

Abstract
Please cite this paper as: Svindland et al. The mucosal and systemic immune responses elicited by a chitosan‐adjuvanted intranasal influenza H5N1 vaccine. Influenza and Other Respiratory Viruses DOI:10.1111/j.1750‐2659.2011.00271.x.
Background Development of influenza vaccines that induce mucosal immunity has been highlighted by the World Health Organisation as a priority (Vaccine 2005;23:1529). Dose‐sparing strategies and an efficient mass‐vaccination regime will be paramount to reduce the morbidity and mortality of a future H5N1 pandemic.
Objectives This study has investigated the immune response and the dose‐sparing potential of a chitosan‐adjuvanted intranasal H5N1 (RG‐14) subunit (SU) vaccine in a mouse model.
Methods Groups of mice were intranasally immunised once or twice with a chitosan (5 mg/ml)‐adjuvanted SU vaccine [7·5, 15 or 30 μg haemagglutinin (HA)] or with a non‐adjuvanted SU vaccine (30 μg HA). For comparison, another group of mice were intranasally immunised with a whole H5N1 (RG‐14) virus (WV) vaccine (15 μg HA), and the control group consisted of unimmunised mice.
Results The chitosan‐adjuvanted SU vaccine induced an immune response superior to that of the non‐adjuvanted SU vaccine. Compared with the non‐adjuvanted SU group, the chitosan‐adjuvanted SU vaccine elicited higher numbers of influenza‐specific antibody‐secreting cells (ASCs), higher concentrations of local and systemic antibodies and correspondingly an improved haemagglutination inhibition (HI) and single radial haemolysis (SRH) response against both the homologous vaccine strain and drifted H5 strains. We measured a mixed T‐helper 1/T‐helper 2 cytokine response in the chitosan‐adjuvanted SU groups, and these groups had an increased percentage of virus‐specific CD4+ T cells producing two Thelper 1 (Th1) cytokines simultaneously compared with the non‐adjuvanted SU group. Overall, the WV vaccine induced higher antibody concentrations in sera and an HI and SRH response similar to that of the chitosan‐adjuvanted SU vaccine. Furthermore, the WV vaccine formulation showed a stronger bias towards a T‐helper 1 profile than the SU vaccine and elicited the highest frequencies of CD4+ Th1 cells simultaneously secreting three different cytokines (INFγ+, IL2+ and INFα+). As expected, two immunisations gave a better immune response than one in all groups. The control group had very low or not detectable results in the performed immunoassays.
Conclusion The cross‐clade serum reactivity, improved B‐ and T‐cell responses and dose‐sparing potential of chitosan show that a chitosan‐adjuvanted intranasal influenza vaccine is a promising candidate vaccine for further preclinical development.
Keywords: Chitosan, dose, H5N1, influenza, intranasal, vaccine
Introduction
The influenza A virus is annually responsible for substantial morbidity and mortality, 1 and three pandemics caused by this virus resulted in millions of deaths during the twentieth century. 2 , 3 , 4
The virus undergoes two types of antigenic change that can make us vulnerable to infection: antigenic drift leading to annual outbreaks 5 , 6 , 7 and antigenic shift due to reassortment of influenza strains causing occasional pandemics. 8 The H5N1 virus has already caused zoonotic infections in man, resulting in at least 318 deaths (60% mortality rate). 9 As there is little pre‐existing natural immunity to H5N1 in the human population, 10 a pernicious new pandemic could result if the H5N1 virus gains the ability to undergo efficient and sustained transmission amongst humans. Indeed, a recent cluster of human cases of avian influenza H5N1 in Egypt indicates that the virus may now be adapting to man, 11 and as of 6 April 2011, Egypt reported 137 human cases of H5N1. 12 Vaccination is the best method to minimise the morbidity, mortality and socio‐economic effects of the influenza virus. 13 However, owing to the continuous antigenic variation of the influenza A virus, 14 the usefulness of pandemic vaccine stockpiling may be compromised. In addition, H5N1 vaccines have been shown to be poorly immunogenic and need to be adjuvanted. 15
The availability of antigen 13 , 16 and medical staff are likely to be important limiting factors for an efficient mass‐vaccination programme during a pandemic. To prepare for a possible H5N1 pandemic, more knowledge about the effects and safety of different adjuvants and vaccine formulations must be generated.
Therefore, this study has evaluated the effects of an intranasal H5N1 vaccine adjuvanted with chitosan. We have several reasons for investigating the intranasal route of administration. Importantly, intranasal vaccination can be self‐administered and is needle‐free, thus facilitating an efficient mass‐vaccination regime and reducing the risk of accidental infection by blood‐borne pathogens. 17 Also, intranasal vaccination with influenza antigen resembles natural infection and can elicit both systemic and local immune responses. 18 , 19 , 20 The local mucosal antibodies limit epithelial contact and protect mucosal surfaces from invading infectious agents. 21 Thus, the local immune response is important for protection against novel strains and can stop the virus at its point of entry 22 and prevent transmission between individuals. In contrast, conventional parenterally injected vaccines induce a poor IgA response and therefore are unable to prevent the initial replication in the airways 18 , 23 , 24 , 25 and have shown reduced efficacy against drifted viruses. 26
As the uptake of vaccines from the nasal cavity is poor because of rapid clearance from the absorption site and poor transport across the nasal mucosa, intranasal H5N1 vaccines need to be adjuvanted to ensure retention and absorption in the nasal cavity. 15 , 27 Chitosan (a proprietary nasal delivery system of Archimedes Development Ltd.) is a mucoadhesive that enhances drug absorption through tight junctions via the paracellular route. 27 Structurally, chitosan is a cationic hydrophilic biopolymer obtained by hydrolysing the aminoacetyl groups of chitin.
In this study, our candidate H5N1 vaccine adjuvanted with chitosan was evaluated in a mouse model. We investigated in detail the B‐ and T‐cellular responses to the homologous vaccine strain and the antibody response to homologous and heterologous strains.
Furthermore, this study will help us better understand mucosal defence mechanisms and contribute towards the development of mucosal vaccines against a number of infections. Mucosal infections are a major health problem worldwide, and approximately 10 million children succumb to these infections every year 21 ; hence, development of effective vaccination strategies against these infections is an urgent priority.
Materials and methods
Animals and vaccination
Six‐ to 8‐week‐old female BALB/c mice (Taconic M&B, Ejby, Denmark) were housed at the Vivarium, University of Bergen. The study was approved and conducted according to the Norwegian Animal Welfare Act. The animals were anaesthetised with ketalar (10 mg/ml) (Pfizer, Lysaker, NO, USA) and rompun (1 mg/ml) (Vetalar; Pfizer Limited, Kent, UK), and the mice were intranasally immunised once or twice with an RG‐14 (derived from A/Vietnam/1194/2004 (H5N1)) subunit (SU) vaccine [7·5, 15 or 30 μg haemagglutinin (HA), 15 mice for each dose] formulated with chitosan glutamate or with 30 μg HA alone (15 mice). A further group of 15 mice was intranasally immunised with a whole RG‐14 virus vaccine (15 μg HA), and a control group consisted of nine unimmunised mice. The maximum volume administered to each nostril was 5·5 μl. Chitosan (ChiSys®, Nottingham, UK), which is Archimedes’ proprietary intranasal delivery system, was kindly provided by Archimedes Development Ltd.
