cytosolic protease 45. The Deg1-Sec62 protein is targeted for degradation after it persistently and aberrantly engages the translocon, a channel that facilitates protein translocation into the ER lumen or membrane 43. Similarly, mammalian apolipoprotein B, a protein component of low-density lipoproteins, persistently engages the ER translocon and subsequently undergoes degradation by a related mechanism when its lipid binding partners are absent 46-48. Thus, analyses of Deg1-Sec62 degradation in yeast cells may yield insight into the degradation of proteins that persistently or aberrantly engage the translocon, provisionally termed ERAD-T (for translocon-associated) substrates 43.
Previous studies have indicated that the ER-resident ubiquitin ligase Hrd1 functions with the ubiquitin-conjugating enzyme Ubc7 to catalyze degradation of Deg1-Sec62 16,42-44. The transmembrane protein Cue1 anchors Ubc7 to the ER membrane and activates the enzyme 49-51. However, a requirement for Cue1 in the degradation of Deg1-Sec62 has not been directly investigated. To test the hypothesis that Cue1, like Hrd1 and Ubc7, is required for efficient degradation of Deg1-Sec62, wild-type and mutant yeast cells expressing Deg1-Sec62 were subjected to cycloheximide chase and western blot analysis (Figure 2A). The Deg1-Sec62 protein migrates on SDS-PAGE as multiple species. This is consistent with earlier studies indicating extensive post-translational modification of the protein 43,44,52. In wild-type yeast cells, Deg1-Sec62 was readily degraded. As previously observed, loss of either Hrd1 or Ubc7 substantially stabilized the Deg1-Sec62 protein 42-44. As predicted, Deg1-Sec62 was stabilized in the absence of Cue1 (to a similar extent as in the absence of Ubc7), confirming a role for the Ubc7-interacting protein in ERAD-T.
The proportion of Deg1-Sec62 protein remaining at each time point was quantified and is plotted in Figure 2B. We note that the apparent rate of disappearance of Deg1-Sec62 in wild-type cells may be an underestimation of the rate of degradation of nascent Deg1-Sec62 (i.e., the protein's half-life, which can be most directly determined by pulse chase analysis 43). Because wild-type yeast actively degrade the Deg1-Sec62 protein prior to cycloheximide addition, steady state abundance of Deg1-Sec62 is substantially reduced in these cells (compared to cells in which degradation is impaired). This can be appreciated by comparing the abundance of Deg1-Sec62 at the initial time points for each strain in Figure 2A. Further, any degradation-resistant subpopulation of the substrate protein (e.g., if the protein accumulates in intracellular pools from which degradation is prevented) might appear relatively stable in wild-type cells over the time course of the experiment.
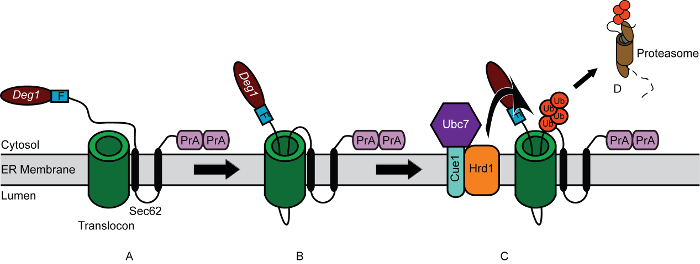 Figure 1. Model for Degradation of Deg1-Sec62 following Aberrant Engagement of Translocon. (A) Schematic depiction of Deg1-Sec62. Deg1-Sec62 consists of the following elements, in sequence: Deg1 (the amino-terminal 67 amino acids from the yeast transcriptional repressor MATα2), a Flag (F) epitope, the 2-transmembrane protein Sec62, and two copies of the Staphylococcus aureus Protein A (PrA). (B) Following normal co-translational insertion of its two transmembrane segments into the endoplasmic reticulum (ER) membrane, persistent association of Deg1-Sec62 with the translocon triggers abnormal, Deg1-dependent, post-translational translocon engagement. A portion of the cytosolic amino terminus aberrantly enters-and likely remains within-the translocon. (C) Following translocon engagement, the Hrd1 ubiquitin ligase ubiquitylates Deg1-Sec62. Hrd1 is assisted by the ubiquitin-conjugating enzyme Ubc7, which is anchored in the ER membrane by Cue1. Ub, ubiquitin protein monomers. (D) Ubiquitylated Deg1-Sec62 is extracted from the ER membrane and degraded by the proteasome, relieving translocon obstruction. Please click here to view a larger version of this figure.
