

Abstract
A protocol is described for sequencing the transcriptome of a cell nucleus. Nuclei are isolated from specimens and sorted by FACS, cDNA libraries are constructed and RNA-seq is performed, followed by data analysis. Some steps follow published methods (Smart-seq2 for cDNA synthesis and Nextera XT barcoded library preparation) and are not described in detail here. Previous single-cell approaches for RNA-seq from tissues include cell dissociation using protease treatment at 30 °C, which is known to alter the transcriptome. We isolate nuclei at 4 °C from tissue homogenates, which cause minimal damage. Nuclear transcriptomes can be obtained from postmortem human brain tissue stored at −80 °C, making brain archives accessible for RNA-seq from individual neurons. The method also allows investigation of biological features unique to nuclei, such as enrichment of certain transcripts and precursors of some noncoding RNAs. By following this procedure, it takes about 4 d to construct cDNA libraries that are ready for sequencing.
INTRODUCTION
Methods for carrying out RNA-seq from single cells1–5 are dramatically affecting many research fields, including the study of cellular development, the identification of cell types and states, the exploration of human disease and the development of stem cell technologies. The gene expression repertoires of individual cell types are revealed as opposed to the averaging of all transcrip-tomes obtained from bulk tissue. However, cells of the central nervous system (CNS) have been under-studied, partly because of the difficulty of isolating intact whole cells. Neurons are highly interconnected, and considerable damage must be done to their extensions to separate them by physical means such as laser-capture microdissection. An intracellular tagging method called TIVA uses RNA extracted from single cells, but it is limited to small numbers of cells6. Extraction of cytoplasmic content by a glass microcapillary7,8 or by laser-capture microdissection9 is of low throughput. An alternative, high-throughput approach is to disperse the cells and to isolate them by FACS. This approach has been recently reported for neurons isolated from brain tissue10,11. However, dispersion of cells by proteolytic degradation of surface proteins places the cells under stress, which substantially alters gene expression12.
We have developed an alternative approach that takes advantage of the low levels of mRNA contained in the nucleus of the cell13, and it avoids harsh treatment that would perturb gene expression. Through extensive comparisons of nuclear and cellular transcriptomes, we demonstrated that nuclei can substitute for whole cells in most RNA-seq applications13. For the majority of genes, nuclei yielded expression signatures that were very similar to those obtained from whole-cell controls. Furthermore, some transcripts that are known to be enriched in the nucleus on the basis of earlier bulk RNA studies14–17 were also confirmed to be enriched in single nuclei, adding confidence to the accuracy of data. Here we provide a detailed protocol based on our previously published method13 for RNA-seq using nuclei from brain tissue or cells, which can be used to obtain global transcriptomes from neurons, glia and other cell types. Although it is described here for brain tissue, it should also be applicable to any tissue type in which dissociation of whole cells would require harsh treatments and the consequent alteration of the transcriptome.
Development of the protocol
Many methods are available for the isolation of nuclei; however, the literature spans decades, and it typically lacks detailed information on the quality of RNA obtained, focusing instead on accessing intact DNA for chromatin preparation or for assaying the nuclear protein content18. We therefore developed an approach to meet the need for isolating individual nuclei for use in RNA analysis, which we have successfully applied to cultured neuroprogenitor cells and fresh mouse brain tissue13. The protocol detailed here includes two main modifications to the published method. First, we now consider cleanup by sucrose-iodixanol gradient centrifugation18 to be necessary only if cell debris is likely to interfere with immunostaining; it is therefore included in the PROCEDURE as an optional step with the default approach to subject the filtered crude homogenate directly to FACS19. Second, we now use Smart-seq2 for cDNA synthesis3 (instead of the method by Tang et al.5), which is reported to improve synthesis of full-length cDNA via a template switching mechanism for synthesis of the second-strand cDNA4.
Overview of the procedure
Our experimental workflow (Fig. 1) begins with tissue homogenization in the presence of a detergent to lyse the cell membrane, determination of the number of nuclei obtained with a hemocytometer (Steps 1–5) and FACS (Steps 13–18). The nuclei are lysed and cDNA is synthesized, amplified (Step 19) and tested in quantitative PCR (qPCR) quality control assays to indicate successful capture of the transcriptome by assaying several housekeeping and tissue-specific genes (Steps 20–23). Samples that pass quality control assays are used in downstream sequencing library preparation (Step 24) and RNA-seq (Step 25). A series of bioinformatic analyses then follow to assess sequence quality (Steps 26–28), mapping and expression (Steps 29–34), variation (Steps 35–38), gene coverage (Steps 39–41), intron and exon coverage (Steps 42–46), and the classification of cell types (Step 47). The main stages of the protocol are discussed in more detail below.
Figure 1.

Single nuclei isolation experimental workflow. Dounce homogenization in lysis buffer is used to disrupt cellular membranes for fresh or frozen tissue (a). Nuclei quality and yield is determined by hemocytometer count (b). (c–e) Nuclei and cellular debris are filtered for optional purification and immunostaining steps (density gradient centrifugation (c) or staining for neuronal enrichment (d)), or for FACS sorting (e). (f,g) Subsequently, lysis of the nuclei and cDNA synthesis is carried out using either published methods3 or commercial kits (SMARTer, Clontech) (f), and it is quality-controlled for size distribution using a Bioanalyzer (Agilent) and the presence of several transcripts by qPCR (g). (h) Sequencing and data analysis confirm single nucleus transcriptome capture. Step numbers indicate the corresponding step numbers in the PROCEDURE section. Graphs in g and h are for illustrative purposes only.
Tissue handling and homogenization to release nuclei
In general, the initial quality and methods used to handle postmortem brain specimens will affect the quality of the RNA-seq data. RIN scores (RNA integrity number20 ranging from 1 to 10) for specimens are often provided by the brain banks; however, we also determined RIN scores in our laboratory and sometimes found differences, possibly because the specimens had been stored for long periods of time and then taken through a thawing step in our laboratory. The RIN scores that we determined were used to evaluate the starting quality of the frozen specimens. We selected specimens with a RIN value of ≥7.
We chose Dounce homogenization to handle the very small tissue dissections often required to investigate various brain regions. Dounce homogenization21 with a nonionic surfactant, Triton X-100, is used to lyse the cell membrane and release nuclei. The detergent can also permeabilize and lyse the nuclear envelope, but only under harsh conditions for an extended period of time22. Sufficient Triton X-100 is included in the homogenization step to facilitate the release of nuclei, allowing them to remain intact, and to permit optimal antibody18 staining and isolation by FACS without forming aggregates. Hoechst stain is added to the homogenization lysis buffer to identify nuclei during FACS.
Before proceeding with FACS, the overall quality of the nuclei and number obtained should be determined using fluorescence photomicrography (after Dounce homogenization and again after the sucrose-iodixanol gradient centrifugation if that optional step is performed). High-resolution electron microscopy has been used for assessing the integrity of the nuclei and purity of the preparation, but it will be impractical for most laboratories19. Light microscopy can be used to assess whether the outer cell membranes are lysed, and whether the suspension contains encumbering amounts of non-nuclear material. A phase-contrast light microscope should be used at each stage of the nuclear isolation procedure to evaluate the yield, purity and integrity of nuclei, which can be visualized and scored with a hemocytometer (Fig. 2a). Nuclei will stain with trypan blue, and the nucleolus can often be identified (Fig. 2b). Fluorescent labels, Hoechst for DNA and a neuronal nuclei marker, NeuN, can be used together for facile detection of nuclei derived from neurons (Fig. 2c–h).
Figure 2.

Quality control of nuclei isolation. (a,b) Nuclei were obtained from the human prefrontal cortex and extracted via Dounce homogenization; they were stained with 0.2% (vol/vol) trypan blue, counted on a hemocytometer (a), placed on a slide and microscopically examined for morphological quality and yield (b). (c,d) By using epifluorescence microscopy, nuclei were stained with DNA intercalating dye Hoechst 33342 (10 ng μl−1) (c), with blue fluorescent nuclei images overlaid with the bright-field image to identify intact nuclei (d). (e) After cell strainer filtration, nuclei were stained with NeuN-Alexa Fluor 488–conjugated antibody (0.01 mg ml−1) to identify intact neuronal nuclei. (f) The fluorescent image was overlaid with the bright-field image to further distinguish nuclei derived from neuronal versus non-neuronal cells. (g,h) By using FACS, cells were sorted onto a microscope slide and imaged for NeuN fluorescence (g) and overlaid in bright field (h) to confirm FACS sorting conditions.
Staining and FACS
To enable sorting of nuclei derived from neurons, nuclei can be immunostained with an antibody specific to NeuN, a nuclear membrane protein (Supplementary Fig. 1), before filtering the homogenate to remove large aggregated debris and subjecting it to FACS. Software gating on the FACS (Fig. 3) uses a series of doublet discrimination gates (Fig. 3a–c) to isolate single nuclei from any remaining aggregated nuclei, followed by a nuclear staining gate using Hoescht and NeuN labeling to isolate single neuronal nuclei (Fig. 3d,e). Alternatively, nuclei from all cell types can be sorted by using nuclear staining with either Hoechst or propidium iodide (PI) (Fig. 3d,f). Single nuclei are sorted into lysis buffer containing ERCC (External RNA Consortium Control) spike-in RNA standards (Ambion), which allow the sensitivity of transcript detection to be determined. Following FACS, single nuclei can be verified to be free of the debris particles and aggregated nuclei by microscopic observation (Figs. 2g,h and 3h,i).
Figure 3.

FACS of single nuclei. Nuclei triple-stained with NeuN-Alexa Fluor 488–conjugated antibody (0.01 mg ml−1; EMD Millipore), Hoechst 33342 (10 ng ml−1) and PI (1 μM) were filtered through a 35-μm cell strainer and loaded onto a custom FACS ARIA II flow sorter (Becton Dickinson) equipped with a forward scatter photomultiplier tube. (a–d) Doublet discrimination gating was used to isolate single nuclei (a–c) and intact nuclei determined by subgating on Hoechst 33342 (d). (a) Particles smaller than nuclei (black dots) are eliminated with an area plot of forward scatter (FSC-PMT-A) versus side scatter (SSC-A), with gating for nuclei-sized particles inside the gate (box). (b,c) Plots of height versus width in the side scatter and forward scatter channels, respectively, are used for doublet discrimination with gating to exclude aggregates of two or more nuclei. (e,f) Subsequent plots and gating discern NeuN-Alexa Fluor488–conjugated antibody (e) and PI-stained nuclei (f). The resultant hierarchical color key ensures that only single nuclei that are positive or negative for staining with the NeuN antibody (NeuN+ and NeuN−) are passed through each gating condition. (g) Yellow fluorescent 10- to 14-μm polystyrene microspheres (Spherotech) were used to determine the accuracy and precision of microplate targeting, and they were confirmed by microscopic imaging of single spheres in a 384-well microplate. (h,i) Subsequent FACS gating of labeled nuclei (arrows) was confirmed via imaging on a microscope slide (h), as well as within individual wells of a 384-well microplate (i).
Lysis of nuclei, cDNA preparation and quality control
We do not provide detailed procedural information for nuclear lysis and cDNA preparation. Instead, we refer users to the Smart-seq2 protocol3, which we now use because it generates a higher percentage of full-length cDNAs4. We follow the protocol exactly for lysis of the nuclei, but we have made two modifications for cDNA preparation: first, the cDNA is amplified by PCR for 21 cycles instead of 18 to compensate for the lower amount of RNA in a nucleus compared with a whole cell; second, the template-switching oligonucleotide (TSO) primer described in Picelli et al.3 is modified by 5′ biotinylation11. We have recently confirmed (M.N. and R.S.L., unpublished data) observations by others that this modification reduces non-specific amplification caused by synthesis of TSO concatemers.
Before investing time and funds in RNA-seq, we carry out quality control assays by qPCR for targeted gene products. We use reporter housekeeping genes (ACTB and GAPDH), as well as high-, medium- and low-copy ERCC spike-in control qPCR assays (Thermo Fisher). In addition, assays targeting genes specific for neuronal nuclei of interest are recommended.
Preparation of sequencing library and sequencing
For procedural details for preparing sequencing libraries, we refer users to the Fluidigm C1 manual (C1 System for mRNA-Seq, part no. 100–7168 available at https://www.fluidigm.com/documents; select ‘C1 System for mRNA Seq’ to download the PDF automatically). We use the Illumina Nextera XT library preparation kit and perform multiplexed paired-end sequencing of barcoded libraries using an Illumina MiSeq system. Figure 4 shows an example of the quality of the cDNA and sequencing library. Supplementary Table 1 shows a summary of a typical sequencing experiment. The cDNA insert size of the sequencing library is 250–500 bp, and the read-length of paired-end sequences is 150 bases. A read-depth of 1.5–2.0 × 106 has been previously shown to be adequate for the detection of saturating levels of RNA expression in single cells23.
Figure 4.

