

Abstract
Multiple sclerosis is presumed to be an inflammatory autoimmune disease, which is characterized by lesion formation in the central nervous system (CNS) resulting in cognitive and motor impairment. Experimental autoimmune encephalomyelitis (EAE) is a useful animal model of MS, because it is also characterized by lesion formation in the CNS, motor impairment and is also driven by autoimmune and inflammatory reactions. One of the EAE models is induced with a peptide derived from the myelin oligodendrocyte protein (MOG)35-55 in mice. The EAE mice develop a progressive disease course. This course is divided into three phases: the preclinical phase (day 0 - 9), the disease onset (day 10 - 11) and the acute phase (day 12 - 14). MS and EAE are induced by autoreactive T cells that infiltrate the CNS. These T cells secrete chemokines and cytokines which lead to the recruitment of further immune cells. Therefore, the immune cell distribution in the spinal cord during the three disease phases was investigated. To highlight the time point of the disease at which the activation/proliferation/accumulation of T cells, B cells and monocytes starts, the immune cell distribution in lymph nodes, spleen and blood was also assessed. Furthermore, the levels of several cytokines (IL-1β, IL-6, IL-23, TNFα, IFNγ) in the three disease phases were determined, to gain insight into the inflammatory processes of the disease. In conclusion, the data provide an overview of the functional profile of immune cells during EAE pathology.
Keywords: Immunology, Issue 111, Experimental autoimmune encephalomyelitis, MOG, multiple sclerosis, flow cytometry, T cell, B cell, neutrophil, monocyte, macrophage
Introduction
Multiple sclerosis (MS) and its corresponding animal model, experimental autoimmune encephalomyelitis (EAE), show autoimmune neuroinflammation changes in the central nervous system (CNS). Early active MS and EAE lesions are characterized by the presence of infiltrated immune cells. The etiology of MS remains unknown, but is widely considered to involve the destruction of myelin mediated by autoreactive T cells. These autoreactive T cells secrete pro-inflammatory cytokines and chemokines which attract other immune cells such as B cells, monocytes and neutrophils from the circulation. Monocytes differentiate into macrophages. Interferon gamma (IFNγ) secreted by autoreactive T cell polarizes the macrophages into pro-inflammatory macrophages. The pro-inflammatory macrophages release cytokines and reactive oxygen species that promote apoptosis in oligodendrocytes. The death of the oligodendrocytes leads to demyelination. Furthermore, B cells differentiate into plasma cells and release autoantibodies against the myelin sheath, ultimately resulting in degradation of myelin. The loss of myelin leads to degradation of axons and neurons and thereby to the formation of lesion sites in the CNS which represent the main characteristic of MS1. In the periphery, T cells and B cells are activated in the lymph nodes, they proliferate in the spleen and migrate through the circulation into the central nervous system. Monocytes and neutrophils proliferate in the bone marrow and also migrate through the circulation into the central nervous system.
Leukocyte extravasation from bone marrow, spleen and lymph nodes into the blood or from the bloodstream into the CNS is a multistep process that depends on several factors, including molecular interactions between leukocytes and endothelium mediated by chemokines and chemokine receptors. The production of chemokines by various cell types can be induced during the immune reaction by cytokines like tumor necrosis factor-α (TNFα), IFNγ and interleukin-6 (IL-6), which subsequently recruits immune cells to the site of inflammation2,3. Immune cells present a subset of chemokine receptors on their surface, depending on cell type and migration pathway to the inflammatory site. Thus, CXCR2, CCR1 and CXCR1 are expressed on mature neutrophils in bone marrow and blood4, and binding of its ligands, CXCL2, CCL5 or CXCL6, respectively, activates neutrophils and promotes their adhesion to the endothelium and subsequently, the migration of the cells into tissues5-9. CCL2 and CCL20 attract monocytes and Th1/Th17 cells10, which express CCR211 and CCR612, respectively. CCR1 and CCR5, expressed by different cell types, including T cells, monocytes and macrophages13, bind CCL3, CCL5 and CCL7 and are upregulated during MS14. CXCR3 is expressed on T cells and binds CCL9, CCL10 and CCL1115.
One main strategy in MS treatment is the depletion of immune cells or the prevention of immune cell infiltration into the CNS. Therefore, the blockade of specific chemokine receptors has been investigated in EAE. Antagonism or genetic deletion of CCR116, CCR217, CCR718 or CXCR219 reduces EAE pathology, whereas antagonism or genetic deletion of CCR120, CCR520 or CXCR321 did not reduce the pathology. Hence, the expression of specific chemokine receptors on leukocytes is crucial for the infiltration of the latter into the CNS and dictates the course of EAE.
The depletion of immune cells is an effective treatment strategy for MS patients, because infiltrated immune cells release cytokines, such as TNFα, IL-6 and IL-1β, which, in turn, promote the inflammatory process or the degradation of neurons22. Furthermore, auto-reactive Th1 cells release IFNγ, which in turn stimulates macrophages to release TNFα, IL-1β and IL-23.
This manuscript describes the induction of EAE, the determination of the immune cell distribution and the cytokine levels (mRNA) in various tissues in EAE mice. Cells were isolated at different time points during the disease course to provide a time-dependent overview of the inflammatory processes which finally lead to lesion formation in the CNS.
Protocol
ETHICS STATEMENT: Our experimental procedures are approved by the Ethics Committee of the Regierungspräsidium Darmstadt (Germany) and confirm to National and European regulations. All efforts were made to minimize animal suffering and reduce the number of animals used.
1. EAE Model
- Induction of the EAE model
- Use 10- to 13-week-old female 129S4/SvJae×C57BL/6 mice for the induction of EAE.
- Give the mice a subcutaneous injection, into the upper and lower back, of the encephalitogenic MOG35 - 55 (myelin oligodendrocyte glycoprotein) peptide (200 µg), emulsified in 200 µl complete Freund`s adjuvant (CFA) containing 400 µg Mycobacterium tuberculosis. Note: As a negative, non-disease-inducing sham control, incomplete Freund's adjuvant containing Mycobacterium tuberculosis, in the same volume and by the same route, can be used.
- Thereafter, and again 24 hr later, give the mice an intraperitoneal injection of pertussis toxin (in total 0.2 µg, diluted in 200 µl phosphate-buffered saline, PBS). Use untreated mice as the control group, to be able to compare diseased with healthy animals. Note: It is also possible to induce EAE with 200 - 300 µg MOG35 - 55 peptide, 300 - 500 µg of Mycobacterium tuberculosis in the CFA and 0.2 - 0.3 µg pertussis toxin in a volume of 0.1 - 0.2 ml per injection.
