

Abstract
Cell migration is key to many physiological and pathological conditions, including cancer metastasis. The cellular and molecular bases of cell migration have been thoroughly analyzed in vitro. However, in vivo cell migration somehow differs from in vitro migration, and has proven more difficult to analyze, being less accessible to direct observation and manipulation. This protocol uses the migration of the prospective prechordal plate in the early zebrafish embryo as a model system to study the function of candidate genes in cell migration. Prechordal plate progenitors form a group of cells which, during gastrulation, undergoes a directed migration from the embryonic organizer to the animal pole of the embryo. The proposed protocol uses cell transplantation to create mosaic embryos. This offers the combined advantages of labeling isolated cells, which is key to good imaging, and of limiting gain/loss of function effects to the observed cells, hence ensuring cell-autonomous effects. We describe here how we assessed the function of the TORC2 component Sin1 in cell migration, but the protocol can be used to analyze the function of any candidate gene in controlling cell migration in vivo.
Keywords: Developmental Biology, Issue 110, Zebrafish, cell migration, mosaic, cell transplantation, actin, live imaging, developmental biology, micro-injection
Introduction
In multicellular organisms, cell migration is essential both for the development of the embryo where it ensures the organization of cells into tissues and organs, and for adult life, where it takes part to tissue homeostasis (wound healing) and immunity. In addition to these physiological functions, cell migration is also involved in diverse pathological situations, including, in particular, cancer metastasis.
Cell migration has been analyzed in vitro for decades, providing an overall understanding of the molecular mechanisms ensuring cell movements on flat surfaces. In vivo however, cells are confronted by a more complex environment. It clearly appeared in the past years that migration within an organism may be influenced by external cues such as the extracellular matrix, neighboring cells or secreted chemokines guiding migration, and that the mechanisms driving cell migration may vary from what has been described in vitro1,2. The mechanisms ensuring in vivo cell migration have received less attention so far, mainly because of the increased technical difficulty, compared to in vitro studies. In vivo analysis of cell migration in particular requires direct optical access to migrating cells, techniques to label unique cells in order to see their dynamics and morphology, as well as gain or loss of function approaches to test the role of candidate genes. So far, only a few model systems harboring these characteristics have been used to dissect in vivo cell migration3.
We recently used the migration of the prospective prechordal plate in early zebrafish embryos as a new convenient model system to assess the function of candidate genes in controlling in vivo cell migration4,5. Prospective prechordal plate (also known as anterior mesendoderm) is a group of cells forming at the onset of gastrulation on the dorsal side of the embryo. During gastrulation this group collectively migrates towards the animal pole of the embryo6-8, to form the prechordal plate, a mesendodermal thickening, anterior to the notochord, and underlying the neural plate. The anterior part of the prechordal plate will give rise to the hatching gland, while its posterior part likely contributes to head mesoderm9. Thanks to the external development and optical clarity of the fish embryo, cell migration can be directly and easily observed in this structure.
Cell transplantation is a very potent technique that allows for the rapid and easy creation of mosaic embryos10. Expressing fluorescent cytoskeletal markers in transplanted cells results in the labeling of isolated cells, the morphology and dynamics of which can be easily observed. Combining this to loss or gain of function approaches permits the analysis of cell-autonomous functions of a candidate gene.
The presented protocol describes how we assessed the function of the TORC2 component Sin1 in controlling cell migration and actin dynamics in vivo5. But, as mentioned in the results and further discussed, it could be used to analyze the potential implication of any candidate gene in controlling cell migration in vivo.
Protocol
Note: Figure 1 presents the outline of the protocol.
1. Preparation of the Needles for Injection and Transplantation
Note: Needles can be prepared at any time and stored. Keep them in a Petri dish, on a band of modeling clay. Seal the dish with parafilm to protect from dust.
For injection needles, pull a glass capillary (outside diameter 1.0 mm, inside diameter 0.58 mm, without filament (see list of Materials)) with a micropipette puller (see list of Materials). Prefer short and thin needles (tapered part of about 5 mm) as they are more efficient for piercing the chorion. Note: The injection needles are single-use.
- Needle for Cell Transplantation
- Pull a glass capillary (outside diameter 1.0 mm, inside diameter 0.78 mm, without filament (see list of Materials)) with a micropipette puller.
- Using a microforge (see list of materials), cut the tip of the needle at the point where the inside diameter is 35 µm (slightly more than the diameter of cells to be transplanted).
- Bevel the cut extremity of the capillary with a microgrinder (see list of Materials) with an angle of 45°.
- Optionally, pull a barb on the end of the needle using a microforge. For this, touch the hot filament with the tip of the bevel and rapidly pull it away, creating a barb that can help penetrating the embryo.