qPCR and Bioanalyzer quality control analysis of total mRNA, single-nucleus cDNA synthesis and a single-nucleus NexteraXT RNA-seq library. Total RNA from ~2–3 mm3 section of total human prefrontal cortex tissue was purified using a Qiagen RNeasy mini kit, quantified by Nanodrop spectrophotometry and diluted to 5 ng μl−1. (a) The mRNA quality was determined using RIN values by loading 1 μl onto an Agilent RNA pico chip and run on the Agilent Bioanalyzer. (b,c) Representative example using a single nucleus for Smart-seq2 cDNA synthesis followed by PCR amplification (b; 1 μl) and a Nextera XT sequencing library (c; 1 μl) were also analyzed. (b) After AMPure bead purification of the cDNA, a size range of ~150 bp to 7 kbp is expected, with the majority of fragments in the 1–3 kb range. After AMPure bead purification of each Nextera XT library, a size range of ~200 bp to 1 kbp is expected. The hash marks on the x axis are 35, 50, 100, 150, 200, 300, 400, 500, 600, 700, 1,000, 2,000, 3,000, 7,000 and 10,000, with lane marker peaks seen at 35 and 10,380 bp. Separately, Smart-seq2 synthesis of cDNA and PCR was performed on single nuclei (n = 24), and on pools of 8 nuclei (n = 4), 24 nuclei (n = 4), 48 nuclei (n = 2), 96 nuclei (n = 2) and duplicates of 100 pg, 10 pg and 1 pg total RNA from the prefrontal cortex, to serve as technical replicates to reveal artifactual noise level due to technical causes such as variation in pipetting and temperature differences between PCR block wells. NTCs are used to detect nonspecific cDNA amplification derived from contaminants in the reaction components or introduced during handling. (d) Quality control qPCR of cDNA was performed in 10-μl reactions using ABI TaqMan gene expression assays for GAPDH, ACTB and ERCC-00077. qPCR cycle threshold (Ct) values were plotted for comparison with single nuclei Cts, typically ranging between 15 and 25. Note that Cts increase by about 3 cycles per tenfold increase in input RNA template, as expected from the doubling rate of DNA in PCR.
Data analysis
The sequence reads are analyzed for quality and pre-processed to remove artifacts that fail to map to the genome (Box 1). A substantial number of reads contain Smart-seq2 primer and adapter sequences and their concatemers. In addition, deep sequencing yields many duplicate sequences of abundant transcripts that will reduce the ability to detect low-copy transcripts. Duplicate sequences cannot be removed, as removal would preclude accurate quantification of RNA expression. However, it is imperative that the levels of sequence duplication across samples are evaluated to examine its potential impact on the detection of low-copy transcripts.
Box 1. Sequence analysis: evaluation of sequence quality and preprocessing.
(A) Assessment of sequence quality
Illumina sequences obtained from each sample (nucleus) are analyzed by fastQC tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) to evaluate sequence yield, base quality, GC profile, k-mer distribution and primer contamination. A computer grid environment or a multiprocessor (CPU) Unix workstation is required for processing large numbers of samples simultaneously.
(B) Evaluation of sequence duplication
To assess the extent of unique transcript representation and any skewed PCR bias in the fragments represented in cDNA libraries, the degree of read duplication is analyzed. However, the duplicated RNA-seq reads are not removed, as it will preclude the accurate estimation of transcript abundance (expression). The fastx_collapser tool (http://hannonlab.cshl.edu/fastx_toolkit/commandline.html) is used with Phred 33 base quality score offset to calculate the absolute number of identical reads (duplicates) in the input sample .fastq sequences. The program accepts only one sequence file as input. Multiple sequence files require iterative processing by a shell script.
(C) Trimming of adapters, primers and low-quality bases
The Trimmomatic tool (http://www.usadellab.org/cms/?page=trimmomatic) is used to trim the adapter and/or primer sequences present in adapters_primers.txt (Supplementary Note) from the ends of PE input.sample.fastq sequences to facilitate their successful mapping to the reference transcriptome. The program, executed using eight threads per job, performs the following: trims the end bases below a Phred quality score of 3 or any bases in a 4-base-wide sliding window when the average quality per base drops below 15; clips adapters/primers from the sequences by allowing two seed mismatches, requiring a minimum of 30 matches in palindromic mode and a minimum of ten matches in nonpalindromic (simple) mode between the read sequence and the adapters/primers. Any sequences trimmed from the original length of 150 bases to shorter than 60 bases are removed from the output.
After quality assessment and trimming, we perform analysis of RNA expression using the RSEM package24, as described in Box 2 and Steps 29–31. The trimmed sequencing reads are mapped to the human and ERCC spike-in transcript reference sequences. The sequencing depth observed for a given transcript quantitatively reflects the number of mRNA template molecules obtained from the lysed nucleus. The total number of genes detected for each nucleus and the percentage of reads mapped to the genome and ERCC spike-in controls is determined (Fig. 5). The sensitivity of the detection of RNA expression across different samples is analyzed by evaluating the expression of both ERCC spike-in control transcripts (Fig. 6) and the human mRNA at different levels of abundance (Fig. 7). All sequencing data—even from high-quality RNA—will show some level of 3′ bias in the coverage, because the reverse transcriptase (RT) will fail to produce full-length cDNA for some proportion of the transcripts, resulting in little or no coverage for the 5′ end of these RNAs. Even though the Smart-seq2 method disfavors incomplete cDNA strand synthesis, some cDNA that is only partially extended is still generated. In addition, 3′ bias will be indicative of mRNA damage due to RNase degradation, shearing or hydrolysis, which might occur during tissue handling, storage or processing of the nuclei. Partially degraded RNA will result in deeper sequence coverage for the 3′ end of transcripts, as only those degradation products that contain the 3′ polyA tail will be converted to cDNA (Fig. 8a). To confirm that any 3′ bias observed in cDNA from nuclei is not due to RNA degradation, we compare the sequence coverage with that of a high-quality control RNA from the same tissue (Fig. 8b). Sequence coverage of introns and exons is used to ensure that the sequences are derived from mRNA rather than from genomic DNA (Fig. 9), which is not removed from the nuclear extracts.
Box 2. Sequence mapping and RNA expression analysis.
(A) Preparation of the reference genome
The trimmed sequencing reads are mapped to the transcripts derived from the human reference genome (GRCh37). The reference .fasta is prepared by the concatenation of GRCh37 human genome .fasta, the ERCC RNA spike-in .fasta and .fasta files for other marker (GFP) genes (RSEM_GRCh37_ERCC_GFP_RNASpikes.fa). The reference index files required by Bowtie2 mapping program and the transcript- specific reference sequences are generated from the GRCh37_ERCC_GFP_RNASpikes.fa and the corresponding annotation (GRCh37_ERCC_GFP_RNASpikes.gtf) files by the ‘rsem-prepare-reference’ command available in RSEM expression analysis software (http://deweylab.biostat.wisc.edu/rsem/).
(B) Mapping and the calculation of expression values
The ‘rsem-calculate-expression’ command from the RSEM expression analysis software is used to map paired-end reads to the reference transcripts (RSEM_GRCh37_ERCC_GFP_RNASpikes.transcripts.fa). The RNA expression values at gene and isoform levels are calculated using the expectation-maximization (EM) algorithm as implemented by the RSEM program. Multiple threads of eight or more are used to generate alignments mapped to genomic coordinates (sample_name.genome.bam), while tagging reads with nonunique alignments (--tag), calculating 95% credibility intervals (--calc-ci) and posterior mean estimates (--calc-pme), allowing insertions in the range of 1–500 bases (--fragment-length-min/max) and estimating the read start position distribution (--estimate-rspd). The text entries shown in parentheses in the preceding lines indicate the command’s options. The output files are prefixed with sample_name.
(C) Determination of the sensitivity of expression analysis
The ERCC spike-in transcripts available from Life Technologies (https://www.lifetechnologies.com/order/catalog/product/4456740) are added to the reverse transcriptase mix along with sample RNA before the cDNA amplification. The individual ERCC spike-in mRNAs (http://tools.lifetechnologies.com/content/sfs/manuals/cms_095046.txt), which are present at a wide range of low to high molar concentrations in the reaction mixture, facilitate the determination of the lower threshold of detection sensitivity of transcript expression in terms of copy numbers.
Figure 5.

Overall characteristics of mapping and expression. The sequencing reads for ten individual nuclei were split into three groups: ‘ERCC’, ‘Genome’ and ‘Unmapped’ on the basis of their mapping using the RSEM software. On average, 417,964, 183,278 and 941,644 reads were mapped to the genome for each neuronal nucleus, non-neuronal nucleus and total RNA sample, respectively. The numbers in the parenthesis indicate the number of genes with a TPM value >0 for the sample. It is clear that our sequencing did not reach saturation for some samples, as there is a high correlation between the number of reads mapped to the genome and the number of genes expressed. The high number of genes detected for Total RNA also reflects the pooling of RNA from multiple cells, which captures all genes expressed in the population.
Figure 6.
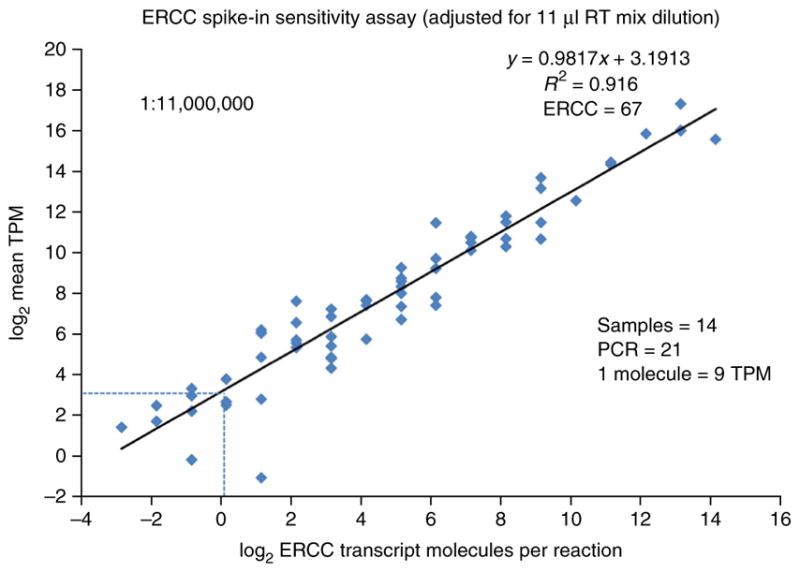
Behavior of ERCC spike-in controls, sensitivity and detection limit estimation. The number of ERCC spike-in transcript molecules, diluted 1.1 × 107 fold from the original stock in the final RT-mix, are plotted against the average TPM expression values across all 14 samples using log2 scale for both axes. The 1.1 × 107-fold dilution (PROCEDURE Step 13 and INTRODUCTION) is greater than that recommended by the ERCC spike-in manufacturer, who had optimized it for use with nanogram quantities of RNA in microarray studies. The low levels of RNA in a single nucleus necessitate the greater dilution in order to avoid high percentages of sequencing reads devoted to ERCC spike-ins. However, some of the lower-copy transcript species present in the ERCC spike-in stock are consequently diluted to <1 copy per Smart-seq2 reaction tube. ERCC spike-in transcripts with expression in at least one of the 14 nuclei were considered (ERCC n = 67 of 92) with regression equation y = 0.9817x + 3.1913 and R2 = 0.916. The RNA released from the lysed nuclei plus the added ERCC spike-in controls were amplified to 21 PCR cycles. The detection threshold for a single ERCC spike-in transcript molecule is shown to be approximately equivalent to 9 TPM RNA expression units (1 molecule = 9 TPM, as indicated by the intersection of the dashed lines).
Figure 7.

Biological variation and technical noise stratified by relative expression of genes. The genes that are expressed in bulk Total RNA-100pg-2 (see Supplementary Table 1) were stratified equally into low, mid and high expressers based on their TPM values (4,292 genes per category). Low genes had TPM values between 0.01 and 7.44, mid genes had TPM values between 7.45 and 25.97 and high genes had TPM values >25.98 (a). For the 4,292 genes of each category, the graph shows the fraction found in each sample. By definition, Total RNA-100pg-2 has 100%, 100% and 100% representation for low, mid and high (b). Each gene that is expressed in the sample is labeled by its expression in the bulk RNA sample. The fraction of low-, mid- and high-expressed genes, as well as novel genes that were not found in the bulk control, was quantified.
Figure 8.

The use of 3′ bias as a quality control assay for cDNA. (a) Total (bulk) RNA derived from tissue is confirmed to have a high RIN score before isolation of nuclei. Partial degradation of the RNA might occur during the preparation of nuclei by Dounce homogenization (nuclei prep) or FACS of the individual nuclei. If the mRNA is degraded by hydrolysis, shearing or RNases, truncated mRNA species could be created, and those containing the polyA sequence at the 3′ end of the transcripts might produce cDNA. This would generate greater RNA-seq coverage of the 3′ end of transcripts (3′-bias) compared with the high-quality bulk RNA. Gene body coverage across 4,292 highly expressed genes was calculated by RseqC. The relative coverage is defined as coverage at a base / maximum coverage across the gene. (b) The total RNA samples are indicated (two replicates of 10 pg and 100 pg RNA each; Supplementary Table 1). As these total RNA controls are all from a single RNA purification from bulk tissue, they would have identical coverage profiles in the ideal case. The minor differences indicate the level of technical variation accumulated from all of the reaction steps. The single nuclei have very similar 3′ bias to the total RNA controls, demonstrating that little damage was done to the RNA during the processing of nuclei. Neuronal nucleus 6 (Supplementary Table 1) is indicated, and it diverges from normal behavior. It may be an example of partially degraded mRNA being obtained from the nucleus and the resulting truncated cDNA; however, we believe that it is actually attributable to its low number of reads mapping to the genome, which must be taken into consideration for this analysis. We have recently confirmed that partially degraded total mRNA, which is formed experimentally by heating in the presence of sodium acetate, results in a commensurate increase in 3′ bias, demonstrating that this analysis can quantitatively detect RNA damage (M.N. and R.S.L., unpublished data).
Figure 9.

Read depth across the GAPDH gene. University of California at Santa Cruz (UCSC) genome browser snapshot of custom bedGraph tracks detailing the coverage across the GAPDH gene for neuronal nucleus 2, non-neuronal nucleus 4 and total RNA 100pg-2 samples (Supplementary Table 1). The lack of coverage across introns indicates that most of the GAPDH transcripts sequenced were spliced transcripts for all three types of sample types. The position of exons is indicated by the black rectangles in the genomic map at the bottom.