- Beginning one week after the injection, examine mice daily for clinical symptoms (see step 1.2.1) Note: The day of disease onset varies in different experiments, but under the conditions in our laboratory, this is around day 11 and thus, here day 14 is defined as 3 days after disease onset. All mice in the present study developed clinical symptoms.
- Scoring of the mice
- Classify clinical symptoms by clinical scores as follows: 0) no signs, 0.5) distal paralysis of the tail, 1) complete paralysis of the tail, 1.5) limp tail and mild weakness of hind legs, 2) limp tail and severe weakness of hind legs, 2.5) limp tail and paralysis of one hind leg, 3) limp tail and paralysis of both hind legs, 3.5) paralysis of both hind limbs and weakness of one fore limb (mice achieving this score were euthanized, in keeping with local ethical guidelines).
2. Preparation of Single Cells for Flow Cytometry Analysis
Note: The antibody mixture consists of 1 µl CD45-Vioblue, 2 µl CD8-eFluor650, 0.5 µl CD11b-eFluor605, 0.5 µl F4/80-PE-Cy7, 1 µl CD3-PE-CF594, CD4-V500, 0.5 µl CD11c-AlexaFluor700, 1 µl CD19-APC-H7 and 1 µl Ly6G-APC-Cy7.
Note: If you take blood, lymph node, spleen and spinal cord then the procedure is as follows: Mice are deeply anaesthetized with a combination of isoflurane (2% in carbogen per minute) and ketamine (100 mg/kg body weight). Next open the thorax, remove the blood with an intracardial stick and perfuse the mice intracardially with cold PBS. Then remove the lymph nodes followed by spleen and finally the spinal cord. If you do not need to perfuse the mice intracardially then euthanize the mice under deep anesthesia by luxation of the neck.
- Isolation of splenocytes
- Anaesthetize mice with a combination of isoflurane (2% in carbogen per minute) and ketamine (100 mg/kg body weight).
- Wet incision area with 80% isopropanol to avoid any contamination with hairs and open the thorax longitudinally, without puncturing deeper tissues, using scissors.
- Perfuse mice intracardially with cold PBS pH 7.023. Note: The spleen is located in the left superior abdominal quadrant under the rib cage. If only the spleen is to be studied, this organ may be removed before perfusion in order to retain all cell types of interest.
- Remove the spleen and cut off approximately 1/8 and weigh it. Store the sample in PBS on ice.
- Squeeze the spleen tissue through a 70 µm mesh sieve (placed over a 50 ml tube), using the plunger of a 2 ml syringe.
- Wash the mesh with 5 ml PBS. Centrifuge at 1,800 x g for 3 min. Resuspend the pellet in 500 µl lysing solution. Note: At this stage, it may be necessary to make a differential cell count if, later, an alternative FACS analysis method is used that does not employ beads (see section 3).
- Incubate for 10 min at room temperature (RT) and add 500 µl PBS. Centrifuge for 6 min at 650 x g at room temperature (RT).
- Repeat the washing step with 500 µl PBS. Discard the supernatant, resuspend the cell pellet in 100 µl 0.2% bovine serum albumin (BSA)/PBS, add 2 µl Fc receptor-1 (FcRI) blocking buffer and incubate for 15 min at RT in the dark.
- Add 13 µl antibody mixture, incubate 15 min at RT in the dark, add 500 µl PBS and centrifuge for 6 min at 650 x g at RT. Discard the supernatant. Note: The manufacturer's staining procedure recommends a single wash, but additional reduction of background staining may be achieved by optionally repeating the washing step one or two more times.
- Resuspend the cell pellet in 500 µl PBS (or possibly running buffer for protein separation, to potentially reduce cell clumping) and transfer it to the flow cytometry tube. Keep cells on ice.
- Isolation of lymph node cells
- Anaesthetize mice with a combination of isoflurane (2% in carbogen per minute) and ketamine (100 mg/kg body weight).
- Wet incision area with 80% isopropanol and open the thorax longitudinally using scissors. Perfuse mice intracardially with cold PBS pH 7.023.
- To isolate the inguinal lymph node, carefully remove the skin in the region of the hip and pick the lymph node carefully out of the fat tissue with forceps. Weigh the inguinal lymph node and store the sample in PBS on ice. Note: We used inguinal lymph nodes in our studies because O`Connor et al. found that high numbers of activated monocytes, macrophages, neutrophils and T cells were all present in the inguinal lymph nodes after EAE induction24.
- Squeeze the lymph node through a 70 µm mesh sieve (placed over a 50 ml tube) using the plunger of a 2 ml syringe.
- Wash the mesh with 5 ml PBS. Centrifuge at 2,400 x g for 8 min. Resuspend the cell pellet in 100 µl 0.2% BSA/PBS, add 2 µl FcRI blocking buffer and incubate for 15 min at RT in the dark.
- Add 13 µl antibody mixture, incubate 15 min at RT in the dark, add 500 µl PBS and centrifuge for 6 min at 650 x g at RT.
- Discard the supernatant, resuspend the cell pellet in 300 µl PBS (or a suitable running buffer) and transfer it to a flow cytometry tube.
- Isolation of blood cells
- Add 50 µl 20 mM HEPES to 50 µl blood (stabilized with EDTA) and add 500 µl lysing solution. Incubate for 10 min at RT and add 500 µl PBS. Centrifuge for 6 min at 650 x g at RT. Discard supernatant and repeat the wash step.
- Resuspend the cell pellet in 100 µl 0.2% BSA/PBS, add 2 µl FcRI blocking buffer and incubate for 15 min at RT in the dark.
- Add 13 µl antibody mixture, incubate 15 min at RT in the dark, add 500 µl PBS and centrifuge for 6 min at 650 x g at RT. Discard the supernatant, resuspend the cell pellet in 300 µl PBS and transfer it to the flow cytometry tube.
- Isolation of spinal cord cells
- Anaesthetize mice with a combination of isoflurane (2% in carbogen per minute) and ketamine (100 mg/kg body weight).
- Wet incision area with 80% isopropanol and open the thorax longitudinally using scissors. Perfuse mice intracardially with cold PBS pH 7.023.
- Wet the incision area on the back and make again a longitudinal cut with a scalpel and remove the skin.
- Cut out the lumbar part of the spine (which innervates the hind limbs) and flush it with a PBS-filled syringe to extract the spinal cord. Cut out approximately 1/3 of the lumbar cord, weigh this and store the sample in PBS on ice.