- Mount the needle on a needle holder attached to a syringe. Using the syringe, rinse the inside of the needle three times with 2% hydrofluoric acid to eliminate glass residues, and then rinse three times with acetone. Caution: hydrofluoric acid is toxic, manipulate under the hood, with gloves. Note: The cell transplantation needles can be used several times.
2. Preparation of the Dishes for Injection and Cell Transplantation
- Heat 50 ml of embryo medium11 containing 0.5 g of agarose in a microwave for 1 min at full power to get 1% agarose in embryo medium. Cool the agarose gel to about 60 °C and pour the 50 ml in a 90 mm Petri dish. Place at the surface of the gel a specially designed mold (see list of material), either with lines for the injection dish or with wells for the cell transplantation dish.
- Observe the mold float on the agarose, if the agarose is not too hot. After the agarose gel is set, remove the mold with forceps (Figure 2A-B). Note: dishes can be stored at 4 °C, up to a few weeks. Keep them in a closed wet box, to avoid drying.
Place the dish in a 28 °C incubator 30 min before use.
3. Collection of Embryos and Injection
- Injection Solution Preparation Note: Actin binding domain of the yeast actin binding protein 140 (ABP-140) such as Lifeact bound to mCherry (referred to as ABP140-mCherry) is used to visualize polymerized actin within the cell, in order to analyze the protrusive activity. Add another compound to test the function of a candidate gene (e.g. mRNA of dominant negative or constitutively active construct, or Morpholino oligonucleotides). To assess the functions of Sin1 as described in the results (Figure 3), we used Morpholino oligonucleotides. As a control, we used a morpholino identical to the sin1 morpholino but for 5 nucleotides distributed along the sequence so that this control morpholino doesn't match the target RNA.
- Thaw sin1 and 5-mismatch control morpholinos (2 mM stock solutions), and ABP140-mCherry mRNAs on ice. Add 375 ng of mRNAs (75 ng/µl final) and 0.75 µl of either sin1 or 5-mismatch control morpholino (0.3 mM final) in Danieau buffer (58 mM NaCl, 0.7 mM KCl, 0.4 mM MgSO4, 0.6 mM Ca(NO3)2, 5.0 mM HEPES pH 7.6), to reach a total volume of 5 µl.
- Embryo Collection Note: for this experimental set up both donor and host embryos are transgenic, expressing the GFP under the control of the goosecoid (gsc) promoter in order to visualize the prospective prechordal plate11.
- Collect Tg(gsc:gfp) eggs. Place about 80 eggs in a 90 mm Petri dish filled with embryo medium and incubate at 28 °C.
- Inject Donor Embryos
- Under a stereomicroscope, gently squeeze collected embryos with forceps into the lines of an injection dish filled with embryo medium. Using forceps, orient the embryos with the cells towards the top. Place the injection dish in the incubator at 28 °C until the embryos have reached the 4-cell stage (1 hr post fertilization).
- Load an injection needle with 2 µl of injection solution. Insert the needle in a needle holder (see list of materials) that is placed in a micro-manipulator (see list of materials) and connected with polytetrafluoroethylene (PTFE) tubing (see list of Materials) to an air transjector (see list of materials).
- Open the capillary by gently touching the tip with fine forceps. If necessary, cut the capillary open by pinching its extremity.
- Enter one of the four cells with the needle and inject the solution within the cell in order to fill 70% of the cell volume (2 nl). Inject 30 embryos. Note: Getting the needle in the cell may be difficult, as the plasma membrane is soft. Aiming at the junction between the cell and the yolk facilitates needle penetration.
- Place the embryos in the 28 °C incubator until they have reached the sphere stage (4 hours post fertilization).
4. Preparation of the Embryos for the Cell Transplantation
Prepare 20 ml of embryo medium with 200 µl penicillin/streptomycin (see list of materials) (Pen/Strep EM).
- Dechorionation of the embryos at the sphere stage (4 hr post fertilization) Note: Dechorionated embryos are very fragile and will explode in case of contact with air or plastic. They thus need to be placed in agarose-coated dishes, and transferred with fire-polished glass Pasteur pipettes.
- Using a plastic Pasteur pipette, transfer host embryos collected at step 3.2 and donor embryos injected at step 3.3 into two 35 mm Petri dishes coated with 1% agarose in embryo medium and filled with Pen/Strep EM. Keep host embryos and donor embryos in two separate dishes.
- Manually extract the embryos from their chorions with fine tweezers (see list of materials).
- Place the embryos in the 28 °C incubator until they have reached the shield stage (6 hr post fertilization).
5. Cell Transplantation
- Arrange the Embryos for the cell Transplantation.
- Under a fluorescent stereomicroscope, select donor embryos expressing ABP140-mCherry (red) within the shield (green).