The primary goal of many single-cell or single-nuclei sequencing pipelines is the classification and characterization of known and potentially novel cell types, and several strategies have been presented for such analyses of hundreds to many thousands of cells11,25,26. For the small number of nuclei analyzed here, we developed an approach based on a straightforward application of dimensionality reduction (principal coordinate analysis), k-means clustering and manual inspection of canonical cell type marker genes (Fig. 10), which can be reproduced using the code provided as Supplementary Methods.
Figure 10.

Nuclei captured from several neuronal and glial cell types. Nuclei cluster into four discrete groups. (a) Multidimensional scaling (MDS) plot of 10 nuclei (Supplementary Table 1) based on the first two principal coordinates (PC, x and y axes). Labels 1–6 are the NeuN+ cells 1–6, and A–D correspond to NeuN– cells 1–4, and they are color-coded based on k-means clustering with n = 4. (b) Venn diagram showing the number of genes expressed in at least one cell in each group. The number of cells expressed in all cells of one cluster and no cells in any other cluster are shown in parentheses, and they are color-coded as in a. Cell clusters correspond to discrete cell types based on known marker genes. (c) Average expression of marker genes for glutamatergic neurons, GABAergic neurons36, astrocytes and oligodendrocyte precursor cells37 is shown for each cell, and it is color-coded as in a. Cells to the right and left of the vertical bar are the NeuN+ and NeuN− cells collected by FACS, respectively. (d) Canonical marker genes for glutamatergic neurons (SLC17A7), GABAergic neurons (GAD1), astrocytes (AQP4) and oligodendrocyte precursor cells (NKX2.2) are expressed as expected, based on cell type. Axes and colors for d are the same as those in c.
Advantages and limitations
The key strengths of our protocol are as follows:
The use of nuclei for RNA-seq avoids the difficulties involved in obtaining undamaged whole neurons.
Alteration of the transcriptome by treatment with proteases is avoided. The clinical samples and isolated nuclei are maintained at 4 °C until they are ready for use in cDNA synthesis.
We have demonstrated RNA-seq from nuclei isolated by micro-manipulation13 and FACS.
The technical and biological variation is similar for whole cells and nuclei13. For most transcripts, the nuclear and whole-cell expression profiles were similar, and therefore nuclei can generally be substituted for whole cells to define cell lineage, state or type populations, for example, by principal component analysis.
Nuclear transcriptomes will provide insights into how they differ from cytoplasmic transcriptomes such as enrichment of certain transcripts in nuclei13 and regulatory processes controlling the rate of transcription27.
The main limitations are as follows:
Cytoplasmic mRNA concentrations are directly rate limiting for protein synthesis, and thus whole cells may possibly give a more direct indication of downstream biological functions dependent on the proteome. Use of nuclei might result in loss of some information contained in cytoplasmic mRNA; however, for frozen brain tissue, whole cells have tended to generate poor-quality cDNA, and they may not be an option.
Nuclei are generally fragile compared with whole cells, and some loss can be expected at each stage of an isolation procedure18.
The small amounts of mRNA present in nuclei may necessitate optimization of the number of PCR cycles required to obtain sufficient cDNA for use in sequencing depending on the experimental needs. We amplified the nuclear cDNA with 21 cycles because of low amounts of RNA in the nucleus, compared with 18 cycles for whole cells3. However, some low-copy transcripts may still be more difficult to detect in nuclei. Furthermore, increasing the cycle number could introduce some amplification bias in the library by compressing expression values for high-copy transcripts.
Cytoplasmic transcripts are not detectable, nor are small noncoding RNAs (ncRNAs) and other short sequence mRNAs lacking polyA tails. The low amounts of RNA contained in a nucleus may also prevent the detection of some ncRNAs.
Applications
Nuclear and cytoplasmic transcriptomes are likely to differ in many ways, and a more comprehensive analysis is needed to determine the advantages and limitations of using nuclei for transcriptomic studies. Some studies of specific nuclear functions may be enhanced by directly accessing nuclei—for example, studies of the regulation of transcriptional activation mediated by transcription factors, promoters, enhancers, epigenetic modifications and other mechanisms that control synthesis of mRNA. Critical control of cellular development and function occur at this level of regulation. Some processing of ncRNAs may also require analysis via nuclei such as initial rates of primary miRNA synthesis. The polyA tail of this ncRNA species allowed measurement of cDNAs produced by polyT priming13, whereas the polyA tail is removed before transport of this RNA to the cytoplasm. In general, we anticipate that the nuclear transcrip-tome will have some advantages for investigating the regulatory processes controlling transcription rates. In contrast, the concentration of cytoplasmic mRNA reflects transport from the nucleus and various rates of mRNA processing and degradation. The cytoplasmic mRNAs serve as the template for ribosomes and the formation of the proteome, and thus they may have advantages in some studies.
RNA-seq analysis of human neurons is particularly challenging. For acute surgically derived tissues, the isolation of intact living neurons has been proven to be difficult, although a recent report demonstrated feasibility10. Similarly, technical challenges including cell isolation, RNA quality and glial transcript contamination have hindered progress in profiling single neurons from frozen postmortem tissues (R.H. and E.S.L., unpublished data). The use of nuclei avoids these obstacles. Furthermore, protease treatment to disperse whole cells, as done in recent studies of single neurons10,11, is known to profoundly alter gene expression12. We have recently observed additional examples in which protease treatment altered gene expression. Unexpected Fos activation was found in almost all of the cells dissociated by protease from a mouse brain region that is reported to have low Fos expression and which lacked Fos protein based on antibody staining before pro-tease treatment. No such activation was observed using the nuclei isolation protocol, which is performed at 4 °C and without the use of proteases. Importantly, we are able to detect Fos activation using the nuclei isolation protocol when mice have been exposed to environmental stimuli, which are known to induce Fos28. These observations suggest that caution is needed in interpreting transcriptomes from protease-treated cells. As the majority of accessible human brain specimens are obtained from frozen archives and collections, the use of nuclei may provide the best option that is currently available for RNA-seq from neurons.
The number of different cell types in the brain remains poorly understood. Cell ‘type’ implies stable characteristics, such as the synthesis of a particular neurotransmitter, and these cells have generally arisen by differentiation through developmental pathways, although the steps in these processes and their reversibility are not completely understood. It will be important to identify the abundance and functions of all the cell types in the brain. It also remains unclear how to define cell ‘states,’ which may simply reflect a range of intermediate functional activities rather than being discrete cell types. RNA-seq from individual brain cells will be crucial in resolving these questions. Moreover, RNA-seq will be a powerful new method for investigating the genomics and biochemistry of individual brain cells in a way that is not possible with bulk RNA. New computational methods are rapidly being introduced that will enable discovery of metabolic and regulatory pathways and investigation of brain function at the most basic levels of cell and systems biology. The use of nuclei to obtain transcriptomes from large numbers of cells has the potential to be a powerful new tool in neuroscience to investigate both normal and disease processes.
Experimental design
Starting material
We have used cultured neuroprogenitor cells and fresh mouse brain tissue13, and we include an example using frozen human brain (ANTICIPATED RESULTS), as the source for nuclei. When brain tissue can be used fresh without freezing (as for laboratory animals or when fresh human biopsies are available), we have elected to cool the sample to 4 °C and to use it for isolation of single nuclei as soon as possible. However, frozen brain tissue performed well, by producing full-length cDNAs and informative transcriptomes (ANTICIPATED RESULTS). Methods that cross-link mRNA, such as paraformaldehyde fixation of tissues, will severely limit the ability to produce full-length cDNA. When a sufficient quantity of tissue specimen is available for extraction of bulk RNA, we suggest determining the RNA quality before proceeding with single-nuclei isolation (Box 3). We selected tissues with RIN values ≥7, as these can be obtained from many brain archives. We have not carefully evaluated RNA of poorer quality. However, if RNA with a RIN score of <7 is all that is available, it should be tested and it may still yield valuable data. In general, we selected samples with the highest RIN available.
Box 3. Sample quality assessment of tissue and cultured cells. ●TIMING 1 h.
▲ CRITICAL Sample processing procedures vary widely depending on the sample type, and they can affect the quality of the RNA that can be obtained. For human postmortem brain, fresh mouse brain or cultured cells, we recommend determining the RNA quality by assessing the integrity of the bulk sample before proceeding with single nuclei isolation. If sufficient sample is not available, the tissues can be used directly for nuclei isolation.
For tissue samples, place a sterile Petri dish and scalpel on dry ice to chill. Transfer the brain sample to the Petri dish using sterile and RNase-free forceps. Remove a section of ~2–3 mm3 using the scalpel. For cultured cells, collect them by trypsinization and centrifugation. Remove the supernatant and resuspend the cells in 1× cold PBS. Pellet the cells with centrifugation at 2,000g for 15 min. Repeat resuspension and centrifugation two more times. The pelleted cells can be kept at −80 °C for up to 3 months or they can be processed immediately.
Follow the Qiagen RNeasy mini kit’s recommended protocol to isolate total RNA from either tissue or pelleted cells.
Assess the integrity of the total RNA on an Agilent Bioanalyzer (or similar device) using an RNA 6,000 pico chip as per the manufacturer’s recommendation.
▲ CRITICAL STEP Where possible, it is recommended to proceed with single nuclei isolation using samples that have a RIN value of ≥7.
Homogenization
Nuclei were obtained by Dounce homogenization of ~2–3 mm3 of human brain tissue for use in FACS sorting. In general, the Dounce step does not give quantitative recovery of nuclei because they are fragile and easily damaged. Some large pieces of tissue remained after this step; however, additional Dounce strokes appeared to destroy free nuclei even as more were released from the tissue. About 60,000 intact nuclei were obtained based on a hemocytometer count. If smaller amounts of tissue must be used, micromanipulation can be considered as a means to isolate a small number of nuclei13.
The Dounce homogenization of tissues should be optimized for each specimen. Samples containing a mixture of cell types or samples from connective tissues and intracellular fibrous material may require more strokes. However, note that although more thorough homogenization (by increasing the number of strokes) will release more nuclei, it will also increase the number of damaged nuclei. The Triton X-100 used in this protocol is compatible with RNA-seq methods that use specific cell type enrichment via surface protein labeling. When immunostaining is not required, substitution of NP-40 for Triton X-100 has been suggested as a means to reduce loss of nuclei21, although we have not verified this for use in single-nuclei RNA-seq.
Immunostaining
We immunostain nuclei with an antibody specific to NeuN, a nuclear membrane protein that is specific for neurons. In combination with Hoescht stain, which stains all nuclei, this allowed separation of nuclei by FACS into neuronal and non-neuronal populations. We have not found suitable alternative neuronal markers for FACS; staining for proteins within the nucleus would require permeabilization and fixation steps, which is incompatible with RNA-seq.
Isolating individual nuclei
We isolated individual nuclei by FACS; however, other methods can be used. We have also demonstrated the use of micromanipulation to isolate individual nuclei for use in RNA-seq13. Micromanipulation has the advantage of allowing inspection of nuclear morphology and fluorescent labeling with a microscope, and of providing confirmation that a single nucleus was added to the reaction well for cDNA synthesis. Micromanipulation may be an advantage for confirming the identity of nuclei from rare cell types that are not easily enriched by FACS. Another option is a microfluidic approach such as the C1 Single-Cell Autoprep System (Fluidigm), which can be used to isolate single nuclei from bulk preparations of adult human neurons (M.N., R.S.L. and M. Ray (of Fluidigm), unpublished observations). Similar to intact cells, some optimization of the nuclei loading conditions, including varying concentration, for each tissue type may be needed to maximize the nuclei captured per run. This instrument generally requires that at least 2,000 cells or nuclei be loaded onto the integrated fluidic circuit for optimal performance.
RNA-seq cDNA synthesis and sequencing platform
Smart-seq23 was used here to synthesize double-stranded cDNA; however, other methods can be used1,4,5. Previously13, we successfully used the method by Tang et al.5. Any sequencing method is acceptable if it is well suited for the short cDNA library inserts. We have tested SOLiD sequencing (Life Technologies)13 and Illumina sequencing (ANTICIPATED RESULTS) with comparable results.
Sample controls
It is important to include no-template controls (NTCs) in each experiment. Very low amounts of contaminating DNA or RNA, which are present in the Smart-seq2 reagents, for example, can be sufficient to compete with the small amount of targeted material from a single cell or nucleus. NTCs, which receive water instead of the sorted nucleus, should not support cDNA synthesis. If some bacterial reads are obtained, they are possibly derived from contaminants in the reagents. If human sequence is obtained from the NTCs, contamination introduced in the laboratory is likely. We also use an aliquot of the FACS effluent (lacking a nucleus) as a negative control13 to demonstrate that the sort buffer cannot support cDNA synthesis owing to free RNA or DNA released from the homogenized tissue. Any robust cell line easily maintained in the laboratory can be used as a positive control to demonstrate typical performance and to detect a loss of efficiency due to poor reagents, for example. The use of the same positive control in all experiments is helpful, as typical RNA content and the number of genes expressed may differ among cell types. Also consider using cell lines that express specific marker genes of interest as positive controls for comparison with the brain tissues. It is helpful to include technical replicates in experiments in which purified RNA is used as the template. Technical sources of variation include degree of success in synthesizing cDNA and constructing Nextera libraries. The technical variation contributes noise that interferes with the desired detection of biological differences.