Sample collection
Blood samples were collected weekly post‐vaccination from the hind leg or by cardiac puncture at the time of euthanasia (Figure 1). Sera were separated and stored at −80°C until used in the antibody assays. Spleens were collected at the time of sacrifice (21, 25 and 42 days post‐vaccination), and lymphocytes separated by density gradient centrifugation, as previously described. 28
Figure 1.
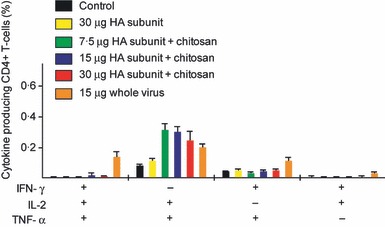
The multifunctional influenza‐specific Th1 CD4+ cytokine‐secreting response. Groups of five mice were intranasally immunised twice (21 days apart) with a subunit (SU) influenza A H5N1 vaccine. The control group consisted of unimmunised mice. Three groups were vaccinated with different antigen doses (7.5, 15 or 30 μg HA) of the chitosan‐adjuvanted (+) SU vaccine. One group was vaccinated with a non‐adjuvanted (−) SU vaccine with 30 μg HA, and a further group was immunised with a non‐adjuvanted 15 μg HA whole virus (WV) vaccine. Splenocytes were activated in vitro with homologous H5N1 antigen and were intracellularly stained for cytokine products and analysed by flow cytometry. The bars show the mean frequencies of multifunctional cells expressing combinations of IFN‐γ, IL‐2 and TNF‐α. HA, haemagglutinin.
Haemagglutination inhibition (HI) assay
One volume of serum was diluted with three volumes of receptor‐destroying enzyme (Denka Seiken CO, Tokyo, Japan) and used in the HI assay with eight haemagglutinating units (HAU) of the homologous vaccine strain RG‐14 or the heterologous strains RG‐6 (A/Anhui/1/05 clade 2·2·4) and RG‐88 (A/Cambodia/R0405050/2007 clade 1) and an equal volume of 0·7% turkey erythrocytes. The serum HI titre was expressed as the reciprocal of the highest dilution at which haemagglutination was inhibited, and titres less than eight were assigned a value of four for calculation purposes. An HI titre ≥40 is considered a surrogate correlate of protection in man, whereas no correlate of protection has been established in mice.
Single radial haemolysis (SRH)
Single radial haemolysis was performed at the University of Siena, Italy, against homologous strains included in the vaccine. Single radial haemolysis was based on a modified reference method standardised by Schild et al. 29
Two different sets of SRH plates were prepared using turkey erythrocytes at a final concentration 10%, to which was added 2000 haemagglutinin units/ml (HAU/ml) inactivated WV provided by US FDA CBER.
All samples were heat‐inactivated at 56°C for 30 minutes before the test, and 6 μl was added to the plates. Antibody titration was made in duplicate in two different assays, and pre‐ and post‐vaccination sera were titrated simultaneously.
The diameters of the haemolytic zones were measured using a Transidyne Calibrating Viewer (Transidyne General Corporation, Ann Arbor, MI, USA). A haemolysis area <4 mm2 was considered negative, between 4 and 25 mm2 was considered positive but not protective, greater than 25 mm2 was considered protective, as shown in EMEA CHMP guidelines.
In each test, we ran a negative control sample, and a positive control serum (sheep hyperimmune sera provided from National Institute for Biological Standards and Controls) was included.
Enzyme‐linked immunosorbent assay (ELISA)
The influenza‐specific serum IgG, IgG1, IgG2a, IgA and IgM antibodies were quantified using the ELISA, as previously described. 30 Briefly, ELISA plates were coated with inactivated WV influenza H5N1 (RG‐14) antigen or, for the standard curve, capture antibody goat anti‐mouse IgA (catalogue number 1040‐01), IgG (1030‐01), IgG1 (1070‐01) or IgG2a (1080‐01) (Southern Biotechnology, Birmingham, AL, USA). Dilutions of murine sera and antibody standards [mouse IgA (M1421), mouse IgG (I5381), mouse IgG2a (M9144) (Sigma, St. Louis, MO, USA) and mouse IgG1 (0102‐14) (Southern Biotechnology)] were added, and bound antibodies were detected with goat anti‐mouse immunoglobulin class‐ and subclass‐specific biotin‐conjugated antibody (goat 1040‐08, 1030‐08, 1070‐08, 1080‐08; Southern Biotechnology) and extravidin peroxidase. The antibody concentrations (μg/ml) were calculated using the IgG, IgG1, IgG2a, IgA and IgM standards and linear regression of the log‐transformed readings.
Enzyme‐linked immunospot assay (ELISPOT)
The total number of splenic B cells spontaneously secreting influenza‐specific antibodies was enumerated using an ELISPOT assay, as described earlier. 30 ELISPOT plates were coated with the RG‐14 antigen, and B‐cell medium [consisting of RPMI 1640 medium supplemented with l‐glutamine (GIBO 21875‐034, GIBO, Paisley, UK), non‐essential amino acids (GIBO 11140‐035), Hepes (Sigma H0887), sodium pyruvate (Sigma S8636), penicillin/streptomycin/fungizone (PSA) (BioWhittaker 17‐745E, Biowhittaker, Verviers, Belgium), mercaptoethanol (Sigma M‐7522) and foetal bovine serum (BioWhittaker 14‐701F)] containing lymphocytes (2·5 × 105 or 5 × 105) was added. To detect secreted antibodies, goat anti‐mouse immunoglobulin class‐ or IgG subclass‐specific biotinylated antibodies were added. Extravidin peroxidase was added and subsequently the substrate 9‐amino 3‐ethyl carbazole (AEC). Plates were scanned and counted using Immunoscan™ and Immunospot software version 4 (Cellular Technology Ltd, Shaker Heights, OH, USA), and results were standardised to the number of influenza‐specific cells per 500 000 lymphocytes.
Flow cytometry for multifunctional CD4+ T cells
Splenic lymphocytes were isolated 3 weeks after the second immunisation and were incubated overnight (5% CO2, 37°C) with influenza antigen (influenza H5 RG‐14 virosomal vaccine at a final concentration of 10 μg HA/ml) together with B‐cell medium or in B‐cell‐medium only. Lymphocytes from non‐vaccinated control mice were incubated in medium with or without influenza antigen. Cells were then stained for CD3, CD4, CD8 and intracellular cytokine production (IFN‐γ, IL‐2, TNF‐α), and live lymphocytes were acquired using a BD FACSCanto flow cytometer. Data were analysed using FlowJo v8·8·6 software (Tree Star, Ashland, OR, USA), Pestle and spice 4.0 (Mario Roederer, Vaccine Research Centre, NIH, Bethesda, MD, USA), and multifunctional T cells were identified. 30 T cells were classified based on cytokine secretion as single producers (one cytokine), double producers (two cytokines) and multifunctional triple producers (simultaneously secreting IFN‐γ, IL‐2 and TNF‐α).