Figure 1. Model for Degradation of Deg1-Sec62 following Aberrant Engagement of Translocon. (A) Schematic depiction of Deg1-Sec62. Deg1-Sec62 consists of the following elements, in sequence: Deg1 (the amino-terminal 67 amino acids from the yeast transcriptional repressor MATα2), a Flag (F) epitope, the 2-transmembrane protein Sec62, and two copies of the Staphylococcus aureus Protein A (PrA). (B) Following normal co-translational insertion of its two transmembrane segments into the endoplasmic reticulum (ER) membrane, persistent association of Deg1-Sec62 with the translocon triggers abnormal, Deg1-dependent, post-translational translocon engagement. A portion of the cytosolic amino terminus aberrantly enters-and likely remains within-the translocon. (C) Following translocon engagement, the Hrd1 ubiquitin ligase ubiquitylates Deg1-Sec62. Hrd1 is assisted by the ubiquitin-conjugating enzyme Ubc7, which is anchored in the ER membrane by Cue1. Ub, ubiquitin protein monomers. (D) Ubiquitylated Deg1-Sec62 is extracted from the ER membrane and degraded by the proteasome, relieving translocon obstruction. Please click here to view a larger version of this figure.
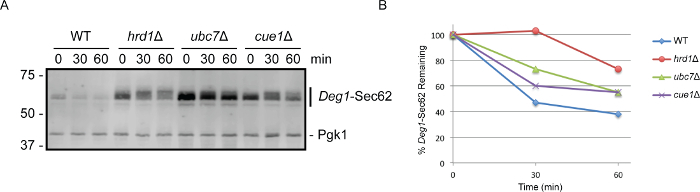 Figure 2. Cycloheximide Chase followed by Western Blotting Indicates that Cue1 is Required for Efficient Deg1-Sec62 Degradation. (A) Yeast cells of the indicated genotypes were transformed with a yeast centromeric plasmid encoding Deg1-Sec62 (pRS416-PMET25-Deg1-Flag-Sec62-2xProtA; AmpR/URA343) and subjected to cycloheximide chase analysis. Proteins from each time point (equivalent to 0.125 OD600 units) were separated on an 8% SDS-PAGE gel and detected by western blotting with rabbit anti-mouse secondary antibodies, which directly bind the C-terminal Protein A epitopes of the Deg1-Sec62 fusion protein. As a loading control, the membrane was subsequently incubated with antibodies specific to Pgk1. This experiment has been replicated three times; representative results are pictured. (B) Quantification of data in (A) was performed using imaging software (see Table of Materials). The total fluorescence intensity of an area encompassing the Deg1-Sec62 protein was determined for each sample. Background fluorescence intensity for each sample was extrapolated from the average fluorescence intensity of pixels near the Deg1-Sec62 protein. This extrapolated background fluorescence intensity was subtracted from the total fluorescence intensity to yield adjusted fluorescence intensity for each sample. Similar quantifications for total, background, and adjusted fluorescence intensities were performed for Pgk1, a loading control protein whose abundance does not vary in the conditions assayed. To compare the abundance of Deg1-Sec62 protein among samples, a ratio of the adjusted signal intensities of Deg1-Sec62 to Pgk1 was determined for each sample. The Deg1-Sec62/Pgk1 ratio was defined as 100% for the first time point (i.e., immediately after cycloheximide addition) for each strain under analysis. The amount of Deg1-Sec62 remaining at subsequent time points (i.e., the Deg1-Sec62/Pgk1 ratios) was calculated as a percentage of the Deg1-Sec62/Pgk1 ratio at the first time point. Please click here to view a larger version of this figure.