Spike-in controls
An extrinsically added spike-in RNA is used as a positive control for the reverse transcriptase (RT) reaction. We used the ERCC spike-ins29, a set of 96 different microbial mRNAs. These are present in a range of concentrations, allowing the determination of the sensitivity and range for the detection of transcripts. The concentration of ERCC spike-ins added is adjusted for various applications so that they will contribute a smaller percentage of the reads compared with the experimental specimen. We have adjusted the dilution of the ERCC spike-in stock commensurate with the small amount of RNA in a human cell nucleus. The dilution can be adjusted if it is found that too many or too few reads are obtained. Changing the dilution will alter which of the 96 mRNA species represent <10 molecules, the lower limit for detection. The dilution of 1:1.1 × 107 in the RT reaction results in ERCC-00077 being present at 2.2 copies per reaction. Approximately 50 of the 92 species are detected by sequencing at this dilution, and the remaining 42 are not detected, as they are added at <1 copy per reaction. Failure to detect ERCC spike-in controls in RNA-seq indicates a failed Smart-seq2 reaction. Detection of ERCC spike-ins but failure to detect cellular transcripts indicates failed recovery of RNA from the nuclei. An unexpectedly high proportion of ERCC spike-in sequencing reads relative to cellular transcripts also indicates poor recovery of RNA from the nucleus or that the cell was relatively quiescent and had low RNA content.
qPCR controls for cDNA quality
The quality of the gene expression information in the cDNA libraries can be assessed before investing time and expense in DNA sequencing by using TaqMan qPCR for a limited number of transcripts. We have found that cycle thresholds for housekeeping genes typically range between 15 and 30 cycles, depending on the amount of available mRNA in the nucleus and the original sample RIN. Control samples with 8, 24, 48 and 96 pooled nuclei should have correspondingly lower cycle thresholds. Total RNA controls from the same tissue sample ranging from 1 to 100 pg should also have progressively lower cycle thresholds. We have generally discarded cDNAs that lack all of the housekeeping genes tested for by qPCR. However, the pass/fail criteria are not easily defined, and they must be developed for each specific study. Caution should be exercised in discarding samples simply because certain transcripts are not detected, as transcription rates are highly variable through time, even for constitutive genes. qPCR for transcripts that are diagnostic for a cell type and other specialized characteristics can also be very useful in prescreening before investing in RNA-seq. However, where an unbiased sampling of a cell population is desired, it is important to weigh the benefits of selecting for specific transcripts against the risk of systematically biasing the selection.
In addition to sorting single nuclei, pools of 8, 24, 48 and 96 nuclei, for example, can serve as positive controls for cDNA synthesis. The pools also reveal the full range of transcripts in a cell population (the pan-transcriptome), and they can serve to validate detection of differentially expressed transcripts in the individual nuclei. The sequencing depth for a given transcript from a single nucleus can be compared with the sequencing depth from a pool of nuclei. For example, a transcript found at high copy number, but only in a small percentage of nuclei, should be commensurately low in the pools.
MATERIALS
REAGENTS
Tissue sample. We have successfully used cultured neuroprogenitor cells and fresh mouse brain tissue13 and frozen human prefrontal cortex brain obtained from the US National Institutes of Health (NIH) NeuroBioBank located at the University of Maryland as an example here (ANTICIPATED RESULTS). The quality of the initial sample can be checked before isolating nuclei, as described in Box 3. ! CAUTION An Institutional Review Board approval may be required to obtain, process and place samples on a flow-sorting instrument. Precautions to protect the user include standard personal protective equipment, but potentially also a protective laminar flow hood for the flow cytometer if biohazardous sample material is to be used.
RNaseZap RNase decontamination solution (Ambion, cat. no. AM9780)
Nuclease-free water (Ambion, cat. no. AM9932)
-
β-Mercaptoethanol, 14.3 M (Sigma, cat. no. M6250-100 ml)
! CAUTION This is a combustible liquid. It is toxic if swallowed or if inhaled. It is very hazardous in case of skin contact (permeator) and ingestion. Severe overexposure can result in death. It causes skin irritation, and it may cause an allergic skin reaction. It also causes serious eye damage.
Avoid contact with skin and eyes. Avoid inhalation of vapor or mist, and handle it while you are wearing appropriate personal protective equipment.
Complete, EDTA-free (Roche, cat. no. 11873580001)
Sucrose (Sigma, cat. no. S0389-500G)
Potassium chloride, 2 M (Ambion buffer kit, cat. no. 9010)
Tris buffer, pH 8.0, 1 M (Ambion buffer kit, cat. no. 9010)
Magnesium chloride, 1 M (Ambion buffer kit, cat. no. 9010)
EDTA, 0.5 M (Ambion buffer kit, cat. no. 9010)
RNase inhibitor, cloned (40 U μl−1; Ambion, cat. no. AM2682)
Hoechst 33342, trihydrochloride, trihydrate (10 mg ml−1; Molecular Probes, cat. no. H3570) ! CAUTION This compound is harmful if swallowed. It causes skin irritation, and it may cause respiratory irritation. It is suspected of causing genetic defects; handle it while you are wearing appropriate personal protective equipment.
-
Propidium iodide (PI; 1.0 mg ml−1; (Molecular Probes, cat. no. P3566)
! CAUTION This compound is harmful if swallowed. It causes skin irritation, and it may cause respiratory irritation. It is suspected of causing genetic defects; handle it while you are wearing appropriate personal protective equipment.
DAPI (1.0mg ml−1; Molecular Probes, cat. no. 62248) ! CAUTION DAPI is harmful if swallowed. It causes skin irritation, and it may cause respiratory irritation. It is suspected of causing genetic defects; handle it while you are wearing appropriate personal protective equipment.
Triton X-100 (Sigma-Aldrich, cat. no. T8787-100ML) ! CAUTION Triton X-100 is harmful if swallowed, and it causes serious eye damage; handle it while you are wearing appropriate personal protective equipment.
dNTP mix (10 mM each; Thermo Fisher, cat. no. 18427-088)
Superscript II reverse transcriptase (Thermo Fisher, cat. no. 18064-014)
Betaine (BioUltra ≥99.0%; Sigma-Aldrich, cat. no. 61962)
KAPA HiFi HotStart ReadyMix (2×; KAPA Biosciences, cat. no. KK26010)
Ethanol, molecular biology grade (Sigma-Aldrich, cat. no. E7023-500 ml)
Agencourt Ampure XP beads (Beckman Coulter, cat. no. A63881)
Adapter oligos (See Synthesis of cDNA, Step 19). All oligos except the LNA-modified TSO were ordered from IDT (https://www.idtdna.com), and they were HPLC-purified. LNA-modified TSO was ordered from Exiqon (http://www.exiqon.com/), and it was HPLC-purified. TSO (5′-biotin-AAGCAGTGGTATCAACGCAGAGTACATrGrG+G-3′); oligo-dT30VN (5′-biotin–AAGCAGTGGTATCAACGCAGAGTACT30VN-3′); ISPCR oligo (5′-biotin-AAGCAGTGGTATCAACGCAGAGT-3′)
UltraPure BSA (50 mg ml−1; Ambion, cat. no. AM2616)
Trypan blue (0.4%; Sigma-Aldrich, cat. no. T8154)
ERCC spike-in mix 1 (Ambion, cat. no. 4456740)
RNase-free PBS, pH 7.4 (Ambion, cat. no. AM9625)
0.5% RNase-free BSA (Ambion, cat. no. AM2616)
RNasin Plus RNase inhibitor (Promega, cat. no. N2615)
Mouse IgG1k (BD Pharmingen, cat. no. 554121)
Mouse monoclonal anti-NeuN antibody (Millipore, cat. no. MAB377)
Goat anti-mouse Alexa Fluor 594–conjugated secondary antibody (Life Technologies, cat. no. A11005)
DAPI (Life Technologies, cat. no. D1306)
Yellow fluorescent polystyrene microspheres, 10 μm (Spherotech, cat. no. FP-10052-2)
Perfecta ROX FastMix (Quanta Bioscience, cat. no. 95077-05K)
TaqMan gene expression real-time PCR assay (Thermo Fisher)
RNeasy Mini Kit (50) (Qiagen, cat. no. 74104)
Quant-iT PicoGreen dsDNA assay kit (Molecular Probes, cat. no. P11496)
Agilent RNA 6000 pico kit (Agilent Technologies, cat. no. 5067-1513)
Agilent high-sensitivity DNA kit (Agilent Technologies, cat. no. 5067-4626)
Nextera XT DNA library preparation kit, 96 samples (Illumina, cat. no. FC-131-1096)
Nextera XT 96-index kit (Illumina, cat. no. FC-131-1002)
MiSeq reagent kit v2, 300-cycles PE (Illumina, cat. no. MS-102-2002)
Software for sequence quality assessment
FASTX (http://hannonlab.cshl.edu/fastx_toolkit/download.html)
fastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/).
RSeQC30,31 (http://rseqc.sourceforge.net/) can be used as an alternative to FASTX and fastQC
Software for sequence trimming
Trimmomatic (http://www.usadellab.org/cms/uploads/supplementary/Trimmomatic/Trimmomatic-0.33.zip)
Alternatively, Cutadapt32 (https://cutadapt.readthedocs.org/en/stable/) can be used
Software for sequence alignment
Software for RNA expression analysis
RSEM (http://deweylab.biostat.wisc.edu/rsem/). Alternatives to RSEM include Tophat2 (ref. 33) (https://ccb.jhu.edu/software/tophat/index.shtml), Cufflinks33 (http://cole-trapnell-lab.github.io/cufflinks/) and Star34 (https://code.google.com/p/rna-star/)
Software for data analysis
Python and related packages (https://www.python.org/)
IPython (http://ipython.org/)
Pandas (http://pandas.pydata.org/)
Matplotlib (http://matplotlib.org/)
Bedtools (http://bedtools.readthedocs.org/en/latest/)
EQUIPMENT
Dounce homogenizer, 1 ml (Wheaton, cat. no. 357538)
Sterile forceps (VWR, cat. no. 89259-946)
Sterile Petri dish (VWR, cat. no. 25384-070)
Sterile scalpel (Miltex, cat. no. 4-410)
BD FACS-ARIA II Flow sorter with an automated cell deposit unit
BD Falcon tube with a cell strainer cap (Becton Dickinson, cat. no. 352235)
Falcon polystyrene conical tube (50 ml, BD Biosciences, cat. no. 352095)
Inverted fluorescence microscope Olympus IX70
Hemocytometer (Hausser Scientific, cat. no. 1483)
Teflon-coated multi-well glass slides (Electron Microscopy Sciences, cat. no. 63430-04)
96-well black Fluortrac micro plate (VWR, cat. no. 82050728)
384-well plates (Phenix Research Products, cat. no. MPC-384HS4NH-C)
96-well plates (Eppendorf, twin.tec PCR plate 96, skirted, colorless, cat. no. D156224K)
8-strip, nuclease-free, 0.2 ml, thin-walled PCR tubes with caps (Eppendorf, cat. no. 951010022)
Microcentrifuge Safe-Lock tubes (Eppendorf, cat no. 022363344)
Multichannel pipettes and filter tips (Rainin LTS pipette set, 1–10 μl; 2–20 μl; 20–200 μl)
DynaMag-96 side skirted magnetic rack (Thermo Fisher, cat. no. 12027)
MicroAmp clear adhesive film (Applied Biosystems, cat. no. 4306311)
MicroAmp optical adhesive film (Applied Biosystems, cat. no. 4311971)
Thermal cycler (Applied Biosystems 9700)
Fluorometer (Molecular Dynamics Flexstation 3)
Spectrophotometer (Thermo Fisher, Model: NanoDrop ND-1000)
Agilent 2100 Bioanalyzer (Agilent Technologies)
Refrigerated centrifuge (Eppendorf, Model: Centrifuge 5804 R)
C1 system for RNA-seq manual: ‘Using C1 to Generate Single-Cell cDNA Libraries for mRNA Sequencing Protocol’ (Fluidigm Part No. 100-7168, https://www.fluidigm.com/documents)
DNA sequencing instrument ▲ CRITICAL A compatible Illumina DNA sequencing instrument (MiSeq, NextGen 500, HiSeq 2000, HiSeq 2500) is necessary to complete sequencing of the Nextera XT libraries, as the barcodes and sequencing adapters are designed for the Illumina sequencing platform.
64-bit computer running Linux with 4 GB of RAM (16 GB preferred)
REAGENT SETUP
Nuclei isolation medium #1 (NIM1)
Combine the following components.
▲ CRITICAL This buffer should be made in advance, and it can be stored in a 50-ml conical tube at 4 °C for up to 6 months.
| Component | Volume (μl) | Final concentration (mM) |
|---|---|---|
| 1.5 M sucrose | 2,500 | 250 |
| 1 M KCl | 375 | 25 |
| 1 M MgCl2 | 75 | 5 |
| 1 M Tris buffer, pH 8.0 | 150 | 10 |
| Nuclease-free water | 11,900 | — |
| Total volume | 15,000 | — |
Nuclei isolation medium #2 (NIM2)
The following reagents should be combined in a 15-ml conical tube and placed at 4 °C or on ice for immediate use and then discarded.
| Component | Volume (μl) | Final concentration |
|---|---|---|
| NIM1 | 4,895 | |
| 1 mM DTT | 5 | 1 μM |
| 50× protease inhibitor | 100 | 1× |
| Total volume | 5,000 |
Homogenization buffer
Combine the following reagents.