Bio‐plex cytokine assay
The Bio‐plex cytokine assay was performed to quantify the concentration of cytokines secreted in the supernatants from stimulated spleen cells as described earlier 30 except that the cytokine secretion was investigated 3 weeks after the second vaccination and that the 7‐plex Group 1 Mouse kit (Bio‐Rad, Cat. #M6000006V7, Bio‐Rad, Gladesville, NSW, Australia) was used to detect cytokines (IL‐2, IFN‐γ, IL‐4, IL‐5, IL‐10 and IL‐17) secreted by spleen cells.
Statistical analysis and figures
A two‐tailed t‐test was used to detect significant differences (P < 0·05) using graphpad Prism for Macintosh version 5.0a (GraphPad Software Inc., La Jolla, CA, USA).
Results
This dose response study investigated the quality, magnitude and kinetics of the B‐ and T‐cell responses in mice intransally immunised once or twice with a chitosan‐adjuvanted SU vaccine at half (7·5 μg HA), standard (15 μg HA) or twice (30 μg HA) the human dose. Another group of mice was intranasally immunised with the SU vaccine (30 μg HA) alone. A further group of mice was intransally immunised with WV vaccine (15 μg HA), and a control group consisted of unimmunised mice.
Chitosan augments the HI and SRH antibody responses against both the homologous vaccine strain and drifted H5 strains
The post‐vaccination HI titres were measured 3 weeks after the first and second immunisation (groups of mice were sacrificed 3 weeks after the first immunisation, 4 days after the second immunisation and 3 weeks after the second immunisation) (Figure 2A,B). No serum HI antibodies were detected (titres <8) in any of the groups after the first immunisation (data not shown). Three weeks after the second vaccination, good HI titres against the homologous vaccine strain and against a drifted H5 strain could be measured in the WV and chitosan‐adjuvanted SU groups. After the booster immunisation, the HI response against the homologous vaccine strain (RG‐14) showed a dose response with the highest titres detected in the adjuvanted 30‐μg HA vaccine group, which was fivefold higher than in the lowest adjuvanted group (7·5 μg) and sevenfold higher than in the non‐adjuvanted group (30 μg) (Figure 2A). No response could be measured in the control group (Figure 2A).
Figure 2.
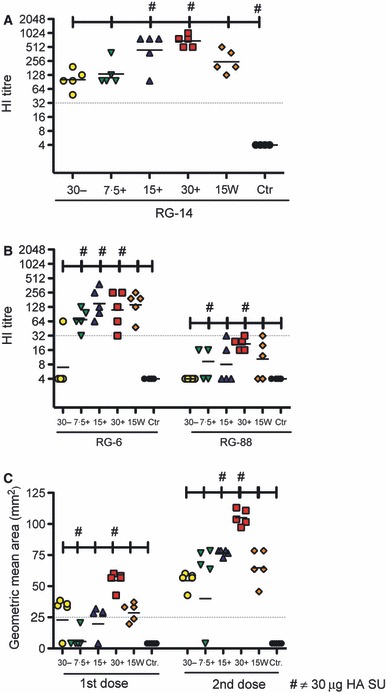
The serological antibody response induced after intranasal influenza vaccination. Groups of five mice were intranasally immunised once or twice (21 days apart) with a subunit (SU) influenza A H5N1 vaccine. The control (Ctr) group consisted of unimmunised mice. Three groups were vaccinated with different antigen doses (measured as μg HA) of the chitosan‐adjuvanted (+) SU vaccine (7.5+, 15+ or 30+). One group was vaccinated with a non‐adjuvanted (−) SU vaccine with 30‐μg HA (30−), and a further group was immunised with a non‐adjuvanted 15 μg HA whole virus (15W) vaccine. The serum haemagglutination inhibition (HI) antibody response was measured 21 days after the second vaccination to the homologous vaccine strain RG‐14 (A/Vietnam/1194/2004) (A) and the cross‐reactive HI response to RG‐6 (A/Anhui/1/05) and RG‐88 (A/Cambodia/R0405050/2007) (B). The single radial haemolysis (SRH) geometric zone area (C) to RG14 was measured 3 weeks after the first and second vaccinations. Each symbol represents an individual mouse, and the line represents the geometric mean titre for each group of five mice (A, B and C). The dotted line represents an HI titre of 32 (A and B) or an SRH zone area of 25 mm2 (C). HA, haemagglutinin.
An effective pandemic influenza vaccine should also induce broad cross‐reactive antibody responses, and we therefore examined the cross‐reactivity of the serum HI response to two antigenically distinct strains of influenza H5N1 [A/Anhui/1/05 (RG‐6) clade 2·2·4 and A/Cambodia/R0405050/2007 (RG‐88) clade 1] (Figure 2B). The highest HI titres were observed to the RG‐6 strain in the chitosan‐adjuvanted or WV vaccine groups. The RG‐6‐specific HI titre was significantly higher in the chitosan‐adjuvanted SU group as compared to the non‐adjuvanted SU vaccine group (Figure 2B). For RG‐88, an increase in the HI titre was detected with increasing antigen concentrations; however, none of the mice achieved an HI titre higher than 32. No response could be measured in the control group (Figure 2B).
In the SRH assay, influenza‐specific antibodies were detected 3 weeks after the first and second vaccination (Figure 2C). A geometric mean area of ≥25 mm2 is considered protective in man, 31 although no correlate of protection has been established in mice. SRH antibody was detected in most mice after the first vaccination, and a significant increase in zone area was observed after the second immunisation (P < 0·05) in all groups. All mice, except one in the 7·5‐μg adjuvanted vaccine group, had zone areas of ≥25 mm2 after the second immunisation. A dose response was observed after both one and two immunisations in the chitosan‐adjuvanted groups, and significantly higher antibody titres were observed in the chitosan‐adjuvanted 30‐μg vaccine group compared with all the other groups (P < 0·05) (Figure 2C)
Chitosan‐adjuvanted SU vaccines augment the ASC response
The influenza‐specific splenic ASC response was enumerated 4 days after the booster vaccination as we have previously observed a peak response at this time point 32 (Figure 3). The chitosan‐adjuvanted vaccine groups and the WV group had significantly (P < 0·05) higher numbers of influenza‐specific IgA, IgG, IgG1 and IgG2a ASCs than the non‐adjuvanted SU group (Figure 3). The chitosan‐adjuvanted SU groups induced comparable numbers of IgA and IgG2a ASCs to the WV group (Figure 3).
Figure 3.