Figure 2. Cycloheximide Chase followed by Western Blotting Indicates that Cue1 is Required for Efficient Deg1-Sec62 Degradation. (A) Yeast cells of the indicated genotypes were transformed with a yeast centromeric plasmid encoding Deg1-Sec62 (pRS416-PMET25-Deg1-Flag-Sec62-2xProtA; AmpR/URA343) and subjected to cycloheximide chase analysis. Proteins from each time point (equivalent to 0.125 OD600 units) were separated on an 8% SDS-PAGE gel and detected by western blotting with rabbit anti-mouse secondary antibodies, which directly bind the C-terminal Protein A epitopes of the Deg1-Sec62 fusion protein. As a loading control, the membrane was subsequently incubated with antibodies specific to Pgk1. This experiment has been replicated three times; representative results are pictured. (B) Quantification of data in (A) was performed using imaging software (see Table of Materials). The total fluorescence intensity of an area encompassing the Deg1-Sec62 protein was determined for each sample. Background fluorescence intensity for each sample was extrapolated from the average fluorescence intensity of pixels near the Deg1-Sec62 protein. This extrapolated background fluorescence intensity was subtracted from the total fluorescence intensity to yield adjusted fluorescence intensity for each sample. Similar quantifications for total, background, and adjusted fluorescence intensities were performed for Pgk1, a loading control protein whose abundance does not vary in the conditions assayed. To compare the abundance of Deg1-Sec62 protein among samples, a ratio of the adjusted signal intensities of Deg1-Sec62 to Pgk1 was determined for each sample. The Deg1-Sec62/Pgk1 ratio was defined as 100% for the first time point (i.e., immediately after cycloheximide addition) for each strain under analysis. The amount of Deg1-Sec62 remaining at subsequent time points (i.e., the Deg1-Sec62/Pgk1 ratios) was calculated as a percentage of the Deg1-Sec62/Pgk1 ratio at the first time point. Please click here to view a larger version of this figure.
| Synthetic Defined (SD) Minimal Yeast Medium | 2% dextrose, 0.67% yeast nitrogen base without amino acids, 0.002% adenine, 0.004% uracil, 0.002% arginine, 0.001% histidine, 0.006% isoleucine, 0.006% leucine, 0.004% lysine, 0.001% methionine, 0.006% phenylalanine, 0.005% threonine, 0.004% tryptophan. For solid (plate) medium, include 2% agar. | 1. Selective medium is prepared by omitting appropriate amino acid(s) or nitrogenous bases. |
| 2. For convenience, these ingredients may be maintained as concentrated stock solutions as follows. Amino acids may be maintained as 100x stock solution containing all desired amino acids. Yeast nitrogen base may be maintained in a 20x stock solution (13.4%). Dextrose may be maintained in a 40% stock solution. Adenine and uracil may be maintained as 1% stock solutions in 0.1 M NaOH. | ||
| 3. Sterilize medium by autoclaving. | ||
| Yeast Extract-Peptone Dextrose (YPD) Rich Yeast Medium | 1% yeast extract, 2% peptone, 2% dextrose (Optional: 0.002% adenine). For solid (plate) medium, include 2% agar. | 1. To prevent the slow growth and red coloration characteristic of particular laboratory yeast strains in the absence (or low abundance) of adenine, adenine may be added to YPD growth medium. |
| 2. For convenience, some ingredients may be maintained as concentrated stock solutions as follows. Dextrose may be maintained in a 40% stock solution. Adenine may be maintained as a 1% stock solution in 0.1 M NaOH. | ||
| 3. Sterilize medium by autoclaving. | ||
| 20x Stop Mix | 200 mM sodium azide, 5 mg/ml bovine serum albumin | Sodium azide is toxic via oral ingestion or dermal contact. Follow manufacturer recommendations when preparing, storing, and handling sodium azide. In the event of accidental exposure, consult material safety data sheet provided by manufacturer. |
| 20 mg/ml Cycloheximide | 1. Prepare in ethanol. Store at -20 °C. | |
| 2. Cycloheximide is a dermal irritant and is toxic via oral ingestion. Follow manufacturer recommendations when preparing, storing, and handling cycloheximide. In the event of accidental exposure, consult material safety data sheet provided by manufacturer. | ||
| 0.2 M Sodium Hydroxide | Prepare in water. Sodium hydroxide reacts with glass. Therefore, for long-term storage, 0.2 M sodium hydroxide should be maintained in plastic containers. | |
| Laemmli Sample Buffer | 2% SDS, 10% glycerol, 5% β-mercaptoethanol, 60 mM Tris HCl pH 6.8, 0.008% bromophenol blue | 1. Laemmli Sample Buffer is often prepared by diluting a more concentrated (e.g., 5x) stock. |
| 2. The dye bromophenol blue may be added to desired intensity. A "pinch" (very small amount tapped from the edge of a spatula) is typically sufficient. | ||
| Laemmli Running Buffer (5x) | 125 mM Tris, 960 mM glycine, 0.5% SDS | To prepare 1 L of 1x Laemmli Running Buffer, dilute 1:5 in dH2O. |
| Tris Acetate-SDS Transfer Buffer (5x) | 125 mM Tris acetate (pH 8.8), 960 mM glycine, 0.05% SDS | To prepare 20 L of 1x Tris Acetate-SDS Transfer Buffer, combine 4 L of 5x stock, 4 L of methanol, and 12 L of dH2O. |
| 10x Tris-Buffered Saline (TBS) | 500 mM Tris, 1.5 M NaCl; pH adjusted to 7.5 | To prepare 1 L of 1x TBS, dilute 1:10 in dH2O. 1x TBS may be supplemented with the detergent Tween-20 and powdered skim milk, as appropriate. |
Table 1. Solutions Used in this Study.