- Using a fire-polished Pasteur pipette, transfer the embryos in the wells of the cell transplantation dish filled with Pen/Strep EM. Place the host embryos in one row and the donor embryos in the neighboring row.
- Using an eyelash, carefully orient the embryos with the shield up. Note: the position of the shield can be assessed by GFP expression or anatomically.
- Transplantation System Set-up. Note: The transplantation system is composed of a needle holder (see list of materials) connected with PTFE tubing (see list of Materials) and a tube connector (see list of Materials) to a Hamilton syringe equipped with a micro-drive (see list of Materials). The needle holder is mounted on a 3-way micromanipulator (see list of Materials).
- Using a needle holder attached to a syringe, rinse the needle for cell transplantation with 70% ethanol. Dry by sucking up air into the needle. Do not dry by blowing, as this may result in dust getting stuck in the tapered part of the needle.
- Insert the needle in the needle holder connected to the Hamilton syringe, with the bevel upwards.
- Shield-to-shield Cell Transplantation
- Enter the shield of a donor embryo with the transplantation needle and draw up about 10-20 cells labeled with ABP140-mCherry. Carefully avoid drawing yolk.
- Transplant the cells into the shield of the host embryo. Repeat until all host embryos have been transplanted.
- Remove any damaged embryos with a fire-polished Pasteur pipette. Place the transplanted embryos in the incubator at 28 °C and let them recover for about 30 min.
- Using a needle holder attached to a syringe, rinse the cell transplantation needle with water and place it back in a Petri dish on a band of modeling clay. Seal the dish with parafilm.
6. (OPTIONAL) Single Cell Transplantation
Note: In the embryo, cells adhere to each other, so that it is difficult to draw only one cell in the transplantation needle. We developed a modified protocol to easily transplant single prechordal plate progenitor cells. The idea is to dissociate cells prior to transplantation. Because isolated prechordal plate progenitor cells tend to lose their identity, we genetically impose them a prospective prechordal plate identity, by activating the Nodal signaling pathway, in absence of the Sox32 transcription factor. We have verified that these induced cells behave like endogenous prechordal plate progenitor cells8. Below are the specific steps to perform single cell transplantations. Cell dissociation is achieved by placing embryos in Calcium-free Ringer11, dissecting an explant and mechanically stirring it.
At step 2.1, prepare the transplantation dish with 1% agarose in Calcium-free Ringer instead of Embryo Medium.
At step 3.1, in addition to ABP140-mCherry RNA and sin1 morpholino, add 3 ng of RNAs encoding the active form of the Nodal receptor Taram-A (0.6 ng/µl final) and 1.25 µl of morpholino against sox32 (stock solution at 2 mM, hence 0.5 mM final).
After step 4, transfer three selected donor embryos in the dish for cell transplantation filled with Pen/Strep Calcium-free Ringer using a fire-polished Pasteur pipette. With fine tweezers, dissect an explant containing the injected cells (ABP140-mCherry labeled cells). Rapidly discard the rest of the embryos.
With an eyelash, gently stir the explant until cells dissociate.
Draw up a single isolated cell in the transplantation needle and transplant it into the shield of a host embryo.
Using a fire-polished Pasteur pipette, transfer the embryos in an agarose-coated dish filled with Pen/Strep EM. Place the embryos in the incubator at 28 °C for 30 min.
7. Embryo Mounting
- Imaging Chamber
- Method 1: use an imaging chamber composed of a plastic slide (75*25*0.5 mm) pierced with 4 holes (5.5 mm diameter), with 3 mm high edges on one side (Figure 2C-D). Glue a glass coverslip on the back side, with cyanoacrylate glue (super glue). Note: At the end of the experiment, place the chamber in water for one night to remove the coverslip and get rid of glue residues with sand paper.
- Method 2: use 35 mm MatTek glass bottom dishes (see list of Materials) with a 10 mm hole.
- Embryo Mounting
- Fill up a glass vial with 1 ml of hot 0.2 % agarose in embryo medium. Keep it at 42 °C in a heat-block.
- Under the fluorescent stereomicroscope, select an embryo in which red transplanted cells are part of the prospective prechordal plate labeled in green (i.e. red transplanted cells are within the green prospective prechordal plate, and have started migrating towards the animal pole).
- Transfer a selected embryo into the agarose solution with a fire-polished Pasteur pipette. Discard the excess of embryo medium from the pipette. Draw the embryo in the pipette with agarose, and transfer the embryo in the imaging chamber, depositing a drop of agarose. Before agarose is set, orient the embryo with an eyelash. Place the prospective prechordal plate upwards for imaging with an upright microscope, or on the glass bottom for imaging with an inverted microscope. Wait until the agarose is set (around 1 min, depending on room temperature) before mounting another embryo in the next well of the chamber. Note: an imaging chamber containing 4 wells allows mounting up to 4 embryos.
