

Abstract
Historically, most cellular processes have been studied in only 2 dimensions. While these studies have been informative about general cell signaling mechanisms, they neglect important cellular cues received from the structural and mechanical properties of the local microenvironment and extracellular matrix (ECM). To understand how cells interact within a physiological ECM, it is important to study them in the context of 3 dimensional assays. Cell migration, cell differentiation, and cell proliferation are only a few processes that have been shown to be impacted by local changes in the mechanical properties of a 3-dimensional ECM. Collagen I, a core fibrillar component of the ECM, is more than a simple structural element of a tissue. Under normal conditions, mechanical cues from the collagen network direct morphogenesis and maintain cellular structures. In diseased microenvironments, such as the tumor microenvironment, the collagen network is often dramatically remodeled, demonstrating altered composition, enhanced deposition and altered fiber organization. In breast cancer, the degree of fiber alignment is important, as an increase in aligned fibers perpendicular to the tumor boundary has been correlated to poorer patient prognosis1. Aligned collagen matrices result in increased dissemination of tumor cells via persistent migration2,3. The following is a simple protocol for embedding cells within a 3-dimensional, fibrillar collagen hydrogel. This protocol is readily adaptable to many platforms, and can reproducibly generate both aligned and random collagen matrices for investigation of cell migration, cell division, and other cellular processes in a tunable, 3-dimensional, physiological microenvironment.
Keywords: Bioengineering, Issue 111, Collagen, microfluidic channels, 3D matrices, ECM, collagen alignment, tumor microenvironment
Introduction
Many cellular processes have been extensively studied in 2 dimensions, thereby forming a collective knowledge of basic cell signaling mechanisms. These studies, however, neglect important cellular cues received from the structural and mechanical properties of the local cellular microenvironment and extracellular matrix (ECM). To better understand how cells interact within a physiological context, it is important to study them in 3-dimensional (3D) assays. The ECM for these 3D assays can either be cell-derived or reconstituted from purified proteins. Regardless of the source of the ECM, 3D matrix assays have proven to be invaluable for understanding how cells navigate and interact within the physiological world. For example, cells grown in 3D matrices display distinct modes of locomotion that depend on the mechanical nature of their surrounding ECM which are not observed in 2D experiments4-6. Moreover, cells cultured in 3D also have fewer and less pronounced stress fibers and focal adhesions than their counterparts grown on hard surfaces such as glass or plastic7.
The importance of contextual 3D assays is not limited to cell migration, however. Some other cell signaling events can only be investigated through the use of 3D assays. During tissue and cell differentiation, the stiffness of the extracellular environment and ECM provides signals that can influence morphogenic events. For example, mammary epithelial tubulogenesis only occurs in low stiffness 3D matrices, but not in stiff matrices nor on 2D substrata8,9. When cultured within stiff 3D matrices, these same epithelial cells take on an aberrant phenotype with increased proliferation and cell membrane protrusions driven through altered FAK and ERK signaling10. Many other signaling pathways and cellular processes are known to be similarly affected by the stiffness of the local cellular environment, and these signaling cascades highlight the importance of investigating signaling events and cellular phenotype in the context of appropriate local mechanical properties of a 3D ECM.
Collagen I is a particularly relevant protein to use for in vitro studies as it is the most abundant component of the ECM and is responsible for many of the mechanical properties of the cellular microenvironment. While it was originally thought of as merely a structural protein, its role is now known to be much more complex. Collagen fiber composition, architecture, orientation, density, and stiffness all provide a concentrated milieu of signaling information5. During the progression of certain diseases, such as chronic inflammation and tumorigenesis, the collagen network is dramatically remodeled2,11. More specifically in breast cancers, increased collagen deposition and tissue stiffness accompany and likely contribute to tumor progression. In these early tumors, the stiffened collagen network appears strained and more aligned, such that most of the fibers encapsulate the growing tumor2. As the tumor progresses, the collagen continues to reorganize, and regions of the fibrillar network become orientated perpendicular to the tumor boundary2,12. Perpendicular alignment serves as a prognostic biomarker where these patients have a poorer disease free progression and overall survival1. One explanation for this correlation is that the poor outcomes are a consequence of increased dissemination of tumor cells via persistent cell migration in aligned collagen networks3.