| Strain Name | Alias | Relevant Genotype | References |
| VJY42 | MHY501 | MATα | Chen et al., 1993 |
| his3-Δ200 | |||
| leu2-3,112 | |||
| ura3-52 | |||
| lys2-801 | |||
| trp1-1 | |||
| gal2 | |||
| VJY47 | YWO55; MHY507 | MATα | Jungmann et al., 1993; Rubenstein et al., 2012 |
| his3-Δ200 | |||
| leu2-3,112 | |||
| ura3-52 | |||
| lys2-801 | |||
| trp1-1 | |||
| gal2 | |||
| ubc7Δ::LEU2 | |||
| VJY56 | YTX105; MHY1364 | MATα | Biederer et al., 1997; Ravid et al., 2006 |
| his3-Δ200 | |||
| leu2-3,112 | |||
| ura3-52 | |||
| lys2-801 | |||
| trp1-1 | |||
| gal2 | |||
| cue1Δ::HIS3 | |||
| VJY172 | MHY6199 | MATα | This study |
| his3-Δ200 | |||
| leu2-3,112 | |||
| ura3-52 | |||
| lys2-801 | |||
| trp1-1 | |||
| gal2 | |||
| hrd1Δ::kanMX4 |
Table 2. Yeast Strains Used in this Study. All strains are congenic with MHY500 53. VJY42, VJY47, and VJY56 were described previously 49,53,54. A detailed strain generation and confirmation strategy for VJY172 (prepared for this study) is available upon request.
Discussion
In this paper, a method for analyzing protein degradation kinetics is presented. This technique can be readily applied to a range of proteins degraded by a variety of mechanisms. It is important to note that cycloheximide chase experiments report on degradation kinetics of the steady state pool of a given protein. Other techniques may be used to analyze the degradation kinetics of specific populations of proteins. For example, the degradative fate of nascent polypeptides can be tracked by pulse chase analysis 55. In such experiments, nascent proteins are briefly pulse-labeled (e.g., with radioactive amino acids). Changes in the abundance of the labeled nascent proteins (which may or may not mirror changes in steady state protein abundance) may be tracked over time.
Cycloheximide chase is suitable for analyzing protein stability over a relatively short time course (i.e., up to two hours). Over longer time courses (i.e., two hours to days), cycloheximide, a global inhibitor of translation, is toxic to cells, likely due to depletion of ubiquitin 56. Further, analyses of protein stability over longer time courses are more likely to be compromised by indirect effects of globally impaired protein synthesis on the degradation of the protein of interest (e.g., degradation of a short-lived protein involved in the degradation of the protein of interest). Other techniques, such as pulse chase metabolic labeling experiments, are therefore better suited for studying the degradation of long-lived proteins and may be performed to corroborate results obtained in cycloheximide chase experiments.
The timing of addition of cycloheximide and collection of cells is an important consideration in this procedure. Precision is especially important in the analysis of very short-lived proteins, as small deviations in the time elapsed from cycloheximide addition to cell harvest can have a substantial impact on apparent degradation kinetics. Further, without a clear plan for executing these time-sensitive steps, an experiment may rapidly devolve into chaos and frustration. For this reason, it is recommended to add cycloheximide to cultures at planned, regular intervals. 30-sec intervals are suggested in this protocol, but the timing may be adjusted to match the comfort level and dexterity of the investigator. Novice investigators may find it useful to write a schedule of sample collection times prior to initiating the experiment and to cross off each scheduled collection as it occurs.
In Steps 1.7 and 1.8 of the protocol described here, yeast cell cultures are concentrated in reduced volumes of growth medium for ease of manipulation during the subsequent cycloheximide chase procedure. However, centrifugation of yeast cells rapidly induces certain cellular stress responses (e.g., signaling pathways that are activated by glucose deprivation) 57,58. Investigators are therefore cautioned against extended periods of centrifugation. When analyzing the stability of proteins that may be sensitive to centrifugation, investigators may wish to add cycloheximide directly to mid-log phase cultures without prior centrifugation. In such cases, cycloheximide should be added to the same final concentration (250 μg/ml), and yeast cell samples may be harvested by methodologies that do not require an initial centrifugation step (e.g., rapid filtration) 57. Further, in Steps 2.3 - 2.5, samples collected at each time point are transferred to a solution containing sodium azide and placed on ice. These conditions inhibit continued protein degradation for the duration of the chase. It should be noted that azide reduces the cellular concentration of ATP 59, thereby specifically inhibiting the activity of ATP-dependent proteases. While reduced temperature should also reduce the rate at which non-ATP-dependent proteolytic processes function, investigators studying proteins degraded by such mechanisms may alternatively choose to snap-freeze cell pellets in liquid nitrogen as each sample is harvested.