- When the agarose is set, fill the chamber with Pen/Strep EM to avoid drying.
8. Live Imaging
Use a 40X water-immersion long-range lens. Use appropriate filter sets to image GFP and mCherry (for GFP: excitation BP 470/40, beam splitter FT 510, and emission BP 540/50; for mCherry: excitation BP 578/21, beam splitter FT 596, and emission LP 641/75). Adjust exposure duration in order to optimize image dynamics.
To image all cells in the plate, acquire z-stacks, starting a few µm above the prospective prechordal plate (in the ectoderm) and ending a few µm below the prospective prechordal plate (in the yolk). Use a z-step of 2 µm. To optimize acquisition speed, switch colors between z-stacks rather than between frames. Note: This may lead to slight differences between the green and red images, but is not a problem since no precise co-localization between the green and red signals is required. To reduce further imaging time and exposure to light, green may be acquired only once every 5 time points, which is sufficient to identify prechordal plate progenitor cells.
Using such settings, image up to 4 embryos within the two-minute time interval which is necessary to analyze protrusion frequency and orientation. For analysis of protrusion lifetime, reduce time interval to 30 sec to get higher time resolution. Image from 60% epiboly to 80% epiboly.
9. Cell Dynamics Analysis
Load 4D images in ImageJ Note: Depending on the software used for acquisition, images are stored in different formats (one file per image, one file per z-stack, one file for the whole experiment). Some ImageJ Plugins are available online to open 4D stacks. Our data are stored as one file per z-stack. Because the dataset can be large, it may be convenient to open only part of it (some time points, or a subpart of the z-stack, or 8-bit converted images…). We use a custom-made ImageJ plugin to do so, which we would be happy to distribute on request.
- Analysis of the Orientation and Frequency of Cell Protrusions
- Use GFP images to determine the main direction of prospective prechordal plate migration. Take this as a reference for cell protrusions angle measurements. Rotate the stack, so that this reference direction is parallel to one side of the image.
- Follow one cell. Select angle tool. For each frame and each protrusion, use the angle tool to measure the angle between the protrusion and the reference direction (Figure 3A). Use the Analyze/Measure command to store the value (or press M on keyboard). Save the results as a txt file. Repeat with the next cell.
- Import the data into R and plot angle distribution as histograms. Use the Kolmogorov-Smirnov test (ks.test in R) to compare different conditions. Note: The angle tool provides values comprised between 0° and 180°, allowing the use of classical statistical tests and not circular tests.
- With this data set, calculate emission frequency as the number of protrusions per cell per minute. Use Student T-test (t.test in R) to compare different conditions.
- Analysis of the Cell Protrusions Lifetime
- For each cell and each protrusion, measure protrusion duration (number of frames during which the protrusion is present x time-interval between frames).
- Import the data into R. Use Student T-test (t.test in R) to compare different conditions. Note: Other migration features can be interesting to analyze in order to characterize the cell migration. These include cell tracks, for speed, direction and persistence measurements, or cell morphology. Some commercial software applications are available to measure these features, but a large number of open-source software applications and ImageJ plugins are also available, as for instance ADAPT to analyze morphodynamics12, DiPer to look at cell trajectories and directional persistence13, CellTrack to track cell boundaries during the migration14.
Representative Results
The presented technique was used to analyze the role of Sin1, one of the core components of the Tor complex 2 (TORC2), in controlling in vivo cell migration. The use of cell transplantation permits labeling of isolated cells and analysis of cell-autonomous effects. Movie S1 shows the migration of transplanted prechordal plate progenitor cells. Actin labeling with ABP140 allows the easy visualization of actin-rich cytoplasmic protrusions. We measured their frequency and orientation. Wild-type cells produce frequent large cytoplasmic protrusions oriented in direction of the animal pole, i.e. in the direction of migration (Figure 3B). Loss of function of Sin1 leads to a drastic reduction of the number of protrusions, and randomization of the remaining ones, demonstrating the importance of sin1 in controlling actin-rich protrusion formation. Interestingly, this phenotype can be rescued by expression of a constitutively active form of Rac115, strongly suggesting that TorC2 controls actin dynamics and cell protrusion formation through Rac1 (Figure 3B).
The presented technique was also used to characterize the role of Arpin, a recently identified inhibitor of the Arp2/3 complex, on cell dynamics4. Loss of Arpin function leads to an increase in protrusion frequency (average rate of cells harboring a protrusion at a given time). This could be due either to more frequent protrusion formation or to an increase in protrusion stability. Measuring protrusion lifetime revealed that, in absence of Arpin, protrusion temporal persistence is doubled (Figure 3C). This is consistent with the role of Arpin as an Arp2/3 inhibitor, which would facilitate protrusion retraction, and suggests that Arpin affects protrusion frequency by modulating protrusion stability, rather than protrusion initiation.