To understand how cells specifically respond to alignment and organization that is observed in tumor progression, it is necessary to generate both random and aligned 3D collagen matrices for experimentation. There are three basic methodologies to induce alignment within fibrillar networks. The first technique utilizes a strain-inducing device where the collagen between two points is contracted or stretched to generate alignment. Fibers parallel to the axis of force are pulled taut while fibers perpendicular to the axis are compressed and buckled. While strain-induced techniques typically offer superb alignment, this approach requires bulky equipment that is not easily adaptable to many platforms3,13. Alternatively, cell-induced strain can be created by placing localized plugs of cells that subsequently contract and align the collagen13. This method has the problem of being variable, as many parameters may be subject to change. The second method utilizes magnetic beads and a magnetic field during polymerization to induce collagen alignment13,14. Good results can be obtained from this method with unsophisticated equipment, but it does require the use of antibodies or some other method to magnetize the polymer. Therefore, it can be somewhat expensive to use, and the stiffness of the collagen gel is potentially modified by the increased connections in the network. Moreover, the magnetic beads used in this process are often autofluorescent, which is problematic for imaging experiments. Lastly, alignment can be generated by PDMS microfluidic channels3,15,16. In this method, collagen alignment is achieved by flowing polymerizing collagen through small microfluidic channels. These microfluidic channels can be made in a multitude of designs, and are easily adaptable to many platforms. Moreover, they are very economical as very small quantities of collagen and other reagents are used due to their diminutive sizes.
Provided here is a simple protocol for embedding cells within a 3-dimensional, fibrillar collagen hydrogel. In addition, a more advanced technique, wherein PDMS microfluidic channels are used to control the organization and alignment of the collagen matrix is also provided. This protocol is readily adaptable to many platforms, and can reproducibly generate both aligned and random collagen matrices for investigation of cell migration, cell division, and other cellular processes in a 3-dimensional, physiological microenvironment.
Protocol
1. Neutralization, Dilution and Polymerization of Collagen Solutions for 3D Investigation and Cellular Contraction Assays
On ice in sterile tissue culture hood, neutralize collagen (1:1) with sterile, ice-cold 100 mM HEPES in 2x PBS, pH 7.4, in a 15 ml conical tube. Mix thoroughly with plastic pipette until solution is homogenous and mixing swirls are no longer visible. Be careful not to introduce air bubbles during the mixing process. Store briefly on ice.
- Dilute neutralized collagen to the appropriate concentration with cell media (such as RPMI, DMEM, etc.).
- To calculate the amount of neutralized collagen required to make up desired collagen concentration, use the equation N = (D*V)/(S/2), where N is amount of neutralized collagen required, D is the desired collagen concentration, V is the final volume of the desired collagen concentration, and S is the starting stock collagen concentration.
- Bring up the amount of neutralized collagen to volume by the addition of a cell and cell media solution. Mix thoroughly and store on ice until ready to cast.
- Example: For a 2 mg/ml gel in a 1 ml gel volume (typical volume for a gel cast in 6 well plate or 50 mm glass bottom dish), first multiply the desired concentration (2 mg/ml) by the desired final volume (1 ml). Take this number (2 mg) and divide it by half of the stock concentration listed on bottle (9.49 mg/ml). In this case, 0.42 ml of the neutralized collagen are diluted with 0.58 ml of media/cell mixture.
Pipet the ice-cold collagen/cell/media mixture into either a non-tissue culture treated 6-well plate or a 50 mm glass bottom dish. Use pipet tip to evenly spread out the solution. NOTE: It is important to use a non-tissue culture treated plate to minimize the attachment or growth of the cells outside of the collagen gel.
Allow to polymerize at room temperature for approximately 10 - 15 min. The gels should turn opaque upon polymerization.
After it has turned opaque, move plate or dish to 37 ˚C for additional 45 - 60 min to finish polymerization.
After 45 - 60 min, add 2 - 3 ml of media and release gels from sides of the well by running a p200 pipet tip around perimeter of well or dish. Swirl dish gently to release the gel. The collagen gel should be floating in media.