- Centrifuge at 250 x g for 2 min at 4 °C. Discard the supernatant, add 500 µl cell detachment solution and HBSS (1:1). Add 3 mg/ml collagenase A and 1 Unit/ml DNase I, decollate the cells by repeated up and down pipetting and incubate for 30 min with shaking at 37 °C at 400 rpm. Note: If preferred, the cell suspension can be cleared of contaminating myelin and cellular debris by density gradient centrifugation which may improve the sensitivity of the flow cytometry measurement, thereby, reducing background autofluorescence and the number of events that need to be acquired (see section 3.5).
- Centrifuge at 250 x g for 2 min at RT. Discard the supernatant, resuspend the pellet in 1 ml 10% fetal calf serum (FCS)/Dulbecco's minimum essential medium (DMEM) and decollate the cells again by repeated up and down pipetting.
- Centrifuge at 250 x g for 2 min at RT. Repeat the washing step.
- Resuspend the cell pellet in 1 ml PBS and squeeze the spinal cord through a 70 µm mesh sieve (placed over a 50 ml tube) using the plunger of a 2 ml syringe.
- Wash the mesh with 4 ml PBS. Centrifuge at 1,800 x g for 3 min at RT. Discard the supernatant, resuspend the cell pellet in 100 µl 0.2% BSA/PBS, add 2 µl FcRI blocking reagent and incubate for 15 min at RT in the dark.
- Add 13 µl antibody mixture, incubate 15 min at RT in the dark, add 500 µl PBS and centrifuge for 6 min at 650 x g at RT.
- Discard the supernatant, resuspend the cell pellet in 500 µl PBS (or a suitable running buffer) and transfer it to the flow cytometry tube.
3. Flow Cytometric Analysis
Titrate all antibodies beforehand to determine optimal concentrations as described by Olesch et al.25.
Use antibody-capturing compensation beads for single-color compensation to create multi-color compensation matrices according to the manufacturer`s instructions.
Control the instrument calibration daily using specific beads according to the manufacturer`s instructions.
Directly before the flow cytometry measurement, add 30 µl flow cytometric absolute count standard to the cells (for isolation see section 2.1, 2.2, 2.3, 2.4) to determine absolute cell counts. Instead of using counting beads the cells can be counted.
- Take up samples (cells from spleen, lymph node and blood: 100,000 events, spinal cord: 500,000 events) into a flow cytometer and analyze using specific flow cytometry software.
- Open the flow cytometry software and add fcs 3.0 files by pressing the button "add samples".
- Open the added file by double clicking. Adjust the channels for the x- and y-axis. To select the cell populations of interest create a gate (see Figure 4).
4. Quantitative PCR Analysis
- Isolation of mRNA and transcription into cDNA
- Extract the mRNA from lumbar spinal cord (1/3), spleen (1/8) and inguinal lymph nodes by phenol-chloroform and precipitate with ethanol26.
- For this, homogenize the tissue in 1 ml guanidinium thiocyanate-phenol mixture, incubate for 10 min. Add 200 µl chloroform and incubate again for 10 min.
- After a centrifugation step (18,000 x g for 15 min at 4 °C), wash the upper aqueous phase is with 500 µl isopropanol and centrifuge (18,000 x g for 8 min at 4 °C).
- Wash the pellet with 500 µl ethanol and after a centrifugation step (18,000 x g for 5 min at 4 °C), dry the pellet in the vacuum concentrator (5 min at 30 °C). Thereafter, resuspend the pellet in 30 µl water.
- To extract the mRNA from blood, centrifuge 500 µl EDTA-stabilized blood (300 x g 10 min 4 °C).
- Take the buffy coat, containing white blood cells, and dilute in 1 ml lysing solution (150 mM NH4Cl, 100 mM NaHCO3, 0.1 mM Na-EDTA pH 7.4).
- After 10 min incubation at RT, centrifuge the cells at 650 x g for 6 min and afterwards wash twice with PBS.
- Extract the mRNA from white blood cells (WBCs) using a kit for RNA extraction according to the manufacturer's instructions.
- Perform cDNA synthesis using a kit for cDNA synthesis including random hexamers according to manufacturer's instructions. Use 200 ng mRNA for the cDNA synthesis.
- Quantitative PCR
- To quantify the amount of a specific mRNA, use 1 µl cDNA, 1 µM forward/reverse primer and a fluorescent DNA binding dye according to manufacturer's instructions27. See Table 1 for the sequences for the primer sets. Measure the CT-values of IL-1β, IL-6, IL-23, IFNγ, TNFα and PPIA (peptidyl prolyl isomerase) using a quantitative PCR system.
- To determine the relative mRNA expression use the comparative CT (cycle threshold) method27.
- For this, normalize CT-values of the target genes to the expression levels of PPIA by subtracting the mean CT-value of PPIA from the target gene, as described in previous studies (Barthelmes et al.28, Schiffmann et al.29, Schiffmann et al.30). Subsequently, calculate the so called ΔΔCT-values, using which, normalize the ΔCT-values of the target genes from EAE mice to the expression level of the target genes of untreated mice, again by subtracting the mean ΔCT-value in the untreated mice from the ΔCT-value in EAE mice.
- To gain the relative mRNA expression of the target gene, calculate the ratio using the following formula: 2-ΔΔct.
Test the specificity of each primer. Determine the amplified fragment size using a 2% agarose gel31. The fragment size is shown in Table 1.
Representative Results
Figure 1 gives a schematic overview of the different methods described in this article. 1) Mice receive an injection of MOG35-55 antigen and develop initial clinical symptoms after 10.7 ± 0.3 days28. A representative disease course of EAE mice is shown in Figure 1. 2) Various tissues (spleen, lymph nodes, lumbar spinal cord) and blood are extracted from control and EAE mice at different time points during the preclinical phase (day 2, day 4, day 6), during the onset of the disease (day 10) and during the acute phase of the disease (day 12, day 14). 3) The mRNA expression profile of cytokines, which regulate EAE development, is determined in the various tissues extracted at different time points during the disease course, using quantitative PCR. 4) Single cells are isolated from various tissues extracted at different time points during the disease course and the immune cell distribution determined using flow cytometry.