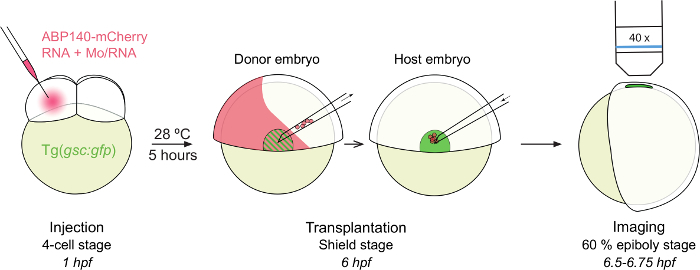 Figure 1: Outline of the Procedure. Tg(gsc:gfp) embryos are injected at the 4-cell stage (1 hr post fertilization). After 5 hr at 28 °C, embryos showing ABP140-mCherry positive cells within the shield (GFP +) are selected as donor embryos. Shield cells are drawn up within the transplantation needle and transferred into the shield of a host embryo. After 0.5 hr of recovery, host embryos are mounted and imaged with an epifluorescent microscope (40X objective). hpf: hours post fertilization. Please click here to view a larger version of this figure.
Figure 1: Outline of the Procedure. Tg(gsc:gfp) embryos are injected at the 4-cell stage (1 hr post fertilization). After 5 hr at 28 °C, embryos showing ABP140-mCherry positive cells within the shield (GFP +) are selected as donor embryos. Shield cells are drawn up within the transplantation needle and transferred into the shield of a host embryo. After 0.5 hr of recovery, host embryos are mounted and imaged with an epifluorescent microscope (40X objective). hpf: hours post fertilization. Please click here to view a larger version of this figure.
 Figure 2: Transplantation Dish and Imaging Chamber. (A) Transplantation dish with individual wells. (B) Mold for cell transplantation dish. (C) Home-made imaging chamber. (D) Schematic drawing of the home-made imaging chamber. Scale bar = 1 cm. Please click here to view a larger version of this figure.
Figure 2: Transplantation Dish and Imaging Chamber. (A) Transplantation dish with individual wells. (B) Mold for cell transplantation dish. (C) Home-made imaging chamber. (D) Schematic drawing of the home-made imaging chamber. Scale bar = 1 cm. Please click here to view a larger version of this figure.
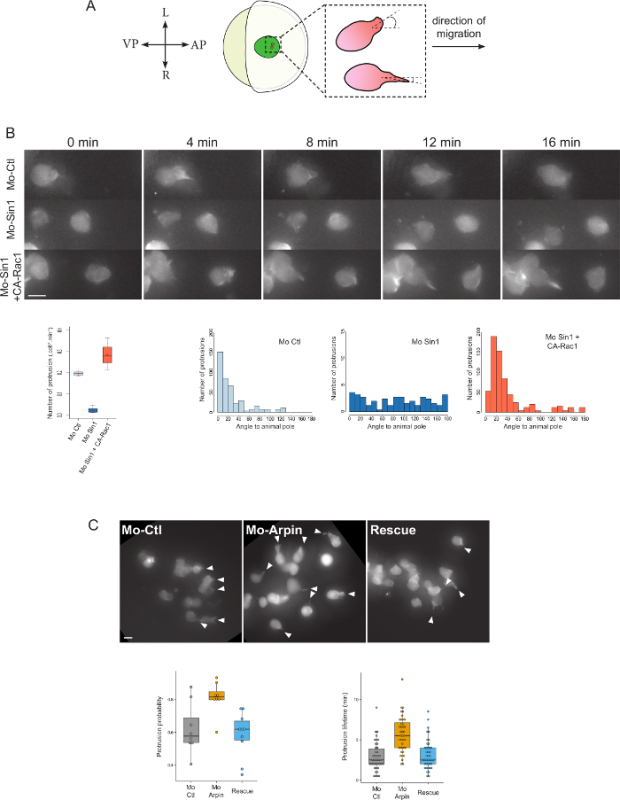 Figure 3: Results from Cell Dynamics Analysis. (A) Scheme of protrusion angle measurement. (B) Analysis of cell protrusion orientation and frequency. In absence of Sin1 (Mo-Sin1), cells emit fewer protrusions and the remaining protrusions are randomly oriented. This phenotype can be rescued by expression of a constitutively active form of the GTPase Rac1 (Mo-Sin1 + CA-Rac1). Modified from Dumortier and David, 20155. (C) Analysis of protrusion lifetime. In absence of Arpin (Mo-Arpin), protrusion frequency is increased. This is due to an increase in protrusion lifetime. Reintroducing an arpin RNA insensitive to the morpholino restores this hyper protrusive phenotype. Modified from Dang et al., 20134. Scale bar = 10 µm. A: Anterior; P: Posterior, L: Left, R: Right. Please click here to view a larger version of this figure.