2. Western Blotting, Cell Morphogenesis and Gel Gontractility Assay
To assess protein levels, morphology or cellular contractility in relation to ECM stiffness, begin by pouring collagen gels, according to 1.1 - 1.6, seeded with cells for a 7 - 10 day assay. Determine each cell line seeding rate empirically depending on growth rate and confluency. For a 10 day assay, the seeding density can range from 20,000 - 100,000 cells/gel.
To measure cell contractility, measure the gel diameter by using a ruler or a camera every 24 hr or at the appropriate time interval. Note: Additionally, corresponding images collected from a microscope can be examined for morphological features characteristic of the cell line seeded in the gel, such as acini-like structures, epithelial tubules, cellular protrusions, and lameliapodia.
- To assess protein levels, lyse the gels in RIPA buffer and process for Western blot analysis of proteins of interest as described in Wozniak et al. 17 and Gallagher et al. 18.
- IMPORTANT: In comparisons of contractility between cell lines, normalize contractility to total DNA, which can be extracted from the gels as outlined by Lui, et al.19, or to an unchanging, housekeeping protein (histones, GAPDH, etc.) by Western blot analysis, as described in Wozniak et al.17 and Gallagher et al.18. If counting cells within the gel using a hemocytometer, make sure to normalize cell number to total gel area as the contracting gel will concentrate cells.
Feed gels every 3 - 4 days by removing 1 ml of media and replacing it with 1 ml of fresh media. Make sure to feed gels after the measurement is taken as the addition of fresh media/serum will cause a spike in contractility.
3. Generation of PDMS Microchannels for Collagen Fiber Alignment
Note: To generate aligned collagen matrices, a mold for PDMS microchannels (Figure 2A) requires a SU-8 silicon master made via soft-lithography15.
To make PDMS channels, mix PDMS thoroughly in a disposable cup with a craft stick. For a 6 inch silicone master, mix 20 g of elastomer base with 2 g of curing agent.
De-gas the PDMS mixture by placing the disposable cup in a vacuum chamber under a vacuum pressure of 550 mm Hg. De-gas for 1 - 1.5 hr.
While de-gassing the PDMS, prepare the silicone master for pouring. Prepare by placing a clean sheet of transparency film onto a hotplate followed by the silicone master. Ensure that the microchannel mold faces up. NOTE: During PDMS casting and curing, the silicone master will be sandwiched between two sheets of transparency film.
After 1 - 1.5 hr, remove the de-gassed PDMS from the vacuum chamber, and slowly pour over the silicone master. IMPORTANT: Avoid air bubbles. Continue to pour in center of master, and allow gravity to spread it evenly. NOTE: The PDMS drop does not need to extend all the way to the edge of the master.
After PDMS has been poured onto the master, apply a second sheet of transparency film on top of the silicone master and PDMS. Carefully, roll the second transparency sheet down on top of the PDMS/silicone master to avoid air bubbles. Do not rush. PDMS should be now evenly spread over master.
Gently place a rubber sheet on top of the transparency, followed by a 1/8" acrylic sheet.
Add three 10 lb weights on top of the acrylic sheet. Initially, the weights will "float". Allow them to settle and stabilize before proceeding further.
Set hotplate temperature to 70 ˚C and cure PDMS for 4 hr. Allow to cool for minimum of 1 additional hr before disturbing.
Carefully peel top sheet of transparency film from the wafer, and remove the channels with a forceps.
Store in dust-free dish until ready to use.
4. Prepping PDMS Microchannels for Use
Place channels upside down on new, clean transparency film. Clean all ports (Figure 2b) using circular motion with a sharp forceps. Remove any fragments of PDMS.
Clean channels by using a piece of packing tape as a tack cloth. Apply tape to surface of bench (sticky side up), and then set channel on top. Press down on channels with round end of forceps to ensure good contact. Remove and repeat on both sides until visible debris is removed.
Transfer cleaned, prepped PDMS channels into a 50 ml conical with 70% EtOH. Vortex at maximum speed for 30 sec. Discard EtOH and replace with fresh 70% EtOH. Store in 70% EtOH until ready to use.
In tissue culture hood and using aseptic techniques, transfer the PDMS channels to a clean and sterile cover glass or glass bottom dishes. Put the channel side up and UV treat until EtOH has evaporated.