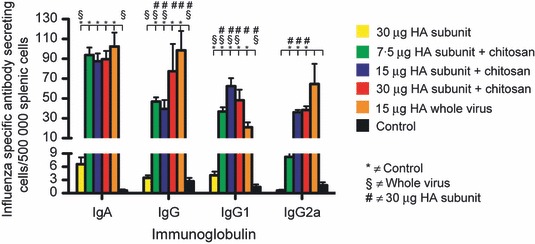
The antibody‐secreting cell response (ASC) induced after intranasal influenza vaccination. Groups of five mice were intranasally immunised twice (21 days apart) with a subunit (SU) influenza A H5N1 vaccine. The control group consisted of unimmunised mice. Three groups were vaccinated with different antigen doses (7.5, 15 or 30 μg HA) of the chitosan‐adjuvanted SU vaccine. One group was vaccinated with a non‐adjuvanted SU vaccine with 30 μg HA, and a further group was immunised with a non‐adjuvanted 15 μg HA whole virus vaccine. Animals were euthanised 4 days after the second immunisation, and splenocytes were used to enumerate the influenza‐specific ASCs by the ELISPOT assay. The data are presented as the mean number of class and IgG subclass influenza‐specific ASCs per 500 000 lymphocytes ± standard error of the mean (SEM). HA, haemagglutinin.
Chitosan‐adjuvanted SU vaccines augment serum IgA and IgG response
Figure 4 shows the kinetics of the influenza‐specific antibody response in serum (IgA and IgG) and nasal wash samples (IgA). At all time points, the nasal wash IgA and the serum IgA and IgG concentrations in the control group were very low or not detectable. After the booster vaccination, the serum IgA response increased significantly (P < 0·05) as compared to the response measured after the first immunisation (Figure 4A). After the second immunisation, the IgA levels were significantly (P < 0·05) elevated in the adjuvanted 30‐μg HA SU vaccine group as compared to the adjuvanted 7·5‐μg and 15‐μg HA SU vaccine groups (Figure 4A). Furthermore, after the second immunisation, the chitosan‐adjuvanted vaccine groups had significantly (P < 0·05) higher serum IgA concentrations than the non‐adjuvanted SU group. Three weeks after the second immunisation, the chitosan‐adjuvanted 30‐μg HA vaccine group was not significantly (P > 0·05) different in serum IgA compared with the WV group (Figure 4A).
Figure 4.
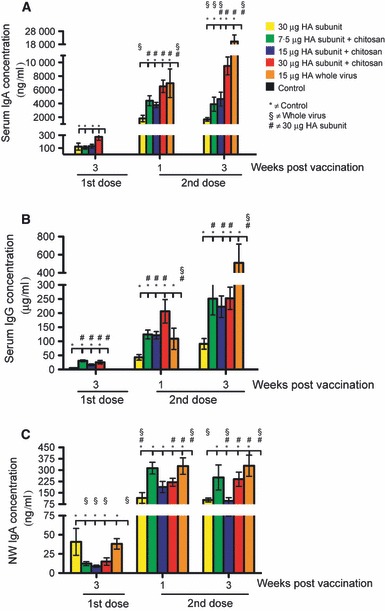
The serum and nasal wash antibody responses induced after intranasal influenza vaccination. Groups of five mice were intranasally immunised twice (21 days apart) with a subunit (SU) influenza A H5N1 vaccine. The control group consisted of unimmunised mice. Three groups were vaccinated with different antigen doses (7.5, 15 or 30 μg HA) of the chitosan‐adjuvanted SU vaccine. One group was vaccinated with a non‐adjuvanted SU vaccine with 30 μg HA, and a further group was immunised with a non‐adjuvanted 15 μg HA whole virus vaccine. Sera were collected at appropriate time points after the first and second immunisation and used to quantify the influenza‐specific serum IgG (B) and IgA (A) concentrations and the nasal wash (NW) IgA (C) concentrations using the ELISA. The data are presented as the mean antibody concentration of serum IgA (ng/ml) (A) and serum IgG (μg/ml) (B) and nasal wash IgA (ng/ml) (C) ± standard error of the mean. HA, haemagglutinin.
For all vaccinated groups, only low levels of IgG could be detected after the first immunisation, but this response was significantly (P < 0·05) elevated after the second immunisation and corresponded with the pattern observed in the HI and SRH assays. After both the first and second immunisations, the groups receiving the chitosan‐adjuvanted SU vaccine produced significantly (P < 0·05) higher levels of IgG antibodies than the non‐adjuvanted SU group, thus demonstrating the immunostimulatory effect of chitosan (Figure 4B).
Figure 4C shows the IgA concentrations measured in nasal wash samples. Although only low levels of IgA antibodies were detected after the first immunisation, the response had increased significantly (P < 0·05) 1 week after the booster immunisation (Figure 4C). Three weeks after the second immunisation, the chitosan‐adjuvanted 30‐μg HA SU vaccine group had significantly (P < 0·05) higher concentrations of nasal wash IgA than the non‐adjuvanted 30‐μg HA SU vaccine group, thus demonstrating the adjuvant effect of chitosan for mucosal IgA antibodies (Figure 4C).
No nasal wash IgG or IgM was detected at any time point throughout the study (data not shown).
The adjuvanted vaccine elicited a mixed Th1/Th2 and a Th17 cytokine response
As shown in Table 1, serum IgG1 and IgG2a were detected in significantly higher (P < 0·05) concentrations in the chitosan‐adjuvanted 30‐μg HA SU group compared with the other SU vaccine groups 1 week after the second vaccination. The IgG2a/IgG1 ratio was <0·3 for all SU vaccine groups and >1 for the WV vaccine group (Table 1). These results indicate that the WV vaccine has a stronger bias towards a Th1 antibody response than the SU vaccine.
Table 1.
The serum IgG subclass response 1, 2 and 3 weeks after the second vaccination
| 1 week IgG2a/IgG1 (ratio) | 2 weeks IgG2a/IgG1 (ratio) | 3 weeks IgG2a/IgG1 (ratio) | |
|---|---|---|---|
| 30 μg HA | 3 ± 1/16 ± 4 = (0·2) | 3 ± 1/36 ± 7 = (0·1) | 1 ± 0·1/0 ± 0 |
| 7·5 μg HA + chitosan | 4 ± 1/37 ± 16 = (0·1) | 5 ± 2/93 ± 27 = (0·1) | 4 ± 2/72 ± 25 = (0·1) |
| 15 μg HA + chitosan | 3 ± 1/36 ± 6 = (0·1) | 5 ± 1/69 ± 12 = (0·1) | 5 ± 2/72 ± 16 = (0·1) |
| 30 μg HA + chitosan | 20 ± 10/216 ± 81 = (0·1) | 12 ± 4/140 ± 23 = (0·1) | 18 ± 8/95 ± 16 = (0·2) |
| 15 μg whole virus | 41 ± 10/34 ± 25 = (1) | 58 ± 10/42 ± 25 = (1) | 65 ± 12/77 ± 42 = (0·8) |
Groups of five mice were intranasally immunised twice (21 days apart) with a subunit (SU) influenza A H5N1 vaccine. Three groups were vaccinated with different antigen doses (7·5, 15 or 30 μg HA) of the chitosan‐adjuvanted SU vaccine. One group was vaccinated with a non‐adjuvanted SU vaccine with 30 μg HA, and a further group was immunised with a non‐adjuvanted 15 μg HA whole virus vaccine. Sera collected 1, 2 and 3 weeks after the second immunisation were used to quantify the influenza‐specific serum IgG1 and IgG2a concentrations using the ELISA. The data are presented as the mean concentration (μg/ml) ± standard error of the mean for IgG2a and IgG1 and the ratio (IgG2a/IgG1) in serum 1, 2 and 3 weeks after the second immunisation.