▲ CRITICAL This buffer should be made in a 5-ml conical tube, protected from light, and it should be placed at 4 °C or on ice for immediate use and then discarded.
| Component | Volume (μl) | Final concentration |
|---|---|---|
| NIM2 | 1,452/1,453.5 (w/woPI) | 1× |
| RNaseIn 40 U μl−1 | 15 | 0.4 U μl−1 |
| Superasin 20 U μl−1 | 15 | 0.2 U μl−1 |
| Triton X-100 10% (v/v) | 15 | 0.1% (v/v) |
| PI (optional for FACS) | 1.5/0 (w/wo PI) | 1 μM |
| DAPI (optional for FACS) | 1.5/0 (w/wo PI) | 1 μM |
| Hoechst 33342 | 1.5/0 (w/wo PI) | 10 ng ml−1 |
| Total volume | 1,500 |
Iodixanol medium (IDM)
The following reagents should be combined in a 50-ml conical tube, and the medium can be stored at 4 °C for up to 6 months.
| Component | 1× volume (μl) | Final concentration (mM) |
|---|---|---|
| 1.5 M sucrose | 2,500 | 250 |
| 1 M KCl | 2,250 | 150 |
| 1 M MgCl2 | 450 | 30 |
| 1 M Tris buffer, pH 8.0 | 900 | 60 |
| Nuclease-free water | 8,900 | — |
| Total volume | 15,000 | — |
Iodixanol dilutions
The following reagents should be combined, according to final concentration, in 50-ml conical tubes, and they can be stored at 4 °C for up to 6 months.
| Component | 1× volume (μl) | Final concentration |
|---|---|---|
| Iodixanol 60% (vol/vol) | 12,500 | 50% vol/vol |
| IDM | 2,500 | — |
| Total volume | 15,000 | — |
| Component | 1× volume (μl) | Final concentration |
| Iodixanol 60% | 7,250 | 29% vol/vol |
| IDM | 7,750 | — |
| Total volume | 15,000 | — |
Nuclei storage buffer (NSB)
The following reagents should be combined in a 50-ml conical tube, and the buffer can be stored at 4 °C for up to 6 months.
| Component | 1× volume (μl) | Final concentration (mM) |
|---|---|---|
| Sucrose | 0.855 g | 166.5 |
| 1 M MgCl2 | 50 | 5 |
| 1 M Tris buffer, pH 8.0 | 500 | 10 |
| Nuclease-free water | 14,450 | — |
| Total volume | 15,000 | — |
EQUIPMENT SETUP
FACS
For high-throughput single-nuclei isolation by flow cytometry, the operator should be familiar with standard doublet discrimination gating and instrument settings for sorting single nuclei events. In preparation for sorting single nuclei into 384-well microplates for cDNA synthesis, accuracy and precision of sorting single events in a plate can be confirmed by targeting the bottom of each microplate well with 10-μm yellow fluorescent polystyrene microspheres and by inverting the plate for direct imaging on an inverted fluorescence microscope. Typically, 16 wells on both ends of the plate are targeted for spatial precision and >95% accuracy for a single bead. For nuclei sorting, staining in 1 μM DAPI, Hoechst 33342 or PI is suitable. The choice of stain depends on the number and type of antibody fluorophores used for the detection of the cell type of interest. Targeting and confirming sorted nuclei on a microscope slide and in microplate wells is also recommended. Figure 3 shows the FACS gating strategy.
Computational requirements
The protocol requires experience in running commands in UNIX (LINUX) shell environment. Experience with running Python and Perl language scripts is also required. C++, Perl, Python, Java and R programs are required to be installed. Prerequisite software is listed in the Reagents section. Users who do not have programming experience can use Galaxy analysis portal (https://usegalaxy.org/), which is an open, web-based platform, to execute most of the programs and commands described in this protocol, including those mentioned under alternate analysis packages. It allows the user to specify parameters and to run tools and workflows almost exactly as described under the PROCEDURE section of this protocol or modify some of the steps in the analysis in accordance with their preference. For more specific details on how to use this software, the user can access the site https://wiki.galaxyproject.org/. Data: requirements vary according to experimental goals. Sequence type: Illumina or other sequencing platforms that generate short reads (50–250 bases). Sequence format: .fastq or .fasta. Reference genome: .fasta, index and .gtf or .gff files.
Directory structure
Choose or create a directory in which analysis is performed (RUNDIR). Save sequence files and reference .fasta, index and annotation (.gtf or .gff) files to SEQDIR and REFDIR, respectively. Trimmed reads are also copied to SEQDIR (these can be symlinks to files located elsewhere). The programs and individual commands described under the PROCEDURE section below are assumed to be available in the RUNDIR either as symlinks to the executables or copies of the installed binary files and scripts.
PROCEDURE
Nuclei isolation ● TIMING 1–2 h
▲ CRITICAL Keep the workstation and tools free of RNases by thoroughly cleaning with RNaseZap solution before the experiment.
-
1|
Prepare nuclei isolation media 1 and 2 (NIM1 and NIM2) and homogenization buffer, and place them on ice.
▲ CRITICAL STEP NIM1 can be prepared and stored at 4 °C for up to 6 months. NIM2 and homogenization buffer should be freshly prepared.
-
2|
Precool the Dounce homogenizer and pestles on ice. Once it is cooled, fill the homogenizer with 1.0 ml of cold homogenization buffer and keep it on ice.
-
3|
If you are using tissue, transfer the sample to a Petri dish (on ice) and cut out a (2–3 mm3) section using a chilled scalpel. Immediately transfer the tissue section into the precooled Dounce homogenizer. If you are using cultured cells, place 250 μl of cells (collected and resuspended in 1 × 106 cells per ml of 1× cold PBS) into the Dounce homogenizer.
-
4|
Homogenize the tissue or cells with five strokes of the loose pestle, followed by 10–15 strokes of the tight pestle.
▲ CRITICAL STEP To reduce heat caused by friction, the Dounce homogenization should be performed on ice with gentle strokes, and care should be taken to avoid foaming. The mortar should be immersed in ice. The precooled homogenization buffer is an important aid in heat reduction during homogenization.
-
5|
Filter the homogenate through a BD Falcon tube with a cell strainer cap; this filters out debris larger than 35 μm. Estimate the number of intact nuclei by staining a 10-μl aliquot of the filtered homogenate with trypan blue (10 μl), by loading it onto a hemocytometer and viewing it under a light microscope. At this point, nuclei can either be immunostained for neuronal markers (Optional Steps 6–12) to enrich for neuronal nuclei during FACS or they can be subjected directly to FACS (Steps 13–18) based on double discrimination only.
▲ CRITICAL STEP We obtained ~6 × 104 nuclei per milliliter from 2–3 mm3 of frozen normal human cortical brain tissue. Figure 2 shows a typical amount of debris present and varying sizes (7–10 μm) of nuclei from prefrontal cortical tissue.
▲ CRITICAL STEP For frozen human brain tissues, we recommend proceeding directly to FACS (Step 13), after filtering the homogenate, without further purification, as the nuclei have been subjected to freezing, and additional purification steps may cause further RNA damage. For fresh brain tissues, an additional iodixanol centrifugation-based purification may be helpful depending on the experiment. In general, each purification step results in lower yields of nuclei, and adjusting the starting material is desirable according to the downstream application.
? TROUBLESHOOTING
(Optional) Neuronal nuclei immunostaining ● TIMING 1–1.5 h
▲ CRITICAL The anti-NeuN antibody can be used to enrich for nuclei originating from neurons. We chose a dual-antibody staining strategy that first tags the nuclei with an unconjugated mouse anti-NeuN antibody, followed by a goat anti-mouse Alexa Fluor 594–conjugated secondary antibody. Mouse IgG1k detected by goat anti-mouse Alexa Fluor 594 serves as an isotype control for FACS to ensure specificity of the NeuN antibody (see Supplementary Fig. 1 for the expected level of staining).
-
6|
After homogenization and filtering (Step 5), concentrate the nuclei by centrifugation (1,000g for 8 min at 4 °C), and remove the supernatant. Resuspend in 500–1,000 μl of staining buffer (RNase-free PBS, pH 7.4, with 0.5% (wt/vol) RNase-free BSA and 0.2 U μl−1 of RNasin Plus RNase inhibitor).
-
7|
Incubate the sample for 15 min on ice to allow for blocking of nonspecific binding with 0.5% (wt/vol) BSA. Remove 100 μl of the sample to a new tube for isotype control staining, and keep the remainder of the sample for staining with mouse anti-NeuN antibody.
-
8|
For the isotype control sample, add purified mouse IgG1k to the tube at a final dilution of 1:5,000. For NeuN staining, add mouse monoclonal anti-NeuN antibody to the tube at a final dilution of 1:5,000. Incubate the samples on a tube rotator for 30 min at 4 °C.
-
9|
Wash the samples by adding 500 μl of staining buffer to each tube and inverting the tubes several times. Spin the samples for 5 min at 400g in a refrigerated (4 °C) centrifuge to pellet nuclei.
-
10|
Resuspend the pelleted nuclei in 500–1,000 μl of staining buffer, and add goat anti-mouse Alexa Fluor 594–conjugated secondary antibody to each tube at a final dilution of 1:5,000. Incubate the samples for 30 min on a tube rotator at 4 °C.
-
11|
Wash the samples by adding 500 μl of staining buffer to each tube and by inverting the tubes several times. Spin the samples for 5 min at 400g in a refrigerated (4 °C) centrifuge to pellet nuclei.
-
12|
Resuspend nuclei in 500–1,000 μl of staining buffer, and add DAPI at a final concentration of 1 μg μl−1 to each tube. Proceed directly to FACS (Steps 13–18).
Nuclei FACS sorting ● TIMING 2–3 h
-
13|
Prepare lysis buffer by adding the following reagents to a 1.5-ml Eppendorf tube, and then place it on ice.
Component 1× volume(μl) Final concentration 10% (vol/vol) Triton X-100 20 0.2% (vol/vol) RNase inhibitor 40 U μl−1 50 2 U μl−1 ERCC spike-in mix 1, 1:2,000 1 1:2 × 106 Nuclease-free water 929 — Total volume 1,000 ▲ CRITICAL STEP The lysis buffer should be freshly made for each experiment.
-
14|
Prepare 96- or 384-well thin-walled PCR plates by adding 2 μl of lysis buffer to each well.
-
15|
Prepare the FACS instrument for daily FACS setup, testing and droplet delay optimization.
▲ CRITICAL STEP We recommend adhering to the FACS manufacturer’s instructions that the droplet stream be optimized for timing delay, with any satellite droplets merged by the fifth drop after the droplet breakoff. Failure to optimize the droplet breakoff may result in a charge placed on the satellite droplet instead of the droplet of interest.
-
16|
Prepare FACS plots for doublet discrimination gating according to the manufacturer’s recommendation to prevent sorting of doublets, triplets and further groupings of attached nuclei. Adjust the instrument software parameters to enable single-cell stringency. Load a small amount of sample into the instrument to confirm gating, and arrange gates on the FACS plots as needed. For samples that have been immunostained, sort populations for both NeuN+ and NeuN− with the NeuN+ population clearly distinguished with Alexa Fluor 488 fluorescence. If an unbiased nuclei population is desired, sorting may be completed using the DAPI+ population.
-
17|
Confirm FACS parameter settings for single nuclei sorting before sorting the actual samples. Confirmation can be achieved by targeting of the plate using 10-μm yellow fluorescent polystyrene microspheres or similar (Equipment Setup).
▲ CRITICAL STEP We recommend that even experienced FACS users complete a series of practice sorts (with single-cell sort instrument parameter settings) of microspheres before the actual sample sorting in order to confirm that the sorting is accurately timed and that the plate is properly targeted. Day-to-day variability in both of these parameters necessitates these precautionary steps to ensure efficient and accurate single nuclei sorting. Accuracy of microsphere sorting is determined by direct imaging of the microspheres at the bottom of the inverted microplate well (Fig. 3g). An accuracy of no less than 95% single microsphere sorting is recommended. For 384-well microplate sorting, the microscope objective often does not possess the dynamic focal range required to image the bottom of the well. A simple loosening of the objective for a few turns will bring the bottom of the well and the microsphere into focus. For 96-well plates, a custom objective with a long working distance for focal range may be required.
-
18|
Proceed to FACS of sample nuclei. We recommend keeping the overall event rate for particles to 200–2,000 events per second on the FACS instrument to prevent swamping of the detectors that may result in a poor sorting accuracy. Depending on the concentration of nuclei, dilution of the sample may be required.
▲ CRITICAL STEP Before microplate sorting, a final confirmation of single nuclei sorting onto a slide for direct imaging of sorted single nuclei is recommended. Sorting into ~1 μl of NSB on a microscope slide can be sufficient to locate, count and image single nuclei. If the nuclei or a subpopulation of the nuclei are found to be difficult to distinguish from other particles, consider performing iodixanol density gradient centrifugation (Box 4) before proceeding with sorting of the rest of the sample.
? TROUBLESHOOTING
■ PAUSE POINT Plates with FACS-sorted nuclei can be sealed with a MicroAmp Thermo-Seal lid, frozen on dry ice and stored at −80 °C. Otherwise, proceed with lysis and reverse transcription immediately (Step 19).
Box 4. Density gradient centrifugation ● TIMING 1 h (optional).
This centrifugation cleanup should be used if the nuclei or a subpopulation of the nuclei are difficult to distinguish from other particles during FACS. However, the added cleanup steps may result in loss of nuclei or potential damage to those recovered. Centrifugation for a lengthy amount of time or at excessive speeds may increase the yield of the nuclei, but it may also promote contamination by non-nuclear material, as aggregates of cell debris sediment faster than nuclei23.
Transfer the homogenate (from PROCEDURE Step 5) into a prechilled 1.5-ml Eppendorf tube and centrifuge at 1,000g for 8 min at 4 °C.