Figure 3: Results from Cell Dynamics Analysis. (A) Scheme of protrusion angle measurement. (B) Analysis of cell protrusion orientation and frequency. In absence of Sin1 (Mo-Sin1), cells emit fewer protrusions and the remaining protrusions are randomly oriented. This phenotype can be rescued by expression of a constitutively active form of the GTPase Rac1 (Mo-Sin1 + CA-Rac1). Modified from Dumortier and David, 20155. (C) Analysis of protrusion lifetime. In absence of Arpin (Mo-Arpin), protrusion frequency is increased. This is due to an increase in protrusion lifetime. Reintroducing an arpin RNA insensitive to the morpholino restores this hyper protrusive phenotype. Modified from Dang et al., 20134. Scale bar = 10 µm. A: Anterior; P: Posterior, L: Left, R: Right. Please click here to view a larger version of this figure.
 Movie 1: Sin1 controls formation of cell protrusions, through Rac1 (Right click to download). Transplanted prechordal plate progenitor cells, injected with ABP140-mCherrry RNAs and a control morpholino, or the sin1 morpholino, or the sin1 morpholino and RNAs for the constitutive form of Rac1. Protrusion frequency and orientation were measured on these images.
Movie 1: Sin1 controls formation of cell protrusions, through Rac1 (Right click to download). Transplanted prechordal plate progenitor cells, injected with ABP140-mCherrry RNAs and a control morpholino, or the sin1 morpholino, or the sin1 morpholino and RNAs for the constitutive form of Rac1. Protrusion frequency and orientation were measured on these images.
Discussion
This protocol presents an easy way to study the role of a candidate gene in cell migration in vivo, by combining the creation of chimeric embryos using cell transplantation with live imaging.
Creation of mosaic embryos
Studying the dynamics of a cell requires the visualization of its contour to analyze cytoplasmic extensions. This can be achieved by labeling isolated cells in an otherwise unlabeled – or differently labeled - environment, thus offering good visual contrast.
An easy way to stochastically label a portion of cells in the embryo is to inject plasmidic DNA at the 1-cell stage16. The injected plasmids form aggregates that are randomly and unequally segregated in cells during cell division. This leads to the expression of the plasmid in a random subset of cells, and thus represents a very fast, easy and non-invasive method for generating mosaic embryos. However, cells expressing the injected plasmid tend to form clusters, and the probability of finding embryos containing few isolated expressing cells in the prospective prechordal plate is quite low. Furthermore, use of plasmidic DNA offers very limited control of the expression level of the injected construct. The creation of chimeric embryos by cell transplantation, although technically more difficult than plasmidic DNA injection, offers a number of advantages: control of the number of transplanted cells, control of the expression level of the constructs (RNA injection), and last but not least, cell transplantations allow to test for cell-autonomous effects, by creating mosaic embryos in which transplanted cells contain loss or gain of function constructs. Conversely, transplantations can also be used to assess the non-autonomous role of the environment by transplanting wild-type cells into embryos that have been modified. This can be of particular interest for testing for instance, the function of extracellular matrix components that are particularly relevant for cell migration analysis.
A detailed protocol for cell transplantation has already been described10. Compared to this protocol, we would like to discuss two main differences in our procedure.
The first difference is related to the stage of donor embryos injection. According to our experience, embryos injected at the 1-cell stage with RNAs of a fluorescent protein do not express homogeneously the fluorescent protein in all cells, due to an incomplete diffusion of the RNAs in the cell. Injection of donor embryos at the 4-cell stage in one of the four cells allows to get a homogeneous clone of cells, which is of particular interest when RNAs of the fluorescent protein are co-injected with other RNAs that are not traceable.
The second main difference is the use of air instead of oil in the transplantation syringe. The main drawback of an air-filled system is the inertia resulting from the elasticity of the air. Oil on the contrary, being incompressible, offers good control of suction and pressure. However, with a little training, and by maintaining the interface between air and embryo medium in the thin, tapered part of the needle, the control of suction and pressure with an air-filled system is sufficient for transplanting cells at the shield stage. For later stages, as cells become smaller and more cohesive, the suction requires a high pressure that needs to be very precisely controlled, which can't be achieved with an air-filled setup. Using air instead of oil eases the setup, as filling the system with oil without any air bubble is tricky. Furthermore, it avoids filling the transplantation needle with oil. This allows transplantation needles to be reused, and hence to be carefully prepared. We feel that a needle without sharp edges and of the correct diameter is key to successful transplantation. This transplantation step is clearly one of the critical steps in the protocol, which requires practice before being easily performed.