A sample protocol for cell lysis and a representative protocol for western blotting included in this manuscript have been adapted from previous reports 16,40. In the post-alkaline lysis method described here, NaOH pretreatment enhances extraction of yeast proteins upon subsequent addition of Laemmli sample buffer. The mechanism by which this enhancement occurs is poorly characterized 40. However, polysaccharides such as those found in yeast cell walls are solubilized in alkali conditions 60. It is therefore tempting to speculate that incubation in the presence of NaOH partially or completely spheroplasts yeast cells (i.e., removes cell wall components), rendering them more sensitive to the SDS detergent present in the Laemmli sample buffer. Because proteins are released under highly denaturing conditions, protease inhibitors need not be included in the Laemmli sample buffer. The specific method of cell lysis may be modified as appropriate for a given experiment. For instance, the post-alkaline lysis method may not be suitable for low-abundance proteins or proteins that are poorly detected by western blot. In such cases, methods that include a trichloroacetic acid precipitation step to concentrate protein samples may be most appropriate 61. Moreover, several important considerations regarding antibody selection, loading control choice, and alternative means of detection (e.g., chemiluminescent western blotting using film or digital imaging systems) have been recently discussed 16. Quantitation of protein abundance following cycloheximide treatment may be performed. Many imaging systems are sold with software packages that facilitate this analysis.
Cycloheximide chase may be implemented to analyze the degradation of a variety of yeast proteins and may be adapted to study protein stability in other eukaryotic cells. As described in this protocol, cycloheximide treatment is followed by cell lysis and detection of protein abundance by western blot analysis. Depending on the application, however, protein abundance following cycloheximide treatment may be assessed by a range of techniques, as appropriate for research objectives. For example, if protein size is not a relevant factor for analysis, cell lysates may be subjected to dot blot or enzyme-linked immunosorbent assay (ELISA) analysis, which report on protein abundance, but not apparent molecular weight 62. For proteins on the cell surface (or internal fluorescently tagged internal proteins), flow cytometry may be used to quantify protein abundance of samples at different times after cycloheximide addition 51. Fluorescence microscopy following cycloheximide treatment would provide information on both protein abundance and localization 18. The range of substrates, organisms, and downstream applications amenable to cycloheximide chase makes the technique a highly versatile and informative means of studying protein degradation.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors thank current and former members of the Rubenstein lab for providing a supportive and enthusiastic research environment. The authors thank Mark Hochstrasser (Yale University) for sharing reagents and expertise. E.M.R. thanks Stefan Kreft (University of Konstanz) and Jennifer Bruns (University of Pittsburgh) for sharing invaluable expertise in kinetic analysis of proteins. This work was supported by a National Institutes of Health grant (R15 GM111713) to E.M.R., a Ball State University ASPiRE research award to E.M.R, a research award from the Ball State University chapter of Sigma Xi to S.M.E., and funds from the Ball State University Provost's Office and Department of Biology.