In spleen and lymph nodes, T cells and B cells are observed as the most prominent cell type. In contrast to lymph nodes, in the spleen, additionally, a high number of monocytes and neutrophils are present. All immune cells show a transient increase in the lymph nodes during the acute phase, whereas only macrophages, B cells, T cells and neutrophils increase in the spleen. In blood, all immune cells increase transiently during the preclinical phase. In the lumbar spinal cord, a disease-dependent increase in all immune cells is observed, apart from monocytes, which presumably differentiate to macrophages after their entrance into the spinal cord (Figure 2). CD4+ and CD8+ T cells show comparable changes in spleen, lymph node and blood during the different disease courses. Among cells infiltrating the spinal cord, the increase in CD4+ T cells was most pronounced (Figure 3). In Figure 4, the gating-strategy for the determination of the different cell populations is shown. In detail, cell populations described in this manuscript were identified as follows: monocytes (CD45hi, CD3-, Ly6G-, CD19-, CD11c-, CD11b+), macrophages (CD45hi, CD3-, Ly6G-, CD11c-, CD11b+, F4/80hi), neutrophils (CD45hi, CD3-, CD11b+, Ly6G+), dendritic cells (CD45hi, CD3-, Ly6G-, CD19-, CD11c+), B cells (CD45hi, CD3-, CD19+), T cells (CD45hi, CD3+), CD4+ T cells (CD45hi, CD3+, CD4+) and CD8+ T cells (CD45hi, CD3+, CD8+). The numbers of cells are related to the amount of tissue (weight of spleen, lymph nodes, lumbar spinal cord) or the blood volume, thus reflecting the absolute cell count. It is, however, possible to express the cell types as percentages of total cells measured.
In lymph nodes, the expression of IL-23 mRNA is transiently increased during the preclinical phase, whereas the expression of IL-6 mRNA is increased in a disease-dependent manner. In the spleen, IL-6 and IL-23 mRNA expression increases transiently in the preclinical phase, while at the onset of the disease, IL-1β mRNA expression is raised. In blood, the expression of TNFα mRNA is increased in a disease-dependent manner. In the lumbar spinal cord, TNFα, IL-1β and IFNγ increase during the acute phase, whereas IL-6 is upregulated during the onset phase (Figure 5).
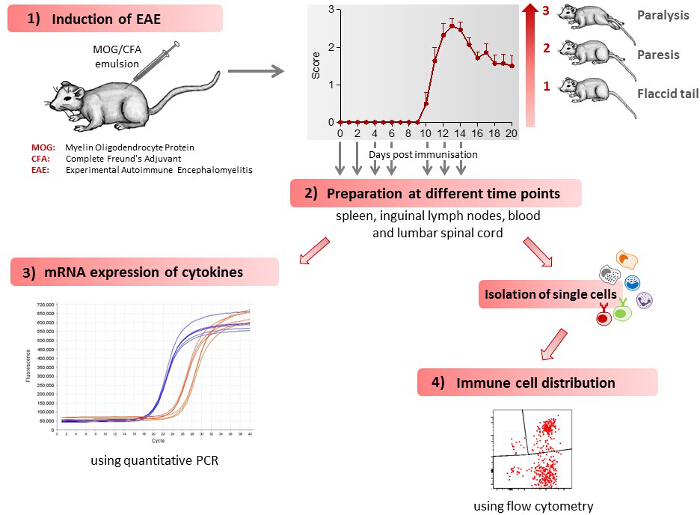
Figure 1: Scheme of the Work Flow for Induction and Immunological Assessment of EAE. 1) EAE is induced in mice leading to the development of clinical symptoms. Clinical symptoms are classified by clinical scores, as follows: 0) no signs, 0.5) distal paralysis of the tail, 1) complete paralysis of the tail, 1.5) limp tail and mild weakness of hind legs, 2) limp tail and severe weakness of hind legs, 2.5) limp tail and paralysis of one hind leg, 3) limp tail and paralysis of both hind legs. The number of mice used in the figure shown was 7. The figure has been modified from Barthelmes et al. 28. 2) The mice are sacrificed at different time points during the disease course, spleen, inguinal lymph nodes, blood and lumbar spinal cord extracted and single cells isolated from these tissues. 3) Immune cell distribution in various tissues, as determined by flow cytometry. 4) mRNA expression for various cytokines in the different tissues, as determined by quantitative PCR. Please click here to view a larger version of this figure.
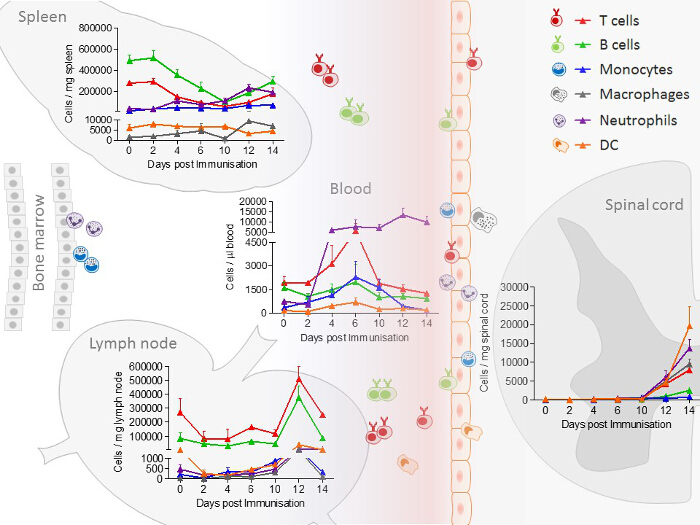 Figure 2: Immune Cell Distribution, Determined by FACS (Fluorescence-Activated Cell Sorting) Analysis in Spleen, Lymph node, Blood and Spinal Cord Samples from EAE Mice at Different Disease Stages. In the experiment shown, the number of mice per group was 2 - 10. The distribution of neutrophils/monocytes in blood and neutrophils/macrophages in the spinal cord are adapted and modified from Barthelmes et al.28. Please click here to view a larger version of this figure.
Figure 2: Immune Cell Distribution, Determined by FACS (Fluorescence-Activated Cell Sorting) Analysis in Spleen, Lymph node, Blood and Spinal Cord Samples from EAE Mice at Different Disease Stages. In the experiment shown, the number of mice per group was 2 - 10. The distribution of neutrophils/monocytes in blood and neutrophils/macrophages in the spinal cord are adapted and modified from Barthelmes et al.28. Please click here to view a larger version of this figure.