Once dry, flip the PDMS so the channels are facing down toward coverglass. Press the PDMS channel down on the glass to make a good seal. Add a patch of sterile PDMS to cover/close the center port (port B, Figure 2b). Allow to thoroughly dry before proceeding.
Pre-coat the inside of the channel with 10 µg/ml collagen in sterile water. To coat, place a 100 µl droplet on the top of a channel, and draw through with vacuum. After 1 hr at 37 ˚C, transfer channels filled with collagen coating solution to a refrigerator. Chill for approximately 15 - 30 min.
Remove collagen coating solution with aspirator or pipet, and begin collagen preparation.
5. Collagen Preparation for Use in Microchannels
On ice, neutralize collagen (1:1) with ice-cold 100 mM HEPES in 2x PBS, pH 7.4. Mix thoroughly until homogenous (For more details, see section 1.1).
Dilute neutralized collagen to the appropriate concentrations with cell media (For more details, see section 1.2). Incubate for 15 min on ice.
At the same time, chill mounted channels on ice. NOTE: The goal is to have all the components for the channel process at 4 ˚C or below. Collagen nucleation temperature and time are key parameters to the polymerization process, and may be the starting point for further optimization, if needed.
Count cells with a hemacytometer and resuspend at the appropriate seeding density at this time. For ease of calculations, resuspend to 1 - 3 million cells/ml. (For more details, also see section 2.1).
Once cells have been counted and 15 min have passed, proceed to collagen pouring.
6. Pouring Aligned and Random Collagen Microchannels
- Before drawing collagen through channels, adjust and set vacuum pressure with an inline vacuum regulator. Vacuum pressure provides the force to induce and control the rate of collagen flow, which determines the degree of alignment.
- For random or unaligned matrices, use a wide channel (3mm wide x 200 µm tall) with a vacuum pressure of 10 mm Hg or less.
- For aligned matrices, use narrow channel (1mm wide x 200 µm tall) with a vacuum pressure of 60 mm Hg or greater.
Remove mounted channel from ice and place on clean, sanitized surface of laminar flow hood.
Work quickly and load 120 - 150 µl of neutralized collagen into port A (Figure 2b).
Draw collagen through the channel by placing a 25 ml pipet attached to the vacuum line over port c (Figure 2b). Draw collagen through in a single, uniform motion. IMPORTANT: For random or unaligned matrices, draw collagen slowly across channel (approximately 0.5 - 1 mm per second), and stop once it reaches the end. For aligned matrices, draw the collagen across more rapidly, but take care to avoid air bubbles.
Carefully remove excess collagen from the port region with p200 pipetman or aspirator.
Place sterile PDMS patches over both ports A and C. All ports should now be covered.
After 2 - 3 mins, remove center PDMS patch (port B) and add 2 - 3 μl of cells (5 - 10 thousand cells) into the center port. Allow to partially polymerize (turn opaque) at room temperature for another 10 - 15 min.
After 10 - 15 min, transfer to 37 ˚C for additional 15 - 30 min to finish polymerization. Remove PDMS covers and add media to completely cover the channel and culture cells as needed. Cells can be fed by removing a ml of old media, and replacing it with a ml of new media.
Representative Results
While 3D assays can be done within the same stiffness of collagen gel, varying the gel stiffness can be used to determine how the cells will respond to mechanical changes in their cellular microenvironment. A stiff collagen hydrogel is defined as a gel where the embedded cells are unable to locally contract the surrounding collagen. The intrinsic contractility of different cell types is unique, and thus it is best to begin with a simple contractility curve to establish the collagen concentration that will be sensed as compliant and stiff.
Begin by holding the cell numbers constant and increasing the concentration of the collagen gel. The stiffness of the gel will scale exponentially with concentration, as shown previously10,20. A representative example of a contractility assay is shown in Figure 1. Here, 50,000 4T1 cells are embedded in either a 2 mg/ml or 3.5 mg/ml collagen gel for 5 days. The 4T1 cell line is a highly contractile, metastatic cancer line, but is only able to contract the high density gel by approximately 10% (Figure 1a, c). This low level of contraction is typical for a stiff gel classification. Comparable low density gels (2 mg/ml, Figure 1b, c) are contracted 57% (n = 4) during the same time frame. It is important to note that increasing the amount of collagen ligand can impact cell signaling pathways, therefore a second means to vary the stiffness is to hold the collagen concentration constant and compare gels that are restrained (stiff) by being left attached to the bottom of the dish or contractile (compliant) because they have been released to float freely in the cell medium. This can serve as a good control to determine whether changes in cellular signaling result from increased collagen ligand or the stiffness of the matrix.