HA, haemagglutinin.
T‐helper cells (Th) can be divided into subsets that produce different cytokines. In mice, the Th1 subtype typically produces higher levels of IL‐2 and IFN‐γ than the Th2 subtype, which produces higher levels of IL‐4, IL‐5 and IL‐10 than the Th1 cells. IL‐17 is specific for the Th17 subtype.
All the vaccinated mice typically produced both Th1 and Th2 cytokines (Figure 5A,B). They had significantly (P < 0·05) higher levels of IFN‐γ than the control, and for this cytokine, the WV vaccine group had significantly (P < 0·05) higher concentrations than all other groups. Furthermore, the WV vaccine group had significantly (P < 0·05) higher concentrations of IL‐2 than the other groups except the chitosan‐adjuvanted 7·5‐μg HA SU group. For the cytokines that characterise the murine Th2 response (IL‐4, IL‐5 and IL‐10), the chitosan‐adjuvanted SU and WV vaccine groups had significantly (P < 0·05) higher concentrations than the control group. IL‐10 is typically produced by Th2 cells; however, it can also to a lesser extent be produced by Th1 cells. The WV vaccine elicited a significantly (P < 0·05) higher IL‐10 production than the SU vaccine groups, except for the 15‐μg HA SU vaccine group and a higher IFN‐γ production than all the SU groups. However, the WV vaccine induced significantly (P < 0·05) lower production of the classical Th2 cytokines IL‐4 and IL‐5 as compared to the chitosan‐adjuvanted SU vaccine groups except the 7·5‐μg SU vaccine group. Taken together, these observations indicate that the WV vaccine has a bias towards inducing a Th1 cytokine response compared with the SU formulation of the vaccine.
Figure 5.
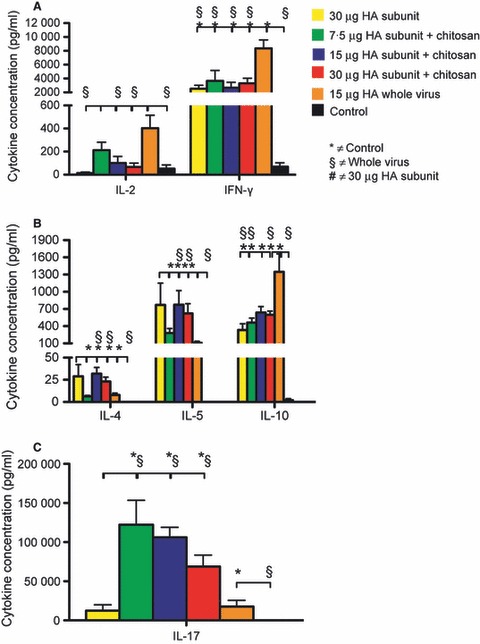
The Th1, Th2 and Th17 cytokine profiles after in vitro activation of spleen cells collected 3 weeks after second vaccine dose. Groups of five mice were intranasally immunised twice (21 days apart) with a subunit (SU) influenza A H5N1 vaccine. The control group consisted of unimmunised mice. Three groups were vaccinated with different antigen doses (7.5, 15 or 30 μg HA) of the chitosan‐adjuvanted SU vaccine. One group was vaccinated with a non‐adjuvanted SU vaccine with 30 μg HA, and a further group was immunised with a non‐adjuvanted 15 μg HA whole virus vaccine. Splenocytes were collected 3 weeks after the second immunisation, and the Bio‐Plex Pro cytokine assay was used to quantify the different cytokines secreted from the in vitro stimulated spleen cells. The data are presented as the mean cytokine concentration (pg/ml) in the supernatant from the stimulated splenocytes ± standard error of the mean for cytokines typically produced by Th1 cells (A), Th2 cells (B) and Th17 cells (C). HA, haemagglutinin.
The chitosan‐adjuvanted vaccine groups had a significantly (P < 0·05) higher IL‐17 response than the non‐adjuvanted vaccine groups and the control group (Figure 5C). Intriguingly, an increase in the HA concentration in the chitosan‐adjuvanted vaccine groups was associated with a corresponding decrease in the IL‐17 response.
Multifunctional CD4+ T‐cell response to vaccination
Recently, multiparameter flow cytometry has allowed the simultaneous assessment of the phenotype and multiple effector functions of single T cells. Therefore, the quality of the T‐cell response can be evaluated by their ability to simultaneously secrete multiple cytokines and thus exert more than one function (hence multifunctionality). We evaluated the CD4+ Th1 cell response by investigating simultaneous secretion of TNF‐α, IFN‐γ and IL‐2 using intracellular staining and flow cytometry 3 weeks after the second immunisation (Figure 6).
Figure 6.
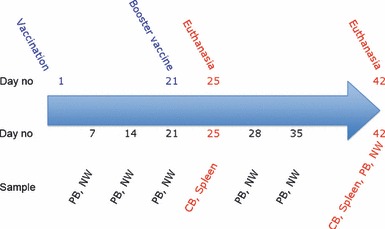
Outline of the study design. Mice were intranasally immunised once or twice (21 days apart) with a subunit influenza A H5N1 vaccine (7.5, 15 or 30 μg HA) with chitosan adjuvant, with 30 μg HA alone or with 15 μg HA whole virus vaccine. Peripheral blood (PB) and nasal wash (NW) samples were collected weekly. Groups of mice were euthanised either at 21 days after the first immunisation or at 4 or 21 days after the second dose. Spleens (SP), cardiac blood (CB) and NW were collected. HA, haemagglutinin.
As shown in Figure 6, mice immunised with WV vaccine had a significantly (P < 0·05) higher percentage of multifunctional (TNF‐α+/IFN‐γ+/IL‐2+) CD4+ T cells than the non‐vaccinated control group. Furthermore, there was a significant (P < 0·05) increase in the percentage of TNF‐α+/IL‐2+ CD4+ T cells in the groups vaccinated with either the chitosan‐adjuvanted SU or the WV vaccine compared with the non‐vaccinated control group and the non‐adjuvanted SU vaccine group.
Discussion
This study was conducted to evaluate the immune response in mice when using chitosan as adjuvant for an H5N1 SU vaccine. In solution, chitosan is positively charged and binds strongly to negatively charged materials such as cell surfaces and mucus, thereby enabling increased antigen‐retention time in the nasal cavity. A g‐scintigraphy technique has demonstrated chitosan’s behaviour as a bioadhesive material in the nasal cavity. 33 The absorption‐promoting effect of chitosan is also enhanced by modification of the paracellular transport system as demonstrated by investigations in cell culture as well as in animal models. 34 Immunohistological studies have shown that chitosan can open the tight junctions between cells through an effect upon F‐actin filaments. 35 Therefore, a combination of bioadhesion and paracellular transport effects of chitosan most likely enable the interaction of the antigen with the subepithelial lymph nodes of the nasal cavity, leading to improved immunological responses.