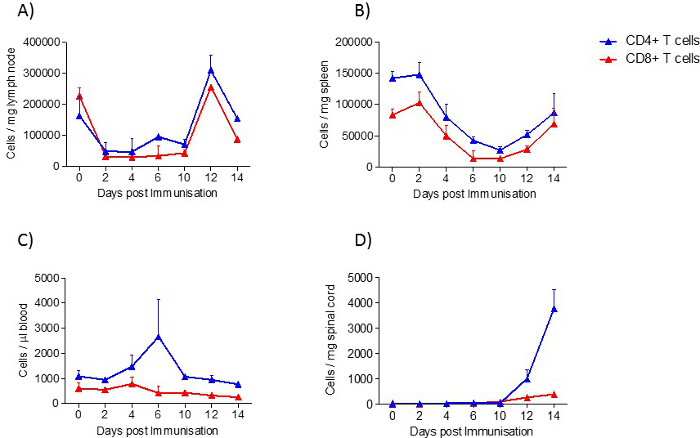 Figure 3: CD4+ T cell and CD8+ T cell Distribution in Samples from EAE Mice at Different Disease Stages, Determined by FACS Analysis. A) lymph node, B) spleen, C) blood and D) spinal cord. In the experiment shown, the number of mice per group was 2 - 10. Results are presented as means ± standard errors (SEM). Please click here to view a larger version of this figure.
Figure 3: CD4+ T cell and CD8+ T cell Distribution in Samples from EAE Mice at Different Disease Stages, Determined by FACS Analysis. A) lymph node, B) spleen, C) blood and D) spinal cord. In the experiment shown, the number of mice per group was 2 - 10. Results are presented as means ± standard errors (SEM). Please click here to view a larger version of this figure.
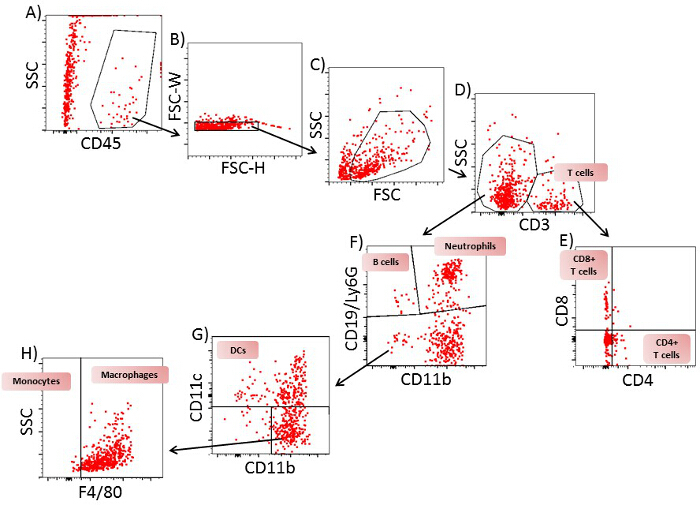 Figure 4: Gating Strategy for Flow Cytometric Analysis of T Cells, B Cells, Neutrophils, Monocytes, Dendritic Cells and Macrophages from Spinal Cord of EAE Mice on Day 12. A) A plot of SSC/CD45 from all cells. Gate: CD45+ cells. B) A plot of FSC-H/FSC-W from CD45+ cells. Gate: single cells. C) A plot of SSC-A/FSC-A from single cells. Gate: viable cells. D) A plot of FSC-H/CD3 from viable cells. Gate: CD3+ and CD3- cells. E) A plot of CD4/CD8 from CD3+ cells. Gate: CD4+CD8- (CD4+ T cells) and CD4-CD8+ (CD8+ T cells). F) A plot of CD19 and Ly6G/CD11b from CD3- cells. Gate: CD19+CD11b- cells (B cells), Ly6G+CD11b+ cells (neutrophils) and CD19-Ly6G- cells. G) A plot of CD11c/CD11b cells from CD19-Ly6G- cells: Gate CD11c+ cells (dendritic cells) and CD11c- cells. H) A plot of SSC/F4/80 from CD11c- cells. Gate: F4/80- cells (monocytes) and F4/80+ cells (macrophages). One representative plot of 9 is shown. Please click here to view a larger version of this figure.
Figure 4: Gating Strategy for Flow Cytometric Analysis of T Cells, B Cells, Neutrophils, Monocytes, Dendritic Cells and Macrophages from Spinal Cord of EAE Mice on Day 12. A) A plot of SSC/CD45 from all cells. Gate: CD45+ cells. B) A plot of FSC-H/FSC-W from CD45+ cells. Gate: single cells. C) A plot of SSC-A/FSC-A from single cells. Gate: viable cells. D) A plot of FSC-H/CD3 from viable cells. Gate: CD3+ and CD3- cells. E) A plot of CD4/CD8 from CD3+ cells. Gate: CD4+CD8- (CD4+ T cells) and CD4-CD8+ (CD8+ T cells). F) A plot of CD19 and Ly6G/CD11b from CD3- cells. Gate: CD19+CD11b- cells (B cells), Ly6G+CD11b+ cells (neutrophils) and CD19-Ly6G- cells. G) A plot of CD11c/CD11b cells from CD19-Ly6G- cells: Gate CD11c+ cells (dendritic cells) and CD11c- cells. H) A plot of SSC/F4/80 from CD11c- cells. Gate: F4/80- cells (monocytes) and F4/80+ cells (macrophages). One representative plot of 9 is shown. Please click here to view a larger version of this figure.
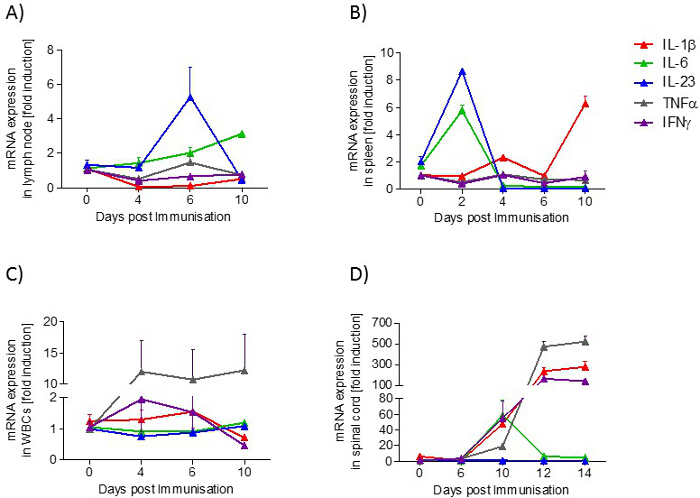 Figure 5: Cytokine Profile (IL-1β, IL-6, IL-23, TNFα, IFNγ) in Samples from EAE Mice at Different Disease Stages. A) lymph node, B) spleen, C) blood and D) spinal cord. The mRNA expression levels were normalized to peptidyl propyl isomerase A (PPIA) and were calculated using the mRNA level of untreated mice of the same age. Measurements were done in triplicate. The number of mice per group was 2 - 3. Results are presented as means ± standard errors (SEM). Please click here to view a larger version of this figure.