While collagen gel contraction assays are straightforward, there are a few items to be aware of when using these types of assays. For one, under high density conditions it is important to verify that there is not a loss of cell viability. We have observed that there is a ceiling for the stiffness of the microenvironment that a cell will tolerate, and have observed increased apoptosis or cell death in some cell lines, which can cause subsequent results to be misinterpreted. There should be no appreciable loss of cell viability between high and low stiffness conditions. This can be determined by Western blotting for caspase activity or some other assay for measuring cell viability. Secondly, collagen gels can also be contracted too much thereby creating a stiffer environment, and diminishing the differences between high and low stiffness conditions. We recommend not exceeding contractions of 60 - 70%. To maximize reproducibility, it is important to optimize the timeframe of the experiment, the initial cell seeding density, and the stiffness of the matrix to the contractility of the cell type in use.
Once mastery of embedding cells within 3D matrices is achieved, assays can be modified to alter the organization and orientation of the collagen fibers. This is achieved by the use of PDMS microfluidic channels (Figure 2). For the purpose of this manuscript, two similar conformations were used, a narrow and a wide channel, with 3 openings or ports (Figure 2b). In a typical experiment to determine the cellular response to random compared to aligned matrices, unpolymerized collagen is drawn through the microfluidic channel from port A to port C with cells being added to port B. The degree of collagen alignment is modulated by the rate of collagen flow such that higher collagen flow rates yield better alignment. The use of narrower channels coupled with higher vacuum pressures enhances the alignment of the collagen network, while a wider channel used in conjunction with low vacuum pressures generates random matrices. The organization of the fibrillar network can be visualized by second harmonic generation, as shown in representative images (Figure 2c, d). In these images, the collagen fibers (indicated by the arrowheads and pseudocolored in purple) align more consistently with the axis of collagen flow in the narrow channels (Figure 2d). The collagen fibers in wide channels have a variety of orientations, and have a more random distribution (Figure 2c).
The difference between wide and narrow channels is discernable to the human eye, but can also be analyzed via software developed specifically for fiber analysis. CT-FIRE, an open source and freely available software, utilizes a curvelet-based algorithm for pre-processing followed by a fiber extraction algorithm to quantify fiber angle, thickness, length, and straightness21. For each condition, more than 500 images were randomly sampled from different locations within the channels, and more than 125,000 fibers were extracted and analyzed by CT-FIRE. The subsequent fiber angle histogram generated for the narrow channels (Figure 2f) shows a clear enrichment of fibers oriented parallel to the direction of flow. In contrast, wide channels have a more equivalent fiber angle distribution (Figure 2e) which is consistent with a more random matrix.
Importantly, the use of microfluidic devices, as a technique to control the organization and orientation of collagen fibers, allows for assessment of various cellular responses to altered matrix conditions. For example, Sung et al. determined that viability of human mammary fibrobasts is dependent on collagen fiber.15 Additionally, recent work from our laboratory demonstrates that breast carcinoma cells migrate more persistently on aligned collagen fibers compared to random collagen matrices.3 These results help to better define the role of the microenvironment within the context of mammary biology and breast cancer. Moving forward, the ability to precisely control the 3D cellular microenvironment will allow for continued investigation of important cellular responses within the context of many different disease states.
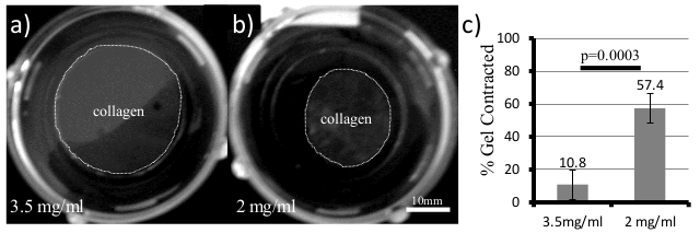 Figure 1. 3D Collagen Gels for Measuring Cell Contractility and Cellular Responses to ECM Stiffness. 4T1 cells were plated in both(a) 3.5 mg/ml and(b) 2 mg/ml collagen gels and allowed to grow for 5 days. Over time, the collagen gel (Gray area within dotted line) will contract and the diameter is measured and quantified in (c, Student's t-test, error bars +/- SD).Please click here to view a larger version of this figure.