References
- Jankowska E, Stoj J, Karpowicz P, Osmulski PA, Gaczynska M. The proteasome in health and disease. Cur Pharm Des. 2013;19(6):1010–1028. [PubMed] [Google Scholar]
- Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006;6(5):369–381. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- Amm I, Sommer T, Wolf DH. Protein quality control and elimination of protein waste: the role of the ubiquitin-proteasome system. Biochim Biophys Acta. 2014;1843(1):182–196. doi: 10.1016/j.bbamcr.2013.06.031. [DOI] [PubMed] [Google Scholar]
- Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426(6968):895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- Guerriero CJ, Brodsky JL. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol Rev. 2012;92(2):537–576. doi: 10.1152/physrev.00027.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagan J, Seto T, Pagano M, Cittadini A. Role of the ubiquitin proteasome system in the heart. Circ Res. 2013;112(7):1046–1058. doi: 10.1161/CIRCRESAHA.112.300521. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443(7113):780–786. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- Turnbull EL, Rosser MF, Cyr DM. The role of the UPS in cystic fibrosis. BMC Biochem. 2007;8 Suppl 1:S11. doi: 10.1186/1471-2091-8-S1-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford L, Lowe J, Dick LR, Mayer RJ, Brownell JE. Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nat Rev Drug Discov. 2011;10(1):29–46. doi: 10.1038/nrd3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar G, Keskin O, Fraternali F, Gursoy A. Emerging role of the ubiquitin-proteasome system as drug targets. Curr Pharm Des. 2013;19(18):3175–3189. doi: 10.2174/1381612811319180002. [DOI] [PubMed] [Google Scholar]
- Paul S. Dysfunction of the ubiquitin-proteasome system in multiple disease conditions: therapeutic approaches. BioEssays: News and Reviews in Molecular, Cellular and Developmental Biology. 2008;30(11-12):1172–1184. doi: 10.1002/bies.20852. [DOI] [PubMed] [Google Scholar]
- Shen M, Schmitt S, Buac D, Dou QP. Targeting the ubiquitin-proteasome system for cancer therapy. Expert Opin Ther Targets. 2013;17(9):1091–1108. doi: 10.1517/14728222.2013.815728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowder JJ, et al. Rkr1/Ltn1 Ubiquitin Ligase-Mediated Degradation of Translationally Stalled Endoplasmic Reticulum Proteins. J Biol Chem. 2015;290(30):18454–18466. doi: 10.1074/jbc.M115.663559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RG, et al. Endoplasmic reticulum degradation requires lumen to cytosol signaling. Transmembrane control of Hrd1p by Hrd3p. J Cell Biol. 2000;151(1):69–82. doi: 10.1083/jcb.151.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger MB, Maurer MJ, Dancy BM, Michaelis S. Degradation of a cytosolic protein requires endoplasmic reticulum-associated degradation machinery. J Biol Chem. 2008;283(47):32302–32316. doi: 10.1074/jbc.M806424200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts SG, Crowder JJ, Coffey SZ, Rubenstein EM. Growth-based Determination and Biochemical Confirmation of Genetic Requirements for Protein Degradation in Saccharomyces cerevisiae. J Vis Exp. 2015. p. e52428. [DOI] [PMC free article] [PubMed]
- Cohen I, Geffen Y, Ravid G, Ravid T. Reporter-based growth assay for systematic analysis of protein degradation. J Vis Exp. 2014. p. e52021. [DOI] [PMC free article] [PubMed]
- Ravid T, Kreft SG, Hochstrasser M. Membrane and soluble substrates of the Doa10 ubiquitin ligase are degraded by distinct pathways. EMBO J. 2006;25(3):533–543. doi: 10.1038/sj.emboj.7600946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmann S, Schafer A, Wolf DH. Ubiquitin ligase Hul5 is required for fragment-specific substrate degradation in endoplasmic reticulum-associated degradation. J Biol Chem. 2008;283(24):16374–16383. doi: 10.1074/jbc.M801702200. [DOI] [PubMed] [Google Scholar]