Figure 5: Cytokine Profile (IL-1β, IL-6, IL-23, TNFα, IFNγ) in Samples from EAE Mice at Different Disease Stages. A) lymph node, B) spleen, C) blood and D) spinal cord. The mRNA expression levels were normalized to peptidyl propyl isomerase A (PPIA) and were calculated using the mRNA level of untreated mice of the same age. Measurements were done in triplicate. The number of mice per group was 2 - 3. Results are presented as means ± standard errors (SEM). Please click here to view a larger version of this figure.
| Gene | forward | reverse | fragement size (Bp) |
| IL-1β | CTGGTGTGTGACGTTCCCATTA | CCGACAGCACGAGGCTTT | 76 |
| IL-6 | TAGTCCTTCCTACCCCAATTTCC | TTGGTCCTTAGCCACTCCTTC | 76 |
| IL-23 | GAGCAGCAACTCTGACTGAGCC | GAACAGCACAAGTCCTAATGGGTTA | 127 |
| IFNγ | CACGGCACAGTCATTGAAAGC | CACCATCCTTTTGCCAGTTCC | 118 |
| TNFα | TGACAAGCCTGTAGCCCACG | GCCTTGTCCCTTGAAGAGAACC | 178 |
| PPIA | GCTGGACCAAACACAAACGG | GCCATTCCTGGACCCAAAAC | 144 |
Table 1: Primer pairs.
Discussion
The EAE model described here has received the most attention as a model of MS and is routinely used in testing therapeutic strategies for MS32. The mouse disease exhibits many clinical and histological features of MS and is caused by the induction of autoimmunity to neuronal antigens. The sensitization to myelin antigens is associated with blood brain barrier dysfunction and thereby, immune cell infiltration into the CNS. Our findings show that immune cells increase transiently in the lymph nodes in the acute phase. Splenic T cells and B cells decrease in the preclinical phase of the disease. In circulating blood, T cells, B cells, dendritic cells and monocytes increase transiently in the preclinical phase, whereas neutrophils increase disease-dependently. Finally, in the spinal cord, an increase in immune cells is detectable (Figure 2). These data indicate that T cells and B cells migrate from the spleen, which acts a reservoir for these cells, to the lymph nodes where they are activated and proliferate. Subsequently, they migrate via the circulation into the spinal cord. Neutrophils, monocytes and dendritic cells, on the other hand, migrate from the bone marrow through the circulation into the CNS.
Our data reveal that in the spinal cord, IFNγ, TNFα and IL-1β are regulated in a disease-dependent manner, whereas IL-6 is transiently upregulated at the onset of disease. These data support the hypothesis that EAE pathology is driven by Th1 and Th17 cells. Th1 cells release IFNγ, which in turn activates macrophages to release TNFα and IL-1β33. Moreover, IL-6 is known to drive differentiation of Th1 and Th17 cells and thereby promote the development of EAE34.
The day of onset of clinical symptoms and the severity of the disease is variable in EAE mice. Based on our experience, there are several reasons for this. Mice exposed to stress during the preclinical phase show a reduced severity of disease and a delay in disease onset. The housing of the mice also influences the severity of EAE and also the day of onset. Recently, it was shown that the commensal microbiota of mice influence the development of EAE35 and different housing situations may influence the microbiome and thereby, the EAE pathology. Furthermore, it is very important always to use freshly prepared pertussis toxin. Pertussis toxin damages the blood brain barrier which is a prerequisite for the development of EAE. Do not use emulsified MOG35-55 peptide after the expiry date. The age of the mice also influences EAE pathology. Thus, there are some rules which should be fulfilled in order to generate a reproducible EAE model. Use mice aged between 10 and 13 weeks and mice from the same supplier, if possible. If the mice are ordered from an external supplier, allow the mice to acclimatize for two weeks in the animal house. Handle the mice gently before EAE induction so that they get used to the handling. Avoid unnecessary handling after EAE induction. Provide the mice with food and water which they can easily reach as soon as they show clinical symptoms. If the mice are to be treated with a drug, it is best to mix it into the food or drinking water, to avoid confounding stress effects. If gavage or an injection is essential for drug administration, it is important to use the same route for the vehicle.
In addition to the primary progressive EAE model described here, relapsing-remitting EAE models also exist. The different disease course is induced by the administration of different antigens in various mouse strains. Thus, the application of MOG35 - 55 in mouse strains, such as C57BL/6, Sv129, B10, NOD and Biozzi mice, leads to a primary progressive disease form. Interestingly, the day of onset differs in the various mouse strains. Whereas C57BL/6, SV129 and B10 mice develop first clinical symptoms from about day 11 to 12, Biozzi mice show first clinical symptoms after 21 days36. In a previous study, it was shown that 129S4/SvJae×C57BL/6 hybrids first exhibit clinical symptoms from about day 1128. The injection of proteolipid protein (PLP)131-151 leads in SJL mice to the induction of a relapsing-remitting disease course37. The best EAE model to use will depend on the focus of the research. Thus, if the relapse phase is of interest, the relapse-remitting model should be employed whereas if disease onset is the main focus, the progressive model should be used. If the role of a specific protein is to be investigated in the EAE model, using knock-out mice, it is important to bear in mind that not all strains can be used to induce EAE and possibly backcrossing of the knock-out strain onto a sensitive strain is necessary.
The three most widely used types of animal models of MS are (1) autoantigen -induced EAE; (2) virally induced spontaneous, chronic demyelinating disease, and (3) toxin-induced demyelination38. EAE models are characterized by the induction of inflammation and autoimmune responses by the application of an autoantigenic peptide such as myelin oligodendrocyte protein or proteolipid protein38. Spontaneous EAE develops in transgenic mice which overexpress a T cell receptor against a peptide of the myelin oligodendrocyte protein39. To induce virally-induced chronic inflammation, a neurotrophic infection of the central nervous system can be induced, for example, with Theiler`s murine encephalomyelitis virus. Cells infected with the virus are attacked by both T cells and a humoral response and lead thereby to chronic demyelination40,41. Toxin-induced demyelination can be induced for example with the copper chelator cuprizone42. This model leads specifically to demyelination because cuprizone attacks the oligodendrocytes and leads, thereby, to activation of astroglia and microglia, whereas inflammatory processes only play a minor role. After clearance of the toxin, new oligodendrocytes are generated and a new myelin sheath is formed. The use of these three models, in a complementary way, allows evaluation of drug effects on different aspects of the pathogenesis of MS, because the models differ with regard to inflammatory, and immune and demyelinating processes. In EAE and the virus-induced model, inflammation and autoimmune processes primarily drive the disease, whereas in the toxin-induced model, demyelinating and remyelinating processes are predominant. For preclinical testing of potential drugs for MS treatment, at least three models, a toxin-induced model, a relapse-remitting EAE and a primary progressive EAE or a virally-induced model should be used to verify the effectiveness of the drug.