Figure 1. 3D Collagen Gels for Measuring Cell Contractility and Cellular Responses to ECM Stiffness. 4T1 cells were plated in both(a) 3.5 mg/ml and(b) 2 mg/ml collagen gels and allowed to grow for 5 days. Over time, the collagen gel (Gray area within dotted line) will contract and the diameter is measured and quantified in (c, Student's t-test, error bars +/- SD).Please click here to view a larger version of this figure.
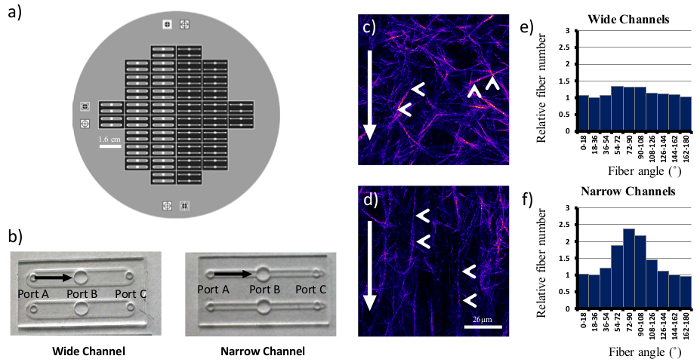 Figure 2. Generating Random and Aligned Collagen Matrices Using PDMS Microchannels. A six inch silicon wafer (a) was used as a template for both wide and narrow PDMS microchannels (b). Identical collagen concentrations were drawn through both types of channels, and individual fibers (arrowheads, c, d) align with the direction of flow (arrows) in narrow microchannels(d). Wide microchannels yielded more random matrices. Fiber angles with respect to the bottom of image were counted and shown in (e) and (f). Scale bar = 26 µm. Please click here to view a larger version of this figure.
Figure 2. Generating Random and Aligned Collagen Matrices Using PDMS Microchannels. A six inch silicon wafer (a) was used as a template for both wide and narrow PDMS microchannels (b). Identical collagen concentrations were drawn through both types of channels, and individual fibers (arrowheads, c, d) align with the direction of flow (arrows) in narrow microchannels(d). Wide microchannels yielded more random matrices. Fiber angles with respect to the bottom of image were counted and shown in (e) and (f). Scale bar = 26 µm. Please click here to view a larger version of this figure.
Discussion
3D collagen gels are a valuable addition to our toolbox to understand how cells interpret and respond to their local microenvironment. This manuscript has provided a very basic protocol for embedding cells within a 3D collagen matrix, and to reproducibly generate matrices with random or aligned collagen fibers. Both protocols work as adaptable platforms where different collagen isoforms, crosslinkers, or other matrix proteins could potentially be added at the time of polymerization. It is also easy to modify the platforms to assess different timeframes and endpoints for gene expression and protein production. Imaging, assessing protein and RNA levels, and reading metabolic profiles are all very compatible with these 3D techniques.
As is important with all techniques, it is important to assess the quality of the gel and assay. A sign of a good quality collagen gel is one that can handle manual manipulation. For example, a 1 ml [1 mg/ml] collagen gel poured into a six well plate should be easily picked up with a forceps and manipulated. If the gel tears or fragments, the quality of the gel is suboptimal, and steps need to be taken to improve the collagen polymerization. Temperature and pH are the most important parameters required for good fiber formation and gelation. Begin the troubleshooting process by making sure all components are kept on ice until ready for gelation to occur. It is also very important to make sure that the neutralization buffer and collagen stock solution are being mixed thoroughly. Next, verify that the pH of the neutralization buffer is at pH 7.4. A HEPES buffered PBS is recommended because it is very compatible to tissue culture, has a broad buffering range, and stock solutions tend to be very stable at 4 ˚C for long periods of time. It also has the added benefit of producing longer, more curved fibers which are more similar to the physiological state15. NaOH is another commonly used neutralizing solution, but needs to be titered more closely. It also appears to have the effect of producing shorter, thicker fibers15. Regardless, the pH of the neutralized collagen should be at a pH of 7.4. If all those conditions are being satisfied, and the integrity of the gels is still not satisfactory, then the problem lies with the stock collagen solution. Although the stock collagen solution has a listed shelf life of 6 months at 4 ˚C, we have found that it is generally shorter than commercial sources claim when used for 3D gels. Discard the old bottle, and order from a new lot. It is not unusual for the quality of collagen from the manufacturer to vary significantly from lot to lot.