- Medicherla B, Kostova Z, Schaefer A, Wolf DH. A genomic screen identifies Dsk2p and Rad23p as essential components of ER-associated degradation. EMBO Rep. 2004;5(7):692–697. doi: 10.1038/sj.embor.7400164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habeck G, Ebner FA, Shimada-Kreft H, Kreft SG. The yeast ERAD-C ubiquitin ligase Doa10 recognizes an intramembrane degron. J Cell Biol. 2015;209(2):261–273. doi: 10.1083/jcb.201408088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran JR, Brodsky JL. Assays to measure ER-associated degradation in yeast. Methods Mol Biol. 2012;832:505–518. doi: 10.1007/978-1-61779-474-2_36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao SH, et al. GSK3beta controls epithelial-mesenchymal transition and tumor metastasis by CHIP-mediated degradation of Slug. Oncogene. 2014;33(24):3172–3182. doi: 10.1038/onc.2013.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY, Rine J. Regulated degradation of HMG-CoA reductase, an integral membrane protein of the endoplasmic reticulum, in yeast. J Cell Biol. 1994;125(2):299–312. doi: 10.1083/jcb.125.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser M, Varshavsky A. In vivo degradation of a transcriptional regulator: the yeast alpha 2 repressor. Cell. 1990;61(4):697–708. doi: 10.1016/0092-8674(90)90481-s. [DOI] [PubMed] [Google Scholar]
- Schneider-Poetsch T, et al. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol. 2010;6(3):209–217. doi: 10.1038/nchembio.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiffen AJ, Bohonos N, Emerson RL. The Production of an Antifungal Antibiotic by Streptomyces griseus. J Bacteriol. 1946;52(5):610–611. doi: 10.1128/jb.52.5.610-611.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obrig TG, Culp WJ, McKeehan WL, Hardesty B. The mechanism by which cycloheximide and related glutarimide antibiotics inhibit peptide synthesis on reticulocyte ribosomes. J Biol Chem. 1971;246(1):174–181. [PubMed] [Google Scholar]
- Ennis HL, Lubin M. Cycloheximide: Aspects of Inhibition of Protein Synthesis in Mammalian Cells. Science. 1964;146(3650):1474–1476. doi: 10.1126/science.146.3650.1474. [DOI] [PubMed] [Google Scholar]
- Kerridge D. The effect of actidione and other antifungal agents on nucleic acid and protein synthesis in Saccharomyces carlsbergensis. J Gen Microbiol. 1958;19(3):497–506. doi: 10.1099/00221287-19-3-497. [DOI] [PubMed] [Google Scholar]
- Chakrabarti S, Dube DK, Roy SC. Effects of emetine and cycloheximide on mitochondrial protein synthesis in different systems. Biochem J. 1972;128(2):461–462. doi: 10.1042/bj1280461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seufert W, Jentsch S. In vivo function of the proteasome in the ubiquitin pathway. EMBO J. 1992;11(8):3077–3080. doi: 10.1002/j.1460-2075.1992.tb05379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky A. Discovery of the biology of the ubiquitin system. JAMA. 2014;311(19):1969–1970. doi: 10.1001/jama.2014.5549. [DOI] [PubMed] [Google Scholar]
- Heinemeyer W, Kleinschmidt JA, Saidowsky J, Escher C, Wolf DH. Proteinase yscE, the yeast proteasome/multicatalytic-multifunctional proteinase: mutants unravel its function in stress induced proteolysis and uncover its necessity for cell survival. EMBO J. 1991;10(3):555–562. doi: 10.1002/j.1460-2075.1991.tb07982.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer T, Jentsch S. A protein translocation defect linked to ubiquitin conjugation at the endoplasmic reticulum. Nature. 1993;365(6442):176–179. doi: 10.1038/365176a0. [DOI] [PubMed] [Google Scholar]
- Hiller MM, Finger A, Schweiger M, Wolf DH. ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science. 1996;273(5282):1725–1728. doi: 10.1126/science.273.5282.1725. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nature Protoc. 2007;2(1):31–34. doi: 10.1038/nprot.2007.13. [DOI] [PubMed] [Google Scholar]
- Bergman LW. Growth and maintenance of yeast. Methods Mol Biol. 2001;177:9–14. doi: 10.1385/1-59259-210-4:009. [DOI] [PubMed] [Google Scholar]
- Biederer T, Volkwein C, Sommer T. Degradation of subunits of the Sec61p complex, an integral component of the ER membrane, by the ubiquitin-proteasome pathway. EMBO J. 1996;15(9):2069–2076. [PMC free article] [PubMed] [Google Scholar]
- Kushnirov VV. Rapid and reliable protein extraction from yeast. Yeast. 2000;16(9):857–860. doi: 10.1002/1097-0061(20000630)16:9<857::AID-YEA561>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Sharma M, Benharouga M, Hu W, Lukacs GL. Conformational and temperature-sensitive stability defects of the delta F508 cystic fibrosis transmembrane conductance regulator in post-endoplasmic reticulum compartments. J Biol Chem. 2001;276(12):8942–8950. doi: 10.1074/jbc.M009172200. [DOI] [PubMed] [Google Scholar]