The EAE model described here can be used to gain further insight into the mechanism of the development of the MS-like disease process, identifying key cellular drivers of the disease, such as Th1, Th17 and B cells43,44. The better the understanding of the inflammatory and immune processes involved in the development of MS and of the processes which promote and resolve the lesions, the more likely it is that new treatment strategies can be developed.
Nevertheless, the EAE model described here merely has some characteristics in common with MS, and features a few clear differences from the human disease. The most prominent difference is that MS patients vary widely in disease presentation, which is not reproduced in the EAE model. This may be due to different lesion patterns in MS patients and EAE mice and to the fact that mice are inbred strains. In EAE mice, homogeneous lesions are most prominent in the spinal cord, whereas in MS patients, primarily heterogeneous lesions are found in the brain45,46. Furthermore, MS and the EAE share the crucial contribution of Th1 cells and Th17 cells for their development, but in contrast to MS, CD8+ T cells play a minor role in EAE pathology33. In conclusion, the EAE model focuses mainly on the combined effects of autoimmunity, inflammatory processes and neurodegeneration whereas demyelinating processes are less amenable to selective assessment. Therefore, other animal models such as the cuprizone model can be used for the distinct study of demyelinating processes47. The EAE model is imperfect, but nevertheless, a useful model of MS33.
Disclosures
MJP is a consultant to Xellia and to Leo Pharmaceuticals.
Acknowledgments
This work was supported by the Else Kröner-Fresenius Foundation (EKFS) Research Training Group Translational Research Innovation - Pharma (TRIP) and by the "Landesoffensive zur Entwicklung wissenschaftlich-ökonomischer Exzellenz (LOEWE), Schwerpunkt: Anwendungsorientierte Arzneimittelforschung" of the State of Hesse.
References
- McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol. 2007;8:913–919. doi: 10.1038/ni1507. [DOI] [PubMed] [Google Scholar]
- Proudfoot AE. Chemokine receptors: multifaceted therapeutic targets. Nat Rev Immunol. 2002;2:106–115. doi: 10.1038/nri722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihara M, Hashizume M, Yoshida H, Suzuki M, Shiina M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin Sci (Lond) 2012;122:143–159. doi: 10.1042/CS20110340. [DOI] [PubMed] [Google Scholar]
- Strydom N, Rankin SM. Regulation of circulating neutrophil numbers under homeostasis and in disease. J Innate Immun. 2013;5:304–314. doi: 10.1159/000350282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerstetter AE, Padovani-Claudio DA, Bai L, Miller RH. Inhibition of CXCR2 signaling promotes recovery in models of multiple sclerosis. Exp Neurol. 2009;220:44–56. doi: 10.1016/j.expneurol.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159–175. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- Fan X, et al. Murine CXCR1 is a functional receptor for GCP-2/CXCL6 and interleukin-8/CXCL8. J Biol Chem. 2007;282:11658–11666. doi: 10.1074/jbc.M607705200. [DOI] [PubMed] [Google Scholar]
- Hartl D, et al. Infiltrated neutrophils acquire novel chemokine receptor expression and chemokine responsiveness in chronic inflammatory lung diseases. J Immunol. 2008;181:8053–8067. doi: 10.4049/jimmunol.181.11.8053. [DOI] [PubMed] [Google Scholar]
- Barcelos LS, et al. Role of the chemokines CCL3/MIP-1 alpha and CCL5/RANTES in sponge-induced inflammatory angiogenesis in mice. Microvasc Res. 2009;78:148–154. doi: 10.1016/j.mvr.2009.04.009. [DOI] [PubMed] [Google Scholar]
- Wojkowska DW, Szpakowski P, Ksiazek-Winiarek D, Leszczynski M, Glabinski A. Interactions between neutrophils, Th17 cells, and chemokines during the initiation of experimental model of multiple sclerosis. Mediators Inflamm. 2014. p. 590409. [DOI] [PMC free article] [PubMed]
- Bose S, Cho J. Role of chemokine CCL2 and its receptor CCR2 in neurodegenerative diseases. Arch Pharm Res. 2013;36:1039–1050. doi: 10.1007/s12272-013-0161-z. [DOI] [PubMed] [Google Scholar]
- Mony JT, Khorooshi R, Owens T. Chemokine receptor expression by inflammatory T cells in EAE. Front Cell Neurosci. 2014;8:187. doi: 10.3389/fncel.2014.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katschke KJ, Jr, et al. Differential expression of chemokine receptors on peripheral blood, synovial fluid, and synovial tissue monocytes/macrophages in rheumatoid arthritis. Arthritis Rheum. 2001;44:1022–1032. doi: 10.1002/1529-0131(200105)44:5<1022::AID-ANR181>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Trebst C, et al. CCR1+/CCR5+ mononuclear phagocytes accumulate in the central nervous system of patients with multiple sclerosis. Am J Pathol. 2001;159:1701–1710. doi: 10.1016/s0002-9440(10)63017-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin N, Wildbaum G. The role of chemokines in adjusting the balance between CD4+ effector T cell subsets and FOXp3-negative regulatory T cells. Int Immunopharmacol. 2015. [DOI] [PubMed]
- Rottman JB, et al. Leukocyte recruitment during onset of experimental allergic encephalomyelitis is CCR1 dependent. Eur J Immunol. 2000;30:2372–2377. doi: 10.1002/1521-4141(2000)30:8<2372::AID-IMMU2372>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J Exp Med. 2000;192:1075–1080. doi: 10.1084/jem.192.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwabara T, et al. CCR7 ligands are required for development of experimental autoimmune encephalomyelitis through generating IL-23-dependent Th17 cells. J Immunol. 2009;183:2513–2521. doi: 10.4049/jimmunol.0800729. [DOI] [PubMed] [Google Scholar]