In addition to identifying quality collagen gels, it is also important to identify the steps that are critical for the generation of good alignment within the PDMS microchannel. The underlying principle of this technique is that the rate of collagen flow through the microchannel subsequently induces filament alignment. Therefore, the most critical step in controlling the degree of fiber alignment is proper modulation of the vacuum pressure. For random collagen alignment in a microchannel, the vacuum pressure must be low, and the collagen must flow slowly down the channel. If the collagen flows through too quickly, the fibers will begin to align. A wider channel makes this proposition easier. Conversely, for good collagen alignment, higher vacuum pressures must be used and the collagen solution must be pulled through more quickly. A second critical step in the generation of aligned matrices is the temperature. It is very important to make sure all components and solutions are chilled on ice prior to use. If the channels are not chilled prior to use, the collagen in the channels will polymerize too quickly, and will result in thinner, smaller fibers. If the neutralized collagen is chilled at 4 ˚C and not on ice, the enhanced nucleation will give rise to larger fiber diameters which are more difficult to align by flow. It is also important to chill the microchannel itself. Without prior chilling, the small volume of collagen will warm very rapidly yielding poor results.
The collagen microchannels are a very useful technique, but there are certain limitations. For one, the alignment in a flow-induced microchannel is never perfect, and will never equal a strain-induced alignment technique. There will always be some fibers oriented perpendicular to the direction of flow, but the reproducibility, adaptability, and ease of use outweigh the small variations in alignment within the channel. More importantly, cells have been shown to effectively sense small changes in alignment, and respond with more persistent migration in these channels3,15. The cells will also enhance the alignment of the channel as their numbers increase and they begin to migrate. If greater alignment is required, the PDMS microchannels can be easily modified to narrower dimensions or to have small constrictions to enhance alignment. Both have been shown to be effective, but have other associated trade-offs16. Another limitation to the microchannel technique is that the PDMS channel has little permeability to drugs or fluorescent dyes. This means that these compounds need to diffuse down the channel from the ports, which is surprisingly slow, and can pose difficulty in washing out treatments.
Although there are certain limitations to this microchannel platform, it does offer several advantages over existing techniques. It is more experimentally accessible as it does not require bulky instruments3,13 or small populations of cells for generating alignment. It also does not require exogenous magnetic beads13,14 which tend to be autofluorescent and could artificially crosslink the collagen network. The collagen microchannel is also simply more cost-effective and economical. In addition to the current advantages, it also has numerous benefits that could be leveraged in future applications. For one, the collagen microchannel can be designed or modified in almost any configuration or experiment. Advanced designs allow the channels to create hollow collagen tubes or 3D structures to mimic the architecture of simplified organs or tumors22. Moreover, the channels can be made of materials other than PDMS. This potentially could convey different mechanical properties to the embedded collagen networks or matrices. Lastly, the small amount of reagents used for this assay might also make it attractive and economical for smaller high throughput screens.
Disclosures
The authors have nothing to disclose
Acknowledgments
The authors would like to acknowledge grant numbers UO1CA143069, R01CA142833, R01CA114462, RO1CA179556, T32-AG000213-24, and T32-GM008692-18 for funding this work. We also acknowledge Jeremy Bredfelt and Yuming Liu of LOCI for the development of and assistance with the CT-FIRE analysis.