- Mayer TU, Braun T, Jentsch S. Role of the proteasome in membrane extraction of a short-lived ER-transmembrane protein. EMBO J. 1998;17(12):3251–3257. doi: 10.1093/emboj/17.12.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein EM, Kreft SG, Greenblatt W, Swanson R, Hochstrasser M. Aberrant substrate engagement of the ER translocon triggers degradation by the Hrd1 ubiquitin ligase. J Cell Biol. 2012;197(6):761–773. doi: 10.1083/jcb.201203061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DC, Schekman R. Role of Sec61p in the ER-associated degradation of short-lived transmembrane proteins. J Cell Biol. 2008;181(7):1095–1105. doi: 10.1083/jcb.200804053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault G, Ng DT. The endoplasmic reticulum-associated degradation pathways of budding yeast. Cold Spring Harb Perspect Biol. 2012;4(12) doi: 10.1101/cshperspect.a013193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher EA, et al. The degradation of apolipoprotein B100 is mediated by the ubiquitin-proteasome pathway and involves heat shock protein 70. J Biol Chem. 1997;272(33):20427–20434. doi: 10.1074/jbc.272.33.20427. [DOI] [PubMed] [Google Scholar]
- Pariyarath R, et al. Co-translational interactions of apoprotein B with the ribosome and translocon during lipoprotein assembly or targeting to the proteasome. J Biol Chem. 2001;276(1):541–550. doi: 10.1074/jbc.M007944200. [DOI] [PubMed] [Google Scholar]
- Yeung SJ, Chen SH, Chan L. Ubiquitin-proteasome pathway mediates intracellular degradation of apolipoprotein. B. Biochemistry. 1996;35(43):13843–13848. doi: 10.1021/bi9618777. [DOI] [PubMed] [Google Scholar]
- Biederer T, Volkwein C, Sommer T. Role of Cue1p in ubiquitination and degradation at the ER surface. Science. 1997;278(5344):1806–1809. doi: 10.1126/science.278.5344.1806. [DOI] [PubMed] [Google Scholar]
- Metzger MB, et al. A structurally unique E2-binding domain activates ubiquitination by the ERAD E2, Ubc7p, through multiple mechanisms. Mol Cell. 2013;50(4):516–527. doi: 10.1016/j.molcel.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazirgan OA, Hampton RY. Cue1p is an activator of Ubc7p E2 activity in vitro and in vivo. J Biol Chem. 2008;283(19):12797–12810. doi: 10.1074/jbc.M801122200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, et al. The Png1-Rad23 complex regulates glycoprotein turnover. J Cell Biol. 2006;172(2):211–219. doi: 10.1083/jcb.200507149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Johnson P, Sommer T, Jentsch S, Hochstrasser M. Multiple ubiquitin-conjugating enzymes participate in the in vivo degradation of the yeast MAT alpha 2 repressor. Cell. 1993;74(2):357–369. doi: 10.1016/0092-8674(93)90426-q. [DOI] [PubMed] [Google Scholar]
- Jungmann J, Reins HA, Schobert C, Jentsch S. Resistance to cadmium mediated by ubiquitin-dependent proteolysis. Nature. 1993;361(6410):369–371. doi: 10.1038/361369a0. [DOI] [PubMed] [Google Scholar]
- Tansey WP. Pulse-chase assay for measuring protein stability in yeast. Cold Spring Harb Protoc. 2007. [DOI] [PubMed]
- Hanna J, Leggett DS, Finley D. Ubiquitin depletion as a key mediator of toxicity by translational inhibitors. Mol Cell Biol. 2003;23(24):9251–9261. doi: 10.1128/MCB.23.24.9251-9261.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson WA, Hawley SA, Hardie DG. Glucose repression/derepression in budding yeast: SNF1 protein kinase is activated by phosphorylation under derepressing conditions, and this correlates with a high AMP:ATP ratio. Curr Biol. 1996;6(11):1426–1434. doi: 10.1016/s0960-9822(96)00747-6. [DOI] [PubMed] [Google Scholar]
- Ashe MP, De Long SK, Sachs AB. Glucose depletion rapidly inhibits translation initiation in yeast. Mol Biol Cell. 2000;11(3):833–848. doi: 10.1091/mbc.11.3.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant CM, MacIver FH, Dawes IW. Mitochondrial function is required for resistance to oxidative stress in the yeast Saccharomyces cerevisiae. FEBS Lett. 1997;410(2-3):219–222. doi: 10.1016/s0014-5793(97)00592-9. [DOI] [PubMed] [Google Scholar]
- Javmen A, et al. beta-Glucan from Saccharomyces cerevisiae Induces IFN-gamma Production In Vivo in BALB/c Mice. In Vivo. 2015;29(3):359–363. [PubMed] [Google Scholar]
- Link AJ, LaBaer J. Trichloroacetic acid (TCA) precipitation of proteins. Cold Spring Harb Protoc. 2011;2011(8):993–994. doi: 10.1101/pdb.prot5651. [DOI] [PubMed] [Google Scholar]
- Hornbeck PV. Enzyme-Linked Immunosorbent Assays. Curr Protoc Immunol. 2015;110:2.1.1–2.1.23. doi: 10.1002/0471142735.im0201s110. [DOI] [PubMed] [Google Scholar]