Aspirate the supernatant (~1,000 μl) and gently resuspend the pellet in 250 μl of homogenization buffer. Strain the mixture through a BD Falcon tube with a cell strainer cap to remove any remaining aggregates, and place it on ice.
Prepare the iodixanol dilution mix (IDM), iodixanol dilutions and NSB by combining and mixing the indicated reagents (see Reagent Setup) and place them on ice.
Gently mix the nuclei with 250 μl of 50% (vol/vol) iodixanol. To a new Eppendorf tube, add 500 μl of 29% iodixanol. Slowly layer 500 μl of the nuclei mixture over the 29% iodixanol and spin it at 13,500g for 20 min at 4 °C. The rotor should be kept at 4 °C throughout the process. If necessary, spin force and time should be optimized for a particular sample type.
Remove and discard the supernatant without disrupting the nuclei pellet.
Add 100 μl of NSB to the nuclei pellet and resuspend gently by pipetting. Estimate the number of intact nuclei by trypan blue staining, as described in PROCEDURE Step 5. Nuclei should be kept on ice while preparing for downstream steps, such as immunostaining (PROCEDURE optional Steps 6–12) or FACS (PROCEDURE Steps 13–18).
cDNA synthesis by Smart-seq2 ● TIMING 1 d
▲ CRITICAL Nuclei lysis, cDNA synthesis and Nextera XT library preparation can be performed using any of the currently available methods for single cells1,4,5,19. We perform nuclei lysis and cDNA synthesis using the SMART-seq2 method3.
-
19|
Perform lysis and cDNA synthesis, starting from Step 5 of the Smart-seq2 protocol3 (addition of oligo-dT primer and dNTPs to the FACS-sorted single nuclei from Step 18) and proceeding through to Step 24, implementing the modifications in the table below. Analyze the quality of the cDNA library, for example, by using the high-sensitivity DNA kit for Agilent Bioanalyzer according to the manufacturer’s recommendations. Accurately quantify the cDNA, for example, with a PicoGreen assay or a similar method.
Smart-Seq2 Procedure step3 Modification Reason 9: Reverse transcription TSO, oligo-dT and ISPCR oligos have a 5′ biotin modification Reduces concatamer formation and increases gene mapping percentage for sequence reads 14: cDNA PCR thermal cycling Increase the number of amplification cycles from 18 to 21 To compensate for the lower amount of RNA in a nucleus compared with a whole cell
qPCR and TaqMan analysis ● TIMING 3 h
▲ CRITICAL Evaluation of cDNA library quality can be achieved by qPCR with selected reporter housekeeping genes (ACTB, GAPDH) as well as high, medium and low-copy ERCC spike-in control qPCR assays. In addition, assays targeting genes specific for neuronal nuclei of interest are recommended.
-
20|
Dilute 2.2 μl of cDNA (from Step 19) in 19.8 μl of nuclease-free water (10-fold dilution). Use 2.5 μl of the diluted cDNA for each qPCR reaction.
-
21|
Add 7.5 μl of qPCR master mix comprising 1× ABI gene expression assay primer-probe mix (housekeeping gene, neuronal gene or ERCC spike-in specific) and 1× Perfecta ROX FastMix in nuclease-free water.
-
22|
Perform qPCR on the diluted stock using the following cycling conditions:
Step Cycle Denature Anneal and extend 1 Holding 95 °C, 2 min — 2 1–50 95 °C, 10 s 60 °C, 30 s -
23|
Plot qPCR data. Typical cycle threshold results for housekeeping genes are between 15 and 30 cycles (Fig. 4d).
? TROUBLESHOOTING
Sequencing library preparation ● TIMING 2 h
-
24|
Use cDNA preparations (from Step 19) that pass quality control (Step 23) to prepare a sequencing library; we use the Illumina Nextera XT library prep kit and follow the instructions in the Fluidigm C1 manual (see INTRODUCTION and MATERIALS). We start at page 35 of the manual with dilution of the cDNA and proceed through tagmentation, PCR amplification and AMPure XP bead cleanup, with the modifications for nuclei indicated in the table below. Determine the quality of the final pooled Nextera XT libraries, for example, by using the high-sensitivity DNA kit for Agilent Bioanalyzer according to the manufacturer’s recommendations.
Modification no. Page and Step in Fluidigm C1 manual Modification Reason 1 Page 41–43, Pool and Cleanup Purify each of the Nextera XT reactions individually (not as a single pool) and Elute each individual reaction in 17 μl of Low TE (10:0.1) and quantify each with PicoGreen Individual purification, elution and quantification of Nextera XT libraries allows for the exclusion of failed sequencing library preps in the final RNA-seq pool 2 Page 43, Repeat Cleanup Step Pool the samples; note the starting volume of the pool. Perform cleanup using AMPure XP beads and elute with the same volume as used when pooled A pool is generated from 3 ng from each individual library. The library should not include libraries that failed amplification
cDNA Sequencing: sequence type and yield ● TIMING 24 h
-
25|
Subject the libraries to paired-end (preferable) or single-end sequencing on a suitable Illumina NGS platform (MiSeq, HiSeq and NextSeq); aim to generate 2–5 million reads per sample with a read length of 100–150 bases. Data are generated in .fastq format. Example sequencing statistics are provided in Supplementary Table 1.
RNA-seq analysis: sequence quality assessment and preprocessing ● TIMING variable
-
26|
Sequence quality assessment. Evaluate sequence files from each nucleus (sample) from Step 25 using the fastQC tool for sequence yield, base quality, GC profile, k-mer distribution, contamination and so on. A computer grid environment should be used for processing a large number of samples simultaneously. The prototype command used is shown below. Note that the fastqc version available to the user can differ from the one shown here.
$ java -Xmx1500m -cp RUNDIR/fastqc_v0.10.1/FastQC/sam- 1.32.jar:fastqc_v0.10.1/FastQC/jbzip2-0.9.jar:fastqc_v0.10.1/FastQC/ -Dfastqc.nogroup=true uk.ac.babraham.FastQC.FastQCApplication SEQDIR/input.sample.fastq.gz
-
27|
Sequence duplication. Determine the degree of sequence duplication in the input data. Use the fastx_collapser tool to calculate the absolute number of identical reads (duplicates) in the input sample fastq sequences (from Step 25). Use correct base quality score offset (-Q). Process multiple sequence files iteratively (the program accepts only one sequence file as input).
$ RUNDIR/fastx_collapser -Q 33 -v -i SEQDIR/input.sample.fastq 1>/dev/null 2>input.sample.fastq.duplicate_summary.txt
-
28|
Sequence trimming. Use the trimmomatic program to perform trimming of input paired-end or single-end .fastq reads (from Step 25) to remove adapter/primer sequences and low-quality end bases. The adapters and primers used in the commands below are shown in the Supplementary Note.
If sequences are paired-end only:
$ java -jar Trimmomatic-0.32/trimmomatic-0.32.jar PE -threads 8 – phred33 -trimlog input.sample.fastq.trim.log input.sample.R1.fastq.gz input.sample.R2.fastq.gz input.sample_trimmed.R1.fastq input.sample_trimmed.S1.fastq input.sample_trimmed.R2.fastq input.sample_trimmed.S2.fastq ILLUMINACLIP:adapters.primers.txt:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:60
If sequences are single-end only:
$ java -jar Trimmomatic-0.32/trimmomatic-0.32.jar SE -threads 8 -phred33 -trimlog input.sample.fastq.trim.log input.sample.single.fastq.gz input.sample_trimmed.single.fastq ILLUMINACLIP:adapters.primers.txt:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:60
RNA-seq analysis: sequence mapping and expression analysis by RSEM ● TIMING variable
-
29|
Preparation of the reference genome. Index the reference genome and transcript fasta files for mapping the trimmed reads to the reference genome using bowtie2 program. Use reference genome annotation file (GTF) for the generation of indexes for individual transcripts. Choose a prefix for naming the index files used in the mapping.
$ RUNDIR/rsem-prepare-reference --gtf REFDIR/GRCh37_ERCC_GFP_RNASpikes.gtf --bowtie2 REFDIR/GRCh37_ERCC_GFP_RNASpikes.fa RSEM_GRCh37_ERCC_GFP_RNASpikes
-
30|
Calculating expression values. Map paired-end reads that survive trimming (Step 28) to the reference transcripts, and calculate gene- and isoform-level expression values using expectation-maximization algorithm, as implemented by the RSEM program.
$ RUNDIR/rsem-calculate-expression --bowtie2 -p 8 --tag MA:i:2 -fragment-length-min 1 --fragment-length-max 500 --output-genome-bam --calc-pme --calc-ci --estimate-rspd --time --paired-end SEQDIR/input.sample.R1.fastq SEQDIR/input.sample.R2.fastq RSEM_GRCh37_ERCC_GFP_RNASpikes sample_name
-
31|
Sensitivity assay of transcript expression. To determine the lower threshold and the dynamic range of detection sensitivity across high to low copy numbers of RNA expression using ERCC spike-in transcripts, first convert the ERCC RNA spike-in molar concentrations (http://tools.lifetechnologies.com/content/sfs/manuals/cms_095046.txt) to number of molecules after adjusting for 1:1.1 × 107 dilutions used in preparing the final reaction mixture. Then, calculate mean transcripts per million (TPM) values from nuclei (samples) for each of the 92 ERCC spike-in transcripts expressed at >0 TPM in at least one sample. Finally, generate a regression plot after transforming the number of ERCC spike-in molecules (x axis) and the mean TPM values (y axis) on log2 scale (Fig. 6 and Supplementary Table 2).
-
32|
Extract supplementary methods and load IPython Notebook. Download the SupplementaryMethods.zip file (Supplementary Methods) and extract its content. It contains files for Steps 32–46 and Step 47 in the folders ‘steps 32–46’ and ‘step47’, respectively. For ease of use, Steps 32–46 are present in the accompanying IPython notebook (data_analysis.ipynb). The notebook also makes calls to the supplementary file (helpers.py) to parse and process the data generated. In what follows, all directions for the notebook appear as IN>. Note that it is not necessary unless directed to change the commands in the notebook; one may execute a code block by pressing control+enter. The commands are duplicated here for completeness and for alternate workflows. This pipeline is also available online at https://github.com/Schork-Lab/np_single_nucleus_rnaseq/
Download and move to directory with the SupplementaryMethods.zip $ unzip SupplementaryMethods.zip $ cd steps32–46 $ ipython notebook In the browser window that opens, click on data_analysis.ipynb
-
33|
Load libraries and change paths. The script begins by loading the necessary libraries. If the libraries cannot be loaded, please use the Python Package Index to download them, and restart the IPython notebook. Before beginning the analysis, it is necessary to set several paths that follow from the Directory Structure. These include paths to the .bam files (bam_dir) and RSEM-generated genes.results file (rsem_dir). These paths follow from the analysis until Step 30. In addition, if tools samtools, bedtools and geneBody_coverage.py are not in the system path, please include full paths to them.
IN> #Python libraries import os # Python packages import pandas as pd import seaborn as sns # User modules import helpers # Figure styles sns.set_context(‘notebook’) sns.set_style(“white”) IN> data_path = “/home/kunal/tscc_projects/lasken/data/” bam_dir = data_path rsem_dir = data_path out_dir = os.path.join(data_path, “out”) if not os.path.exists(out_dir): os.mkdir(out_dir) path_to_samtools = ‘samtools’ path_to_genebody_coverage = ‘geneBody_coverage.py’ path_to_bedtools = ‘bedtools’
-
34|
Calculate and plot overall mapping statistics. Calculate the number of reads mapped to the genome, mapped to the ERCC spike-ins or that remain unmapped using the samtools idxstats tool. Python is used to generate the necessary Unix commands, and they are executed within the IPython environment. Load the resulting files into Python and generate a stacked barplot.
IN> for fn in os.listdir(bam_dir): if fn.endswith(‘.genome.sorted.bam’): out_file = os.path.join(out_dir, 1. fn.replace(‘.bam’,’.idxstats’)) in_file = os.path.join(bam_dir, fn) samtools_cmd = “%s idxstats %s > %s” % \ (path_to_samtools, in_file, out_file) print “Running samtools idxstats for file: %s” % fn !$samtools_cmd mapped_df = helpers.load_mapped_data(out_dir).sort() ax = mapped_df.plot(kind= ‘barh’, stacked=True) ax.set_xlabel(‘Number of Reads’)
? TROUBLESHOOTING
RNA-seq analysis: biological and technical variation ● TIMING variable
-
35|
Load and parse TPM values generated by RSEM. RSEM generates a genes.results file with several quantitative measures of a gene’s expression. For all samples, load and parse these files to extract only the TPM column, and then merge all the files into a single matrix. For this matrix, the rows are gene ids, the columns are sample ids and the value of each cell is the TPM value for that gene in that sample. Filter out all genes that are only expressed in one sample or zero samples. Also, filter out ERCC spike-in contigs’ expression from the TPM matrix.
IN> tpm_df = helpers.filter_df(helpers.load_tpms(rsem_dir), genes_only=True, expressed_in_multiple=False) -
36|
Calculate and plot counts of genes expressed in single nuclei relative to bulk RNA. Divide a chosen control sample’s set of expressed genes into ‘low’, ‘mid’ and ‘high’ designations on the basis of their quantiles of expression. The default values used for the low- expressed genes are those that are in the quantile up to 0.33, the values for mid-expressed genes are: 0.33 to 0.67, and the values for high-expressed genes are: 0.68 to 1. For each sample, count how many genes are designated as low, mid, high, or novel through set intersections.