We have also proposed a specific procedure for transplanting single cells, which consists in dissociating cells prior to transplantation, in order to draw a unique cell in the transplantation needle. This implies that transient cell dissociation will not modify their identity and/or behavior. For prechordal plate progenitor cells, we genetically impose cell identity, and have carefully checked that these cells behave like non-dissociated ones. However, we cannot formally exclude that dissociation and/or genetic induction induce unnoticed modifications in the cells. Transplanting single cells without dissociating them is feasible. Cells should be drawn up carefully in the transplantation needle. However, at least in our hands, success rate is quite low, either because more than one cell gets into the needle, or because the cell shears when drawn up and dies in the needle or once transplanted.
Epifluorescence versus fast confocal imaging
The use of cell transplantation to create mosaic embryos allows for the labeling of isolated cells, separated from other labeled cells. In this context, good imaging of cell morphology and dynamics can be achieved with standard epifluorescence microscopy, as proposed in the protocol. This has the obvious advantage of being a relatively wide-spread equipment and a low-cost alternative to more expensive confocal imaging systems.
For precise subcellular localizations, confocal imaging can be used to improve resolution, in particular axial resolution. To still get sufficient temporal resolution, fast confocal imaging should be used. From our experience, spinning-disc microscopes offer a good compromise between resolution, speed and photo-toxicity. Fast scanning confocal microscopes (like resonant scanning microscopes) are good options as well, but less frequent and more expensive.
Cytoskeleton labeling
Good labeling of actin filaments and their dynamics is crucial to study mechanisms of actin-based cell migration. Although membrane-bound fluorescent markers are more efficient to analyze the cell outlines, actin labeling allows a much better visualization of the cell protrusions. In particular, if three labeled cells get in contact, it is very difficult to identify a cytoplasmic extension located between two neighboring cells as the outline of the extension and the membrane of the neighboring cells can get mixed up. Labeling actin filaments allows the unambiguous detection of actin-based cell protrusions, on which we focused. If cell morphology was to be quantified, a membrane labeling would be preferable. Another option is to use both labeling, either using 3-color imaging (one for the transgenic line, one for actin and one for membrane), or by GFP labeling the membrane. In the transgenic line, GFP is cytoplasmic, and goosecoid-GFP positive cells can thus be identified, even if the membrane is GFP labeled.
Over the past years, a number of probes have been used to label actin in live samples. The first ones were direct fusions to actin monomers. These presented two major drawbacks. First, they labeled all actin and not specifically polymerized actin. Second, they were frequently toxic. The probes currently in use are filamentous actin binding domains fused to fluorescent reporters. In this protocol, we used ABP14017 (actin binding domain of the yeast actin binding protein 140), which is currently the most frequently used marker for filamentous actin, and labels all filamentous actin. Utrophin18 (calponin homology domain) also labels filamentous actin, but appears to label only stable filaments. This difference has been used to identify dynamic actin filaments, as the ones labeled by ABP140 and not Utrophin19.
Recently it has been reported in fly that ABP140 and Utrophin could have toxic effects, in particular over long expression20. Even though we have not noticed any migration defects in ABP140-labeled cells, we cannot exclude that ABP140 expression may perturb endogenous actin dynamics. A third probe for filamentous actin was recently reported21. F-tractin (actin-binding domain from rat inositol triphosphate 3-kinase) has been described as a faithful reporter of filamentous actin, which would not show toxic effects20,22. To our knowledge, the use of F-tractin has not been reported in zebrafish yet.
In addition to actin, many other cell constituents could be labeled to improve our understanding of the cell migration. This includes other cytoskeletal components like myosin, microtubules, centrosomes, as well as known regulators of cell migration (activated forms of the small GTPases Rho, Rac and Cdc42, fluorescent probes for PIP3…) or proteins involved in cell adhesion (integrins, cadherins…).
Overall, this protocol regroups a number of previously described tools and techniques to propose a rapid and easy system to test the involvement of a candidate gene in controlling cell migration in vivo. We used prospective prechordal plate migration as it is an interesting model of directed collective migration, and because of the available transgenic line labeling it. However, a similar protocol could be adapted to analyze other migration events occurring during development.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank F. Bouallague and the IBENS animal facility for excellent zebrafish care. Research reported in this publication was supported by the Fondation ARC pour la recherche sur le cancer, grants N° SFI20111203770 and N° PJA 20131200143.