- Liu L, et al. Myelin repair is accelerated by inactivating CXCR2 on nonhematopoietic cells. J Neurosci. 2010;30:9074–9083. doi: 10.1523/JNEUROSCI.1238-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui M, et al. Treatment of experimental autoimmune encephalomyelitis with the chemokine receptor antagonist Met-RANTES. J Neuroimmunol. 2002;128:16–22. doi: 10.1016/s0165-5728(02)00121-2. [DOI] [PubMed] [Google Scholar]
- Liu L, et al. Severe disease, unaltered leukocyte migration, and reduced IFN-gamma production in CXCR3-/- mice with experimental autoimmune encephalomyelitis. J Immunol. 2006;176:4399–4409. doi: 10.4049/jimmunol.176.7.4399. [DOI] [PubMed] [Google Scholar]
- Lee M, Suk K, Kang Y, McGeer E, McGeer PL. Neurotoxic factors released by stimulated human monocytes and THP-1 cells. Brain Res. 2011;1400:99–111. doi: 10.1016/j.brainres.2011.05.021. [DOI] [PubMed] [Google Scholar]
- Gage GJ, Kipke DR, Shain W. Whole animal perfusion fixation for rodents. J Vis Exp. 2012. [DOI] [PMC free article] [PubMed]
- O'Connor RA, et al. Adjuvant immunotherapy of experimental autoimmune encephalomyelitis: immature myeloid cells expressing CXCL10 and CXCL16 attract CXCR3+CXCR6+ and myelin-specific T cells to the draining lymph nodes rather than the central nervous system. J Immunol. 2012;188:2093–2101. doi: 10.4049/jimmunol.1101118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesch C, et al. MPGES-1-derived PGE2 suppresses CD80 expression on tumor-associated phagocytes to inhibit anti-tumor immune responses in breast cancer. Oncotarget. 2015;6:10284–10296. doi: 10.18632/oncotarget.3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Barthelmes J, et al. Lack of ceramide synthase 2 suppresses the development of experimental autoimmune encephalomyelitis by impairing the migratory capacity of neutrophils. Brain Behav Immun. 2015;46:280–292. doi: 10.1016/j.bbi.2015.02.010. [DOI] [PubMed] [Google Scholar]
- Schiffmann S, et al. Ceramide synthase 6 plays a critical role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2012;188:5723–5733. doi: 10.4049/jimmunol.1103109. [DOI] [PubMed] [Google Scholar]
- Schiffmann S, et al. PGE2/EP4 signaling in peripheral immune cells promotes development of experimental autoimmune encephalomyelitis. Biochem Pharmacol. 2014;87:625–635. doi: 10.1016/j.bcp.2013.12.006. [DOI] [PubMed] [Google Scholar]
- Giglio S, Monis PT, Saint CP. Demonstration of preferential binding of SYBR Green I to specific DNA fragments in real-time multiplex PCR. Nucleic Acids Res. 2003;31:e136. doi: 10.1093/nar/gng135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesterinen HM, et al. Improving the translational hit of experimental treatments in multiple sclerosis. Mult Scler. 2010;16:1044–1055. doi: 10.1177/1352458510379612. [DOI] [PubMed] [Google Scholar]
- 't Hart BA, Gran B, Weissert R. EAE: imperfect but useful models of multiple sclerosis. Trends Mol Med. 2011;17:119–125. doi: 10.1016/j.molmed.2010.11.006. [DOI] [PubMed] [Google Scholar]
- Serada S, et al. IL-6 blockade inhibits the induction of myelin antigen-specific Th17 cells and Th1 cells in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2008;105:9041–9046. doi: 10.1073/pnas.0802218105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berer K, et al. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature. 2011;479:538–541. doi: 10.1038/nature10554. [DOI] [PubMed] [Google Scholar]
- Shetty A, et al. Immunodominant T-cell epitopes of MOG reside in its transmembrane and cytoplasmic domains in EAE. Neurol Neuroimmunol Neuroinflamm. 2014;1:22–22. doi: 10.1212/NXI.0000000000000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz K, et al. R-flurbiprofen attenuates experimental autoimmune encephalomyelitis in mice. EMBO Mol Med. 2014;6:1398–1422. doi: 10.15252/emmm.201404168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Procaccini C, De Rosa V, Pucino V, Formisano L, Matarese G. Animal models of Multiple Sclerosis. Eur J Pharmacol. 2015;759:182–191. doi: 10.1016/j.ejphar.2015.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollinger B, et al. Spontaneous relapsing-remitting EAE in the SJL/J mouse: MOG-reactive transgenic T cells recruit endogenous MOG-specific B cells. J Exp Med. 2009;206:1303–1316. doi: 10.1084/jem.20090299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M, Oleszak E, Leibowitz J. Theiler's murine encephalomyelitis: a model of demyelination and persistence of virus. Crit Rev Immunol. 1987;7:325–365. [PubMed] [Google Scholar]
- Lipton HL. Theiler's virus infection in mice: an unusual biphasic disease process leading to demyelination. Infect Immun. 1975;11:1147–1155. doi: 10.1128/iai.11.5.1147-1155.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima GK, Morell P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 2001;11:107–116. doi: 10.1111/j.1750-3639.2001.tb00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-behi M, Rostami A, Ciric B. Current views on the roles of Th1 and Th17 cells in experimental autoimmune encephalomyelitis. J Neuroimmune Pharmacol. 2010;5:189–197. doi: 10.1007/s11481-009-9188-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann MK, Ray A, Basu S, Karp CL, Dittel BN. Pathogenic and regulatory roles for B cells in experimental autoimmune encephalomyelitis. Autoimmunity. 2012;45:388–399. doi: 10.3109/08916934.2012.665523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann H, Bruck W, Lucchinetti CF. The immunopathology of multiple sclerosis: an overview. Brain Pathol. 2007;17:210–218. doi: 10.1111/j.1750-3639.2007.00064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons SB, Pierson ER, Lee SY, Goverman JM. Modeling the heterogeneity of multiple sclerosis in animals. Trends Immunol. 2013;34:410–422. doi: 10.1016/j.it.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praet J, Guglielmetti C, Berneman Z, Vander Linden A, Ponsaerts P. Cellular and molecular neuropathology of the cuprizone mouse model: clinical relevance for multiple sclerosis. Neurosci Biobehav Rev. 2014;47:485–505. doi: 10.1016/j.neubiorev.2014.10.004. [DOI] [PubMed] [Google Scholar]