IN> control = ‘Total RNA-100pg-2’ # Set control sample name low, mid, high = helpers.get_low_mid_high_genes(tpm_df[control]) expressed_df = helpers.calculate_relative_expression(tpm_df, low, mid, high)
-
37|
Plot relative expression in single nuclei compared with bulk RNA. Create two plots: one plot details which fraction of the control sample’s genes is expressed in each sample (Fig. 7a). The other plot details the relative composition of the genes expressed in each sample to the control sample (Fig. 7b).
IN> cols = [‘Low’, ‘Mid’, ‘High’] control_values = expressed_df[cols].ix[control] fraction_df = expressed_df[cols].astype(float)/control_values ax = fraction_df.plot(kind= ‘barh’) ax.set_title(‘Fraction of %s Genes Expressed’ % control) ax.set_xlabel(‘Fraction of Genes Expressed’) IN> composition_df = expressed_df.apply(lambda x: x.astype(float)/x.sum(), 1. axis=1) cols = [‘Low’, ‘Mid’, ‘High’, ‘Novel’] ax = composition_df[cols].plot(kind= ‘barh’) ax.set_title(‘Composition of Genes Expressed \nRelative to Expression in %s’ % control,loc= ‘left’) ax.set_xlabel(‘Fraction of Genes Expressed’) -
38|
Plot pairwise correlation of expression across all samples. Calculate pairwise Spearman’s correlation for all samples. Stratify the correlation matrices by low, mid and high genes based on their expression in the previously defined control sample. Plot the resulting matrices as heat maps.
IN> for genes, gene_type in zip([low, mid, high], [‘Low’, ‘Mid’, ‘High’]): fig = plt.figure(figsize=(8,8)) ax = fig.add_subplot(111) ax = sns.corrplot(tpm_df.ix[genes.index].sort(axis=1), method= ‘spearman’, ax=ax, diag_names=False, cmap_range=(0, 1), cbar=True) ax.set_title(‘Correlation Stratified by %s Expression in %s’ % (gene_type, control)) sns.despine()
RNA-seq analysis: quality based on coverage across the gene body ● TIMING variable
-
39|
Create bed file of highly expressed genes. To gain a better idea of the quality of the transcripts being sequenced, focus on transcripts that are highly expressed. Create a .bed file to be used in other tools that only has the highly expressed transcripts based on the control sample.
IN> gtf_file = os.path.join(data_path, ‘reference’, ‘GRCh37_ERCC_GFP_RNASpikes.gtf’) high_gtf = gtf_file.replace(‘.gtf’, ‘.high_expressed.gtf’) print “Subsetting %s to only highly expressed genes as %s” % (gtf_file, high_gtf) helpers.subset_gtf_by_genes(high_gtf, gtf_file, list(high.index)) high_bed = high_gtf.replace(‘.gtf’, ‘.bed’) print “Converting %s to %s” % (high_gtf, high_bed) !perl gtf2bed.pl $high_gtf > $high_bed -
40|
Calculate coverage across the gene body. For the highly expressed transcripts, calculate their coverage across the length of the gene body using RseqC’s geneBodyCoverage.py tool. Use Python to generate the command that includes all the sample .bam files, as well as the highly expressed genes .bed file. Run this command through the IPython shell.
IN> rnaseqc_prefix = os.path.join(out_dir, “rnaseqc_high_coverage_control”) sample_files = [os.path.join(bam_dir, fn) for fn in os.listdir(bam_dir) if fn.endswith(‘.genome.sorted.bam’)] in_files = “, “.join(sample_files) rnaseq_c_cmd = “ %s -i %s --refgene %s --out-prefix %s” % \ (path_to_genebody_coverage, in_files, high_bed, rnaseqc_prefix) fns = “, “.join([os.path.basename(fn) for fn in sample_files]) print “Running gene body coverage for sample files: %s” % fns !rnaseq_c_cmd -
41|
Plot relative coverage across the gene body. Load in the previously generated geneBodyCoverage files from Step 40 using a helper function. The helper function defines the normalized coverage as the (coverage – minimum coverage)/(maximum coverage – minimum coverage). Plot the data and set appropriate labels.
IN> rnaseqc_file = rnaseqc_prefix+ ‘.geneBodyCoverage.txt’ normalized_df, coverage_df = helpers.load_gene_body_coverage(rnaseqc_file) ax = normalized_df.plot() ax.set_xlabel(“Gene Body (5′ -> 3′)”) ax.set_ylabel(“Relative Coverage”)
RNA-seq analysis: quality based on intron and exon coverage ● TIMING variable
-
42|
Align reads using TopHat2. As the RSEM program maps sequences to only exons in the annotated reference transcripts, use TopHat2 program for mapping reads to both exons and introns in the reference genomic sequence. Generate the appropriate index files needed for bowtie2 mapper, which is executed by TopHat2. Run the following commands in sequence to generate a .bam alignment file with sequences mapped to exons and introns.
$ RUNDIR/bowtie2-build REFDIR/GRCh37_ERCC_GFP_RNASpikes.fa GRCh37_ERCC_GFP_RNASpikes $ RUNDIR/samtools faidx REFDIR/GRCh37_ERCC_GFP_RNASpikes.fa $ RUNDIR/tophat2 -p 8 --library-type fr-unstranded -G REFDIR/GRCh37_ERCC_GFP_RNASpikes.gtf GRCh37_ERCC_GFP_RNASpikes SEQDIR/input.sample.R1.fastq.gz SEQDIR/input.sample.R2.fastq.gz
-
43|
Inspect in IGV. Open IGV Viewer, and load in the .bam file. Manually zoom in and out of large housekeeping genes such as GAPDH to inspect whether only spliced transcripts are being sequenced.
-
44|
Create intron and exon .bed files. Set paths for the names and locations of the intron and exon .bed files. Use the accompanying create_intron_exon_beds.sh to create intron and exon .bed files based on the provided GTF file.
IN> intronic_bed = os.path.join(data_path, ‘reference’, ‘GRCh37_ERCC_GFP_RNASpikes.gtf.introns.bed’) exonic_bed = os.path.join(data_path, ‘reference’, ‘GRCh37_ERCC_GFP_RNASpikes.gtf.exons.bed’) !sh create_intron_exon_beds.sh $gtf_file $exonic_bed $intronic_bed -
45|
Calculate coverage overlaps with exons and introns. Set the path to the TopHat2-generated .bam file (from Step 42). Use bedtools command bamtobed in conjunction with the bedtool coverage command to look at the coverage across introns and exons of the sample .bam file.
sample_tophat_bam = ‘/path/to/tophat.aligned.bam’ intronic_out = sample_tophat_bam.replace(‘.bam’, ‘.intronic_coverage’) intronic_cmd = “%s bamtobed -splitD -i %s | awk \’BEGIN{OFS=\”\\t\”}$1=\”chr\”$1\’ | %s coverage -a - -b %s > %s” % (path_to_bedtools, sample_tophat_bam, path_to_bedtools, intronic_bed, intronic_out) print “Creating intronic coverage file” !$intronic_cmd print exonic_out = sample_tophat_bam.replace(‘.bam’, ‘.exonic_coverage’) exonic_cmd = “%s bamtobed -splitD -i %s | awk \’BEGIN{OFS=\”\\t\”}$1=\”chr\”$1\’ | %s coverage -a - -b %s > %s” % (path_to_bedtools, sample_tophat_bam, path_to_bedtools, exonic_bed, exonic_out) print “Creating exonic coverage file” !$exonic_cmd -
46|
Load and plot exonic versus intronic coverage. Load the generated bedtools coverage files from Step 45 for the introns and exons. Select regions that have at least 1 read mapping to them and that are at least 1 kb long. Plot the differences between the intronic and exonic regions, as shown in Supplementary Figure 2.
IN> intronic_df = helpers.load_bedtools_coverage(intronic_out, min_reads=1, min_length=100) exonic_df = helpers.load_bedtools_coverage(exonic_out, 1, 100) fig = helpers.plot_bedtools_coverage(intronic_df, exonic_df)
Sample classification ● TIMING variable
-
47|
Cell type classification for assessing how well nuclear and brain cell RNA matches: R code and additional files required to reproduce this step are provided in the folder ‘step47’ (Supplementary Methods). Convert Ensembl Gene identifiers into current gene symbols using BioMart (http://www.ensembl.org/biomart/martview; downloaded 1/26/15 (ref. 26)), and exclude all transcripts without a current gene symbol. Convert TPM values to log scale (offsetting by 1). Cluster cells by identifying the 1,000 genes with the highest variability, finding the Pearson’s correlation distance, performing multidimensional scaling to identify the first four principal coordinates and running k-means clustering with K = 4 on these principal coordinates. Calculate the number of genes expressed in each cluster for comparison. Determine the cell type of each cluster by collecting lists of marker genes for known brain cell types24,35, by determining the expression levels of these sets of genes in each nuclei, assigning cell type based on high expression of markers and confirming cell type classification based on nearly exclusive enrichment of individual canonical marker genes.
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 1.
TABLE 1.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 5 | Low nuclei yield | Poor-quality tissue | Obtain intact tissue with a low number of freeze-thaw cycles |
| Lack of nuclei in tissue | Microscopically assess the density of nuclei in the tissue | ||
| Inadequate cell lysis to release the nuclei | Use appropriate concentration of detergents, salt and sucrose in the cell lysis buffer | ||
| Optimize the number of Dounce strokes | |||
| Use a homogenizer with appropriate clearance level to release the nuclei | |||
| Use chilled buffers and homogenizers, and execute the entire procedure at 4 °C | |||
| Improper centrifugation may cause the cellular debris to sediment, which may alter the yield of pure nuclei | Optimize the density gradient for nuclei isolation and the speed of centrifugation to your tissue type | ||
| 18 | Poor recovery of single nuclei from FACS | Targeting of FACS for single nuclei isolation is compromised | Optimize FACS conditions and determination of sorting gates |
| Sort nuclei onto a glass slide and visualize them under the microscope | |||
| 23 | Failure of qPCR assays | No nuclei in the wells (if using FACS) | Optimize single-nucleus targeting into wells of the microtiter plate prior to FACS |
| Low-quality RNA obtained from the lysed nuclei | Use a sample with a high RIN value | ||
| mRNA degradation | Keep the workstation and tools free of RNases by thoroughly cleaning with RNaseZap. Do this daily or before each experiment | ||
| Inefficient cDNA synthesis | Use fresh dNTPs | ||
| Keep all reagents on ice and minimize the freeze-thaw cycles of sensitive items | |||
| Reverse transcription failure | Check all cDNA synthesis steps using ERCC spike-in as a positive control | ||
| 34 | Excessive DNA sequencing reads failing to map to the reference genome | Concatemer formation from the TSO primer of the Smart-seq2 method | Be certain to use the 5′ biotin–modified TSO primer11 (as done in step 19 and discussed in the INTRODUCTION) rather than the unmodified version used in Picelli et al.3 |
● TIMING
Steps 1–5, nuclei isolation: 1–2 h
Steps 6–12, (optional) neuronal nuclei immunostaining: 1–1.5 h
Steps 13–18, nuclei FACS sorting: 2–3 h
Step 19, cDNA synthesis by Smart-seq2: 1 d
Steps 20–23, qPCR and TaqMan analysis: 3 h
Step 24, sequencing library preparation: 2 h
Step 25, cDNA sequencing: sequence type and yield: 24 h
Steps 26–46, RNA-seq analysis: sequence quality assessment and preprocessing: variable
Step 47, sample classification: variable
Box 3, sample quality assessment of tissue and cultured cells: 1 h
Box 4, density gradient centrifugation: 1 h (optional)
ANTICIPATED RESULTS
This protocol enables the FACS-based isolation of single nuclei suitable for RNA sequencing. The use of a neuron-specific antibody for staining allows comparison of transcriptomes from neurons and other cell types. The RNA-seq data can be used to determine cell types based on the profiles of the genes expressed.
Figures 3–10 are generated from an RNA-seq experiment on single nuclei isolated from frozen normal human cortical brain samples obtained from the NIH NeuroBioBank located at the University of Maryland, where they were stored at −80 °C. The brain specimens had been collected and deposited at NeuroBioBank up to several hours after death. The nuclei were stained with NeuN-Alexa Fluor 488–conjugated antibody and sorted using FACS gating parameters designed to distinguish neurons and non-neurons (Fig. 3a–f). The sorting accuracy and precision for single nuclei was verified by sorting beads into 384-well plates and viewing them under the microscope (Fig. 3g). Figure 3h,i shows PI-stained nuclei.
Bioanalyzer analysis of the quality of cDNA library synthesis and amplification by Smart-seq2 gave typical results (Fig. 4). After AMPure bead purification of the cDNA library, a size range of ~150 bp to 7 kbp is expected, with the majority of fragments in the 1- to 3-kb range (Fig. 4b). Primer dimers in the size range of ~100 bp make up a small minority of the total cDNA, and therefore no further purification after Ampure bead cleanup is necessary before library prep. The primer dimers are further reduced in the purification of the library prep (Fig. 4c) and in a second purification of the pooled library for Illumina sequencing. After Nextera XT purification, the typical size range of the library is 200–1,000 bp (Fig. 4c). If necessary, libraries may be pooled and further purified before sequencing to get the optimal library insert size for maximum read depth, but with the expectation of some loss of material.