Combining chitosan with a potent adjuvant may further optimise vaccine efficacy with the potential to reduce not only the antigen dose but also the adjuvant dose, thereby minimising any irritancy or systemic toxicity. A nasal chitosan‐based vaccine against norovirus, which utilises the co‐adjuvanting concept with monophosphoryl lipid, is currently under investigation. The toxicity evaluation of the intranasal formulation in rabbits showed the vaccine to be well tolerated. 36 Furthermore, a phase II challenge study clearly showed that the intranasally administered vaccine produced statistically significant reductions in the clinical norovirus illness, infection and severity of illness when compared with a placebo treatment. 37 Hence, further studies should be performed to evaluate the effect of combining the mucoadhesive properties of chitosan with novel potent mucosal adjuvants to further enhance the immune response of SU vaccines.
The ability to induce a cross‐protective immune response against drifted strains is an important characteristic of effective influenza vaccines. Elevated mucosal secretory IgA (sIgA) and serum IgG have been reported to be important in providing cross‐clade protection after intranasal administration of low‐dose WV vaccine with potent mucosal adjuvants (cholera toxin or polyI:C). 38 Hence, our finding that both vaccine formulations (SU and WV) induce influenza‐specific IgA and IgG ASC and serum (IgA and IgG), and mucosal IgA antibodies in mice may be indicative of the vaccines’ ability to provide cross‐clade protection after intranasal administration. Further, the IgA response in vaccinated mice may be augmented by elevated levels of IL‐10, which is a cytokine that promotes B cells to switch antibody production to IgA and acts as an anti‐inflammatory and regulatory cytokine controlling the initial inflammation. We also detected elevated serum HI and SRH titres against the homologous H5N1 strain and showed cross‐reactivity against the heterologous H5N1 strains RG‐88 and RG‐6. Interestingly, the SRH assay showed increased sensitivity to detection of serum antibody response after the first immunisation compared with the HI assay. The HI and SRH titres had an r value of 0·809 in the Spearman’s rank test and correlated with a highly significant P value (P < 0·0001) after the second immunisation (Figure 7). Together, our data suggest that using chitosan as an adjuvant for an intranasal SU influenza vaccine improves the vaccines’ ability to induce a protective antibody response against drifted strains.
Figure 7.
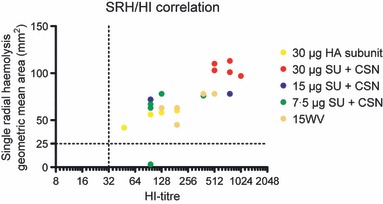
The correlation between single radial haemolysis (SRH) and haemagglutination inhibition (HI) assays. Groups of five mice were intranasally immunised twice (21 days apart) with a subunit (SU) influenza A H5N1 vaccine. The control group consisted of unimmunised mice. Three groups were vaccinated with different antigen doses (7.5, 15 or 30 μg HA) of the chitosan‐adjuvanted SU vaccine. One group was vaccinated with a non‐adjuvanted SU vaccine with 30 μg HA, and a further group was immunised with a non‐adjuvanted 15 μg HA whole virus vaccine. The SRH results 3 weeks after the second immunisation are presented on the x‐axis as the geometric mean area in mm2, and the HI results measured 2 weeks after the second immunisation are presented as the HI titre on the y‐axis. HA, haemagglutinin.
The IgG subclass distribution and the cytokine milieu are indicative of the differences in the Th‐cell response. In mice, a Th1 response is associated with a predominant IgG2a antibody response and elevated IFN‐γ and other cytokines. 39 , 40 This is the typical cell‐associated response to viral infection in mice and is characterised by the recruitment of cytotoxic T cells (CTLs), natural killer cells and macrophages. 41 A Th2 profile in mice is indicative of a humoral immune response and is associated with IgG1 antibodies 39 and a predominant IL‐4 and IL‐5 response. Both the adjuvanted SU and WV vaccine groups induced a mixed Th1/Th2 serum antibody, ASC and cytokine response, suggesting that both vaccine formulations were able to stimulate the humoral and cellular arms of the immune system. However, consistent with our previous studies, 32 we found that mice vaccinated with the WV vaccine had a serum IgG2a/IgG1 ratio, ASC and cytokine response biased towards generating a Th1 response. The Th1 bias may suggest the generation of a better CTL response resulting from efficient endocytosis and presentation of WV antigens on the major histocompatibility complex (MHC) I. A previous study in humans has shown that influenza WV vaccination stimulates IL‐12 from dendritic cells and TNF‐α and IL‐2 from peripheral blood mononuclear cells in vitro, generating a cytokine milieu that supports endocytosis by antigen‐presenting cells and recruitment of CTLs. 42 In contrast to the WV vaccine, non‐replicating exogenous antigens such as the SU vaccine are not generally processed and presented on MHC I to stimulate a CTL response; hence, this may in part explain the bias towards a Th2 response in mice vaccinated with the SU vaccine. Taken together, our results suggest that the vaccine formulation used (WV or SU) is a significant determining factor in polarising the immune response towards a Th1 or Th2 response.
Further, we observed a significant increase in the levels of IL‐17 in mice vaccinated with the chitosan‐adjuvanted vaccines. Both intranasal vaccination 43 and influenza virus challenge 44 , 45 have previously been shown to induce a strong IL‐17 response in mice; however, the exact role of IL‐17 in the pathogenesis of influenza is not yet clear. Recent reports suggest a role for IL‐17 in inducing a protective immune response against influenza viral challenge in mice 45 , 46 and also in patients with severe pandemic H1N1 influenza. 47 In contrast, Crowe et al. reported that IL‐17 may play an important role in neutrophil infiltration leading to acute lung injury in mice following influenza viral challenge. The conflicting data in the current literature suggest that the exact role of IL‐17 in the pathogenesis of influenza merits more investigation.
We further characterised the immune response by evaluating the multifunctional CD4+ T‐cell response. Here, we have shown that the double cytokine producing CD4+ T‐cell response after vaccination was dominated by TNF‐α+/IL‐2+ cells. This is consistent with our previous findings in mice immunised with a pandemic H5N1 virosomal vaccine adjuvanted with matrix M, 30 but differs from other studies where the dominant subtype was TNF‐α+/INF‐α+. 48 , 49 The Th1 cells that secrete IL‐2 or TNF‐α or both can develop into IFN‐γ producers, and these cells can provide a supply of memory CD4+ T cells with effector potential. 50 As very few memory T cells will be sustained from a single IFN‐γ producer, a vaccine that induces mainly this response will probably not elicit protective immunity. 51 Interestingly, intranasal immunisation with the pandemic H5N1 virosomal vaccine adjuvanted with matrix M induced much lower frequencies of TNF‐α+/IL‐2+ CD4+ T cells in the mouse spleen 30 compared with the current study. This may be attributed to the differences in the vaccine formulations and the adjuvants used in the two studies and suggests that chitosan may be a better inducer of a multifunctional CD4+ T‐cell response compared with matrix M following intranasal immunisation. To our knowledge, this study is the first to show that an intranasal pandemic influenza vaccine formulated with a mucosal adjuvant induces high frequencies of multifunctional CD4+ T cells in mice. Multifunctional T cells have been associated with better clinical outcomes of patients infected with human immunodeficiency virus (HIV), 51 reduced HIV replication 52 and protection elicited against smallpox by priming with vaccinia virus. 53 However, the strongest evidence to date for the importance of multifunctional CD4+ T cells in eliciting a protective immune response is in the mouse model of Leishmania major infection. 48 , 54 Therefore, further studies with influenza vaccines are needed to characterise the potential importance of multifunctional T cells in influenza infection.