References
- Charras G, Sahai E. Physical influences of the extracellular environment on cell migration. Nature Reviews Molecular Cell Biology. 2014;15(12):813–824. doi: 10.1038/nrm3897. [DOI] [PubMed] [Google Scholar]
- Friedl P, Wolf K. Plasticity of cell migration: A multiscale tuning model. Journal of Cell Biology. 2010;188(1):11–19. doi: 10.1083/jcb.200909003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nature reviews. Molecular cell biology. 2009;10(7):445–457. doi: 10.1038/nrm2720. [DOI] [PubMed] [Google Scholar]
- Dang I, Gorelik R, et al. Inhibitory signalling to the Arp2/3 complex steers cell migration. Nature. 2013. [DOI] [PubMed]
- Dumortier JG, David NB. The TORC2 Component, Sin1, Controls Migration of Anterior Mesendoderm during Zebrafish Gastrulation. Plos One. 2015;10:e0118474. doi: 10.1371/journal.pone.0118474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solnica-Krezel L, Stemple DL, Driever W. Transparent things: cell fates and cell movements during early embryogenesis of zebrafish. BioEssays. 1995;17(11):931–939. doi: 10.1002/bies.950171106. [DOI] [PubMed] [Google Scholar]
- Ulrich F, Concha ML, et al. Slb/Wnt11 controls hypoblast cell migration and morphogenesis at the onset of zebrafish gastrulation. Development. 2003;130(22):5375–5384. doi: 10.1242/dev.00758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumortier JG, Martin S, Meyer D, Rosa FM, David NB. Collective mesendoderm migration relies on an intrinsic directionality signal transmitted through cell contacts. Proceedings of the National Academy of Sciences of the United States of America. 2012. pp. 1–6. [DOI] [PMC free article] [PubMed]
- Gritsman K, Talbot WS, Schier AF. Nodal signaling patterns the organizer. Development. 2000;127(5):921–932. doi: 10.1242/dev.127.5.921. Cambridge, England. [DOI] [PubMed] [Google Scholar]
- Kemp HA, Carmany-Rampey A, Moens C. Generating chimeric zebrafish embryos by transplantation. Journal of visualized experiments : JoVE. 2009. [DOI] [PMC free article] [PubMed]
- Westerfield M. The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio) 2000.
- Barry DJ, Durkin CH, Abella JV, Way M. Open source software for quantification of cell migration, protrusions, and fluorescence intensities. The Journal of Cell Biology. 2015;209(1):163–180. doi: 10.1083/jcb.201501081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelik R, Gautreau A. Quantitative and unbiased analysis of directional persistence in cell migration. Nature Protocols. 2014;9(8):1931–1943. doi: 10.1038/nprot.2014.131. [DOI] [PubMed] [Google Scholar]
- Sacan A, Ferhatosmanoglu H, Coskun H. CellTrack: An open-source software for cell tracking and motility analysis. Bioinformatics. 2008;24(14):1647–1649. doi: 10.1093/bioinformatics/btn247. [DOI] [PubMed] [Google Scholar]
- Tahinci E, Symes K. Distinct functions of Rho and Rac are required for convergent extension during Xenopus gastrulation. Developmental biology. 2003;259(2):318–335. doi: 10.1016/s0012-1606(03)00206-9. [DOI] [PubMed] [Google Scholar]
- Zhang T, Yin C, et al. Stat3-Efemp2a modulates the fibrillar matrix for cohesive movement of prechordal plate progenitors. Development. 2014;141(22):4332–4342. doi: 10.1242/dev.104885. [DOI] [PubMed] [Google Scholar]
- Riedl J, Crevenna AH, et al. Lifeact: a versatile marker to visualize F-actin. Nature methods. 2008;5(7):605–607. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkel BM, Von Dassow G, Bement WM. Versatile fluorescent probes for actin filaments based on the actin-binding domain of utrophin. Cell Motility and the Cytoskeleton. 2007;64(11):822–832. doi: 10.1002/cm.20226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SK, Deng Q, Cavnar PJ, Wu YI, Hahn KM, Huttenlocher A. Differential regulation of protrusion and polarity by PI3K during neutrophil motility in live zebrafish. Developmental cell. 2010;18(2):226–236. doi: 10.1016/j.devcel.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spracklen AJ, Fagan TN, Lovander KE, Tootle TL. The pros and cons of common actin labeling tools for visualizing actin dynamics during Drosophila oogenesis. Developmental biology. 2014;393(2):209–226. doi: 10.1016/j.ydbio.2014.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson HW, Schell MJ. Neuronal IP3 3-kinase is an F-actin-bundling protein: role in dendritic targeting and regulation of spine morphology. Molecular biology of the Cell. 2009;20(24):5166–5180. doi: 10.1091/mbc.E09-01-0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi J, Wu XS, Crites T, Hammer JA. Actin retrograde flow and actomyosin II arc contraction drive receptor cluster dynamics at the immunological synapse in Jurkat T cells. Molecular Biology of the Cell. 2012;23(5):834–852. doi: 10.1091/mbc.E11-08-0731. [DOI] [PMC free article] [PubMed] [Google Scholar]