Detection of gene expression (Steps 29 and 30)
Of the ten nuclei sequenced in this example, six were identified as neuronal on the basis of FACS for the NeuN protein and four were non-neuronal (Fig. 7). Note that the percentage of reads mapping to ERCC spike-in controls, the genome and unmapped reads can vary widely depending on the starting amount of mRNA derived from the nucleus and the amount of artifactual PCR products such as primer dimers that are created. The number of genes expressed also varies widely among single nuclei, most likely owing to variation in the mRNA content of phenotypically different cell types, as well as technical sources of variation caused by insufficient lysis of the nuclei and suboptimal cDNA synthesis (see ‘Sample controls’ in the INTRODUCTION for comments on use of technical replicates to evaluate experimental noise). The number of genes detected ranged from 1,102 to 6,221 (Fig. 7). The range was higher for total RNA as expected, as a population of different cell types is represented by this RNA template. Failure to detect many genes expressed from a single nucleus may indicate poor yields of cDNA and lack of sensitivity for low-copy transcripts. However, caution must be used in this conclusion, as the cells may simply have been relatively quiescent. More genes will be expressed in pools of multiple nuclei reflecting the full range of genes expressed in the cell population. This can also serve as an important validation for genes detected in single nuclei. In general, the genes expressed in the pools should represent the sum of all genes detected in the individual nuclei. The level of expression should also agree between pools and individual nuclei. For example, a gene that is expressed at a high level but in only a small percentage of nuclei should appear at a commensurately low level in the pools. Expression signatures are nearly identical between nuclei and whole cells over a wide range of RPKM values; however, a subset of transcripts known to be enriched in nuclei, on the basis of bulk-RNA extractions, was confirmed as enriched in the individual nuclei13.
Sensitivity of detection (Step 31)
The detection sensitivity of the RNA expression analysis is determined by adding ERCC spike-in control transcripts of various concentrations to the lysis buffer (Step 13) used to release RNA from the nuclei. The ERCC spike-ins are processed along with sample RNA through the RT reaction and subsequent cDNA amplification and sequencing (Box 2c). The limit of detection for ERCC spike-in transcripts should be <10 copies, as observed in the example provided here (Fig. 6 and Supplementary Table 2). Expression of a single-copy ERCC spike-in transcript can be detected at an approximate threshold value of nine TPM (intersection with the y axis, Fig. 6). A failure to generate cDNA for the ERCC spike-ins would indicate failure of the Smart-seq2 reaction, for example, because of inactive RT. If the ERCC spike-ins generate the expected amount of cDNA but cellular transcripts are not detected, then the transcripts were lost at some stage of the process, probably because of degradation of RNA in the cell resulting from improper handling or storage, failure to successfully sort the nuclei into the wells or failure to completely lyse the nucleus.
When compared with the transcripts expressed at high and medium levels, those with a low level of expression show a greater degree of variation relative to the pattern of expression seen in the control total RNA samples (Fig. 7b and Supplementary Fig. 3). It is possible that the lack of expression for low-copy transcripts reflects real biological phenomena. For example, low-copy transcripts may be more likely to be variably expressed if they tend to be involved in regulatory or other nonconstitutive functions. However, we suspect that at least some of the effect results from variable sensitivity below 10 transcript copies.
Assessing 3′ bias (Steps 40 and 41)
3′ bias can be indicative of damaged RNA, as well as poor activity from the RT, and it is a source of noise in RNA-seq experiments (Fig. 8a). The graph output details the relative coverage across the gene body from the 5′ end to the 3′ end for the highly expressed genes in the example given (Fig. 8b). An almost square wave should be observed, showing uniform relative coverage across the gene body with drop-offs near the 5′ and 3′ end because of end effects in the Nextera tagmentation reaction for library construction. If the plot is highly skewed to the 3′ end relative to the plot for control RNA of high RIN value, it is indicative of poor RNA quality. In this example, a nearly identical 3′ bias was found in cDNA from nuclei and the control-purified RNA (Fig. 8b), confirming that the cDNAs from single nuclei were predominantly full length. Recently, we have confirmed that damaged mRNA controls, generated by heating in the presence of sodium acetate, quantitatively generate 3′ bias in the sequence coverage (M.N. and R.S.L., unpublished data).
Analysis of exon and intron coverage (Steps 42–46)
Most or all of the detected transcripts will be fully spliced with relatively uniform coverage across exon/exon junctions but not intron/exon junctions13. In the example shown here, exons are fully covered by at least one read, whereas only a few intronic regions have their entire length covered fully by reads (Fig. 9 and Supplementary Fig. 2). Although many reads map to intronic regions13, the source of these reads is not clear. The length of an exon does not seem to show a correlation with the extent to which the exon is covered. Only small introns (<10 kb) show full coverage across their entire length. The absence of intronic reads and the relatively even sequence coverage across exon/exon junctions confirms an earlier finding13 that most or all of the transcripts obtained from the nuclear lysates have been spliced. Intronic reads were detected, but these were not present evenly across exon/intron junctions (Fig. 9 and Supplementary Fig. 2), as should be observed if unspliced transcripts were detected.
Cell type classification (Step 47)
The RNA-seq data can be used to verify that specific cell types have been enriched by FACS. In the example shown here, the presence of the NeuN protein (Fig. 5, nuclei labeled neuronal), a neuron-specific nuclear marker, based on antibody labeling during FACS of nuclei, was consistent with the RNA-seq detection of NeuN transcript in half of the nuclei labeled with anti-NeuN antibody and none of the NeuN-negative nuclei (Supplementary Table 1). In cases in which the cell is positive for a protein marker based on FACS, but the transcript is not detected, it is possible that the protein is longer lived than the transcript. Transcription tends to occur in bursts, and it does not exactly reflect protein concentrations. Alternatively, some nuclei may be spuriously identified as positive during FACS.
Gene expression values from nuclei can be used to identify cell types13. In the example given here, gene expression was analyzed from the ten postmortem human nuclei to evaluate the identities and characteristics of the cells. To do so in an unbiased manner, we first identified the 1,000 annotated genes with the highest variability across the 10 nuclei, and we then clustered the nuclei into four groups using k-means clustering (Fig. 10a). All of the NeuN+ nuclei (labeled 1–6 in Fig. 10) and one of the NeuN– (D) nuclei were found in two clusters that contained a large number of overlapping genes (Fig. 10b and Supplementary Table 3), whereas the remaining three NeuN– nuclei (A–C) clustered separately, suggesting that our FACS strategy is highly accurate, but not perfect, at separating nuclei from different cell types. To further characterize these nuclei, we measured the average expression levels of known marker genes for different brain cell types, on the basis of two studies that transcriptionally profiled pure cell populations in mouse (Fig. 10c and Supplementary Table 4). The remaining two clusters of predominantly NeuN+ nuclei both showed high expression for neuronal markers, but they showed different levels of inhibitory and excitatory marker genes36. The remaining two clusters of NeuN– nuclei showed lower expression of neuronal markers but high expression of markers for specific glial populations37: astrocytes and oligodendrocyte precursor cells (Fig. 10c and Supplementary Table 4). Expression patterns of specific marker genes for these cell types confirm these cell type classifications (Fig. 10d). Overall, we found that the ten nuclei profiled by RNA-seq came from four distinct brain cell types.
Acknowledgments
R.S.L. was funded by NIH-1RC1 HG005471, NIH-2R01 HG003647; R.S.L. and F.H.G. by Transformative R01MH095741; F.H.G. by the Jeffry M. and Barbara Picower Foundation, the Mathers Foundation, the McDonnell Foundation and the National Institute of Mental Health (NIMH); and M.J.M. by a Crick-Jacobs Junior Fellowship. K.B. is supported by the National Science Foundation Graduate Research Fellowship under grant no. DGE-1144086. The authors thank the Allen Institute for Brain Science founders, Paul G. Allen and Jody Allen, for their vision, encouragement and support. Research was supported in part by the Allen Institute for Brain Science.
Footnotes
AUTHOR CONTRIBUTIONS R.S.L., R.V.G., M.J.M., S.R.K., F.H.G., E.S.L. and J.L. contributed to design of the project. S.R.K., M.N., B.J.L., J.A.E., R.H. and J.K.M. carried out experiments. P.V., K.B., S.B.L., S.P., M.K., J.M., J.K.M., N.S., B.D.A., F.D.-F. and R.H.S. contributed to sequencing data analysis. R.S.L., R.V.G., S.R.K. and M.N. wrote the manuscript with contributions from all the authors.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
References
- 1.Hashimshony T, Wagner F, Sher N, Yanai I. CEL-Seq: single-cell RNA-seq by multiplexed linear amplification. Cell Rep. 2012;2:666–673. doi: 10.1016/j.celrep.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 2.Kurimoto K, Yabuta Y, Ohinata Y, Saitou M. Global single-cell cDNA amplification to provide a template for representative high-density oligonucleotide microarray analysis. Nat Protoc. 2007;2:739–752. doi: 10.1038/nprot.2007.79. [DOI] [PubMed] [Google Scholar]
- 3.Picelli S, et al. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc. 2014;9:171–181. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- 4.Ramskold D, et al. Full-length mRNA-seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol. 2012;30:777–782. doi: 10.1038/nbt.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tang F, et al. RNA-seq analysis to capture the transcriptome landscape of a single cell. Nat Protoc. 2010;5:516–535. doi: 10.1038/nprot.2009.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lovatt D, et al. Transcriptome in vivo analysis (TIVA) of spatially defined single cells in live tissue. Nat Methods. 2014;11:190–196. doi: 10.1038/nmeth.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Citri A, Pang ZP, Sudhof TC, Wernig M, Malenka RC. Comprehensive qPCR profiling of gene expression in single neuronal cells. Nat Protoc. 2012;7:118–127. doi: 10.1038/nprot.2011.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qiu S, et al. Single-neuron RNA-seq: technical feasibility and reproducibility. Front Genet. 2012;3:124. doi: 10.3389/fgene.2012.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lovatt D, Bell T, Eberwine J. Single-neuron isolation for RNA analysis using pipette capture and laser capture microdissection. Cold Spring Harb Protoc. 2015 doi: 10.1101/pdb.prot072439. [DOI] [PubMed] [Google Scholar]
- 10.Darmanis S, et al. A survey of human brain transcriptome diversity at the single cell level. Proc Natl Acad Sci USA. 2015;112:7285–7290. doi: 10.1073/pnas.1507125112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zeisel A, et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science. 2015;347:1138–1142. doi: 10.1126/science.aaa1934. [DOI] [PubMed] [Google Scholar]
- 12.Huang HL, et al. Trypsin-induced proteome alteration during cell subculture in mammalian cells. J Biomed Sci. 2010;17:36. doi: 10.1186/1423-0127-17-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grindberg RV, et al. RNA-sequencing from single nuclei. Proc Natl Acad Sci USA. 2013;110:19802–19807. doi: 10.1073/pnas.1319700110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barthelson RA, Lambert GM, Vanier C, Lynch RM, Galbraith DW. Comparison of the contributions of the nuclear and cytoplasmic compartments to global gene expression in human cells. BMC Genomics. 2007;8:340. doi: 10.1186/1471-2164-8-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng J, et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science. 2005;308:1149–1154. doi: 10.1126/science.1108625. [DOI] [PubMed] [Google Scholar]
- 16.Schwanekamp JA, et al. Genome-wide analyses show that nuclear and cytoplasmic RNA levels are differentially affected by dioxin. Biochim Biophys Acta. 2006;1759:388–402. doi: 10.1016/j.bbaexp.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 17.Trask HW, et al. Microarray analysis of cytoplasmic versus whole cell RNA reveals a considerable number of missed and false positive mRNAs. RNA. 2009;15:1917–1928. doi: 10.1261/rna.1677409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang Y, Matevossian A, Huang HS, Straubhaar J, Akbarian S. Isolation of neuronal chromatin from brain tissue. BMC Neurosci. 2008;9:42. doi: 10.1186/1471-2202-9-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Birnie GD. Isolation of nuclei from animal cells in culture. Methods Cell Biol. 1978;17:13–26. doi: 10.1016/s0091-679x(08)61131-0. [DOI] [PubMed] [Google Scholar]
- 20.Schroeder A, et al. The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol. 2006;7:3. doi: 10.1186/1471-2199-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dounce AL, Witter RF, Monty KJ, Pate S, Cottone MA. A method for isolating intact mitochondria and nuclei from the same homogenate, and the influence of mitochondrial destruction on the properties of cell nuclei. J Biophys Biochem Cytol. 1955;1:139–153. doi: 10.1083/jcb.1.2.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hymer WC, Kuff EL. Isolation of nuclei from mammalian tissues through the use of Triton X-100. J Histochem Cytochem. 1964;12:359–363. doi: 10.1177/12.5.359. [DOI] [PubMed] [Google Scholar]
- 23.Wu AR, et al. Quantitative assessment of single-cell RNA-sequencing methods. Nat Methods. 2014;11:41–46. doi: 10.1038/nmeth.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Macosko EZ, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Usoskin D, et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci. 2015;18:145–153. doi: 10.1038/nn.3881. [DOI] [PubMed] [Google Scholar]
- 27.Rabani M, et al. High-resolution sequencing and modeling identifies distinct dynamic RNA regulatory strategies. Cell. 2014;159:1698–1710. doi: 10.1016/j.cell.2014.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lacar B, et al. Nuclear RNA-seq of single neurons reveals molecular signatures of activation. Nat Commun. doi: 10.1038/ncomms11022. in the press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang L, et al. Synthetic spike-in standards for RNA-seq experiments. Genome Res. 2011;21:1543–1551. doi: 10.1101/gr.121095.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeLuca DS, et al. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics. 2012;28:1530–1532. doi: 10.1093/bioinformatics/bts196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang L, Wang S, Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012;28:2184–2185. doi: 10.1093/bioinformatics/bts356. [DOI] [PubMed] [Google Scholar]
- 32.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Bioinformatics Action. 2013;17:2. [Google Scholar]
- 33.Trapnell C, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sugino K, et al. Molecular taxonomy of major neuronal classes in the adult mouse forebrain. Nat Neurosci. 2006;9:99–107. doi: 10.1038/nn1618. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Y, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]