During an influenza pandemic, limited antigen availability requires vaccine formulations with dose‐sparing potential. A recent study by Yang et al. 38 demonstrated that immunisation with a low‐dose (1 μg HA content) Anhui H5N1 inactivated vaccine adjuvanted with cholera toxin, polyI:C or MF59 provides cross‐clade protection in mice. In the current study, we have shown a significant increase in local and systemic humoral and cellular immune responses with cross‐strain reactivity in mice immunised with the chitosan‐adjuvanted H5N1 SU vaccine. The immunogenicity of this vaccine indicates potential for significant dose‐sparing and vaccine efficacy; however, this should be tested in a future study using an animal challenge model.
Acknowledgements
We acknowledge funding for this work through the European Commission under the project ‘NASPANVAC FP7 2007‐2.3.3.1‐202083’. We also thank the staff, and in particular Gabriel Pedersen, at the Influenza Centre in Bergen and the staff at the animal house at the University of Bergen for excellent assistance.
References
- 1. Stohr K. Influenza – WHO cares. Lancet Infect Dis 2002; 2:517. [DOI] [PubMed] [Google Scholar]
- 2. Ghendon Y. Introduction to pandemic influenza through history. Eur J Epidemiol 1994; 10:451–453. [DOI] [PubMed] [Google Scholar]
- 3. Beveridge WI. The chronicle of influenza epidemics. Hist Philos Life Sci 1991; 13:223–234. [PubMed] [Google Scholar]
- 4. Ghendon Y. Influenza vaccines: a main problem in control of pandemics. Eur J Epidemiol 1994; 10:485–486. [DOI] [PubMed] [Google Scholar]
- 5. Scholtissek C. Molecular aspects of the epidemiology of virus disease. Experientia. 1987; 43:1197–1201. [DOI] [PubMed] [Google Scholar]
- 6. Scholtissek C. Source for influenza pandemics. Eur J Epidemiol 1994; 10:455–458. [DOI] [PubMed] [Google Scholar]
- 7. Kendal AP. Epidemiologic implications of changes in the influenza virus genome. Am J Med 1987; 82:4–14. [DOI] [PubMed] [Google Scholar]
- 8. Webster RG, Laver WG. Antigenic variation in influenza virus. Biology and chemistry. Prog Med Virol 1971; 13:271–338. [PubMed] [Google Scholar]
- 9. Cumulative number of confirmed human cases of avian influenza A/(H5N1) reported to WHO. 2011. Available at: http://www.who.int/csr/disease/avian_influenza/country/cases_table_2011_04_06/en/index.html (Accessed 7 April 2011).
- 10. Leroux‐Roels I, Leroux‐Roels G. Current status and progress of prepandemic and pandemic influenza vaccine development. Expert Rev Vaccines 2009; 8:401–423. [DOI] [PubMed] [Google Scholar]
- 11. Schroedl A. Age‐based human influenza A virus (H5N1) infection patterns, Egypt. Emerg Infect Dis 2010; 16:161–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Avian influenza – situation in Egypt – update 49. 2011. Available at: http://www.who.int/csr/don/2011_04_06/en/index.html (Accessed 7 April 2011).
- 13. Williams JR, Chen PY, Cho CT, Chin TD. Influenza: prospect for prevention and control. Kaohsiung J Med Sci 2002; 18:421–434. [PubMed] [Google Scholar]
- 14. Williams JP, Lednar W. New developments in influenza vaccine technology: a potential new prevention strategy for employers and managed care organizations. Am J Manag Care 2002; 8(5 Suppl):S143–S154. [PubMed] [Google Scholar]
- 15. Sambhara S, Poland GA. H5N1 Avian influenza: preventive and therapeutic strategies against a pandemic. Annu Rev Med 2010; 61:187–198. [DOI] [PubMed] [Google Scholar]
- 16. Gust ID, Hampson AW, Lavanchy D. Planning for the next pandemic of influenza. Rev Med Virol 2001; 11:59–70. [DOI] [PubMed] [Google Scholar]
- 17. De Filette M, Ramne A, Birkett A et al. The universal influenza vaccine M2e‐HBc administered intranasally in combination with the adjuvant CTA1‐DD provides complete protection. Vaccine 2006; 24:544–551. [DOI] [PubMed] [Google Scholar]
- 18. Muszkat M, Friedman G, Schein MH et al. Local SIgA response following administration of a novel intranasal inactivated influenza virus vaccine in community residing elderly. Vaccine 2000; 18:1696–1699. [DOI] [PubMed] [Google Scholar]
- 19. Garmise RJ, Mar K, Crowder TM et al. Formulation of a dry powder influenza vaccine for nasal delivery. AAPS PharmSciTech 2006; 7:E19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hirabayashi Y, Kurata H, Funato H et al. Comparison of intranasal inoculation of influenza HA vaccine combined with cholera toxin B subunit with oral or parenteral vaccination. Vaccine 1990; 8:243–248. [DOI] [PubMed] [Google Scholar]
- 21. Brandtzaeg P. Mucosal immunity: induction, dissemination, and effector functions. Scand J Immunol 2009; 70:505–515. [DOI] [PubMed] [Google Scholar]
- 22. Tamura S, Hasegawa H, Kurata T. Estimation of the effective doses of nasal‐inactivated influenza vaccine in humans from mouse‐model experiments. Jpn J Infect Dis 2010; 63:8–15. [PubMed] [Google Scholar]
- 23. Cassetti MC, Couch R, Wood J, Pervikov Y. Report of meeting on the development of influenza vaccines with broad spectrum and long‐lasting immune responses, World Health Organization, Geneva, Switzerland, 26–27 February 2004. Vaccine 2005; 23:1529–1533. [DOI] [PubMed] [Google Scholar]
- 24. Muszkat M, Greenbaum E, Ben‐Yehuda A et al. Local and systemic immune response in nursing‐home elderly following intranasal or intramuscular immunization with inactivated influenza vaccine. Vaccine 2003; 21:1180–1186. [DOI] [PubMed] [Google Scholar]
- 25. Clements ML, Betts RF, Tierney EL, Murphy BR. Serum and nasal wash antibodies associated with resistance to experimental challenge with influenza A wild‐type virus. J Clin Microbiol 1986; 24:157–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Potter CW, Jennings R, Nicholson K, Tyrrell DA, Dickinson KG. Immunity to attenuated influenza virus WRL 105 infection induced by heterologous, inactivated influenza A virus vaccines. J Hyg (Lond) 1977; 79:321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Davis SS, Illum L. Absorption enhancers for nasal drug delivery. Clin Pharmacokinet 2003; 42:1107–1128. [DOI] [PubMed] [Google Scholar]
- 28. Cox RJ, Mykkeltvedt E, Robertson J, Haaheim LR. Non‐lethal viral challenge of influenza haemagglutinin and nucleoprotein DNA vaccinated mice results in reduced viral replication. Scand J Immunol 2002; 55:14–23. [DOI] [PubMed] [Google Scholar]
- 29. Schild GC, Pereira MS, Chakraverty P. Single‐radial‐hemolysis: a new method for the assay of antibody to influenza haemagglutinin. Applications for diagnosis and seroepidemiologic surveillance of influenza. Bull World Health Organ 1975; 52:43–50. [PMC free article] [PubMed] [Google Scholar]
- 30. Madhun AS, Haaheim LR, Nilsen MV, Cox RJ. Intramuscular Matrix‐M‐adjuvanted virosomal H5N1 vaccine induces high frequencies of multifunctional Th1 CD4+ cells and strong antibody responses in mice. Vaccine 2009; 27:7367–7376. [DOI] [PubMed] [Google Scholar]
- 31. European Committee for Proprietary Medicinal Products . Note for guidance on harmonisation of requirements for influenza vaccines SCBCN‐p.