References
- Conklin MW, et al. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am J Pathol. 2011;178:1221–1232. doi: 10.1016/j.ajpath.2010.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, et al. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006;4:38. doi: 10.1186/1741-7015-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riching KM, et al. 3D collagen alignment limits protrusions to enhance breast cancer cell persistence. Biophys J. 2014;107:2546–2558. doi: 10.1016/j.bpj.2014.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Even-Ram S, Yamada KM. Cell migration in 3D matrix. Curr Opin Cell Biol. 2005;17:524–532. doi: 10.1016/j.ceb.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Friedl P, Wolf K. Plasticity of cell migration: a multiscale tuning model. J Cell Biol. 2010;188:11–19. doi: 10.1083/jcb.200909003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie RJ, Gavara N, Chadwick RS, Yamada KM. Nonpolarized signaling reveals two distinct modes of 3D cell migration. J Cell Biol. 2012;197:439–455. doi: 10.1083/jcb.201201124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukierman E, Pankov R, Yamada KM. Cell interactions with three-dimensional matrices. Curr Opin Cell Biol. 2002;14:633–639. doi: 10.1016/s0955-0674(02)00364-2. [DOI] [PubMed] [Google Scholar]
- Wozniak MA, Desai R, Solski PA, Der CJ, Keely PJ. ROCK-generated contractility regulates breast epithelial cell differentiation in response to the physical properties of a three-dimensional collagen matrix. J Cell Biol. 2003;163:583–595. doi: 10.1083/jcb.200305010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszek MJ, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Provenzano PP, Inman DR, Eliceiri KW, Keely PJ. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene. 2009;28:4326–4343. doi: 10.1038/onc.2009.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tlsty TD, Coussens LM. Tumor stroma and regulation of cancer development. Annu Rev Pathol. 2006;1:119–150. doi: 10.1146/annurev.pathol.1.110304.100224. [DOI] [PubMed] [Google Scholar]
- Provenzano PP, et al. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008;6:11. doi: 10.1186/1741-7015-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Inman DR, Eliceiri KW, Trier SM, Keely PJ. Contact guidance mediated three-dimensional cell migration is regulated by Rho/ROCK-dependent matrix reorganization. Biophys J. 2008;95:5374–5384. doi: 10.1529/biophysj.108.133116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Kaufman LJ. Flow and magnetic field induced collagen alignment. Biomaterials. 2007;28:1105–1114. doi: 10.1016/j.biomaterials.2006.10.010. [DOI] [PubMed] [Google Scholar]
- Sung KE, et al. Control of 3-dimensional collagen matrix polymerization for reproducible human mammary fibroblast cell culture in microfluidic devices. Biomaterials. 2009;30:4833–4841. doi: 10.1016/j.biomaterials.2009.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P, Lin R, Moon J, Lee LP. Microfluidic alignment of collagen fibers for in vitro cell culture. Biomed Microdevices. 2006;8:35–41. doi: 10.1007/s10544-006-6380-z. [DOI] [PubMed] [Google Scholar]
- Wozniak MA, Keely PJ. Use of three-dimensional collagen gels to study mechanotransduction in T47D breast epithelial cells. Biol Proced Online. 2005;7:144–161. doi: 10.1251/bpo112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher S, Winston SE, Fuller SA, Hurrell JG. Ausubel FM, et al., editors. Immunoblotting and immunodetection. Current protocols in molecular biology. 2008. p. 18. Chapter 10, Unit 10. [DOI] [PubMed]
- Liu X, Harada S. Ausubel FM, et al., editors. DNA isolation from mammalian samples. Current protocols in molecular biology. 2013. p. 14. Chapter 2, Unit 2. [DOI] [PubMed]
- Roeder BA, Kokini K, Sturgis JE, Robinson JP, Voytik-Harbin SL. Tensile mechanical properties of three-dimensional type I collagen extracellular matrices with varied microstructure. J Biomech Eng. 2002;124:214–222. doi: 10.1115/1.1449904. [DOI] [PubMed] [Google Scholar]
- Bredfeldt JS, et al. Computational segmentation of collagen fibers from second-harmonic generation images of breast cancer. J Biomed Opt. 2014;19:16007. doi: 10.1117/1.JBO.19.1.016007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischel LL, Beebe DJ, Sung KE. Microfluidic model of ductal carcinoma in situ with 3D, organotypic structure. BMC Cancer. 2015;15:12. doi: 10.1186/s12885-015-1007-5. [DOI] [PMC free article] [PubMed] [Google Scholar]