- 32. Hovden AO, Cox RJ, Haaheim LR. Whole influenza virus vaccine is more immunogenic than split influenza virus vaccine and induces primarily an IgG2a response in BALB/c mice. Scand J Immunol 2005; 62:36–44. [DOI] [PubMed] [Google Scholar]
- 33. Soane RJ, Frier M, Perkins AC, Jones NS, Davis SS, Illum L. Evaluation of the clearance characteristics of bioadhesive systems in humans. Int J Pharm 1999; 178:55–65. [DOI] [PubMed] [Google Scholar]
- 34. Dodane V, Amin Khan M, Merwin JR. Effect of chitosan on epithelial permeability and structure. Int J Pharm 1999; 182:21–32. [DOI] [PubMed] [Google Scholar]
- 35. Illum L, Jabbal‐Gill I, Hinchcliffe M, Fisher AN, Davis SS. Chitosan as a novel nasal delivery system for vaccines. Adv Drug Deliv Rev 2001; 51:81–96. [DOI] [PubMed] [Google Scholar]
- 36. Richardson C. Product development and clinical plan for a mucosally‐delivered Norwalk vaccine. The 4th International Conference on Vaccines for Enteric Diseases; Lisbon, Portugal. Available at: http://www.meetingsmanagement.com/pdf/VED_2007.pdf2007 (Accessed 1 February 2011).
- 37. El‐Kamary SS, Pasetti MF, Mendelman PM et al. Adjuvanted intranasal Norwalk virus‐like particle vaccine elicits antibodies and antibody‐secreting cells that express homing receptors for mucosal and peripheral lymphoid tissues. J Infect Dis 2010; 202:1649–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang P, Tang C, Luo D et al. Cross‐clade protection against HPAI H5N1 influenza virus challenge in BALB/c mice intranasally administered adjuvant‐combined influenza vaccine. Vet Microbiol 2010; 146:17–23. [DOI] [PubMed] [Google Scholar]
- 39. Stevens TL, Bossie A, Sanders VM et al. Regulation of antibody isotype secretion by subsets of antigen‐specific helper T cells. Nature 1988; 334:255–258. [DOI] [PubMed] [Google Scholar]
- 40. Markine‐Goriaynoff D, van der Logt JT, Truyens C et al. IFN‐gamma‐independent IgG2a production in mice infected with viruses and parasites. Int Immunol 2000; 12:223–230. [DOI] [PubMed] [Google Scholar]
- 41. Tamura S, Kurata T. Defense mechanisms against influenza virus infection in the respiratory tract mucosa. Jpn J Infect Dis 2004; 57:236–247. [PubMed] [Google Scholar]
- 42. Saurwein‐Teissl M, Zisterer K, Schmitt TL, Gluck R, Cryz S, Grubeck‐Loebenstein B. Whole virus influenza vaccine activates dendritic cells (DC) and stimulates cytokine production by peripheral blood mononuclear cells (PBMC) while subunit vaccines support T cell proliferation. Clin Exp Immunol 1998; 114:271–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pedersen G, Major D, Roseby S, Wood J, Madhun AS, Cox RJ. Matrix‐M adjuvanted virosomal H5N1 vaccine confers protection against lethal viral challenge in a murine model. Influenza Other Respi Viruses 2011; 5:426–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Crowe CR, Chen K, Pociask DA et al. Critical role of IL‐17RA in immunopathology of influenza infection. J Immunol 2009; 183:5301–5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hamada H, Garcia‐Hernandez Mde L, Reome JB et al. Tc17, a unique subset of CD8 T cells that can protect against lethal influenza challenge. J Immunol 2009; 182:3469–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McKinstry KK, Strutt TM, Buck A et al. IL‐10 deficiency unleashes an influenza‐specific Th17 response and enhances survival against high‐dose challenge. J Immunol 2009; 182:7353–7363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bermejo‐Martin JF, Ortiz de Lejarazu R, Pumarola T et al. Th1 and Th17 hypercytokinemia as early host response signature in severe pandemic influenza. Crit Care 2009; 13:R201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Darrah PA, Patel DT, De Luca PM et al. Multifunctional TH1 cells define a correlate of vaccine‐mediated protection against Leishmania major . Nat Med 2007; 13:843–850. [DOI] [PubMed] [Google Scholar]
- 49. Forbes EK, Sander C, Ronan EO et al. Multifunctional, high‐level cytokine‐producing Th1 cells in the lung, but not spleen, correlate with protection against Mycobacterium tuberculosis aerosol challenge in mice. J Immunol 2008; 181:4955–4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sui Z, Chen Q, Wu R et al. Cross‐protection against influenza virus infection by intranasal administration of M2‐based vaccine with chitosan as an adjuvant. Arch Virol 2010; 155:535–544. [DOI] [PubMed] [Google Scholar]
- 51. Seder RA, Darrah PA, Roederer M. T‐cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol 2008; 8:247–258. [DOI] [PubMed] [Google Scholar]
- 52. Almeida JR, Price DA, Papagno L et al. Superior control of HIV‐1 replication by CD8+ T cells is reflected by their avidity, polyfunctionality, and clonal turnover. J Exp Med 2007; 204:2473–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Precopio ML, Betts MR, Parrino J et al. Immunization with vaccinia virus induces polyfunctional and phenotypically distinctive CD8(+) T cell responses. J Exp Med 2007; 204:1405–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Darrah PA, Hegde ST, Patel DT et al. IL‐10 production differentially influences the magnitude, quality, and protective capacity of Th1 responses depending on the vaccine platform. J Exp Med 2010; 207:1421–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]