

Abstract
The understanding of developmental processes at the molecular level requires insights into transcriptional regulation, and thus the transcriptome, at the level of individual cell types. While the methods described here are generally applicable to a wide range of species and cell types, our research focuses on plant reproduction. Plant cultivation and seed production is of crucial importance for human and animal nutrition. A detailed understanding of the regulatory networks that govern the formation of the reproductive lineage (germline) and ultimately of seeds is a precondition for the targeted manipulation of plant reproduction. In particular, the engineering of apomixis (asexual reproduction through seeds) into crop plants promises great improvements, as it leads to the formation of clonal seeds that are genetically identical to the mother plant. Consequently, the cell types of the female germline are of major importance for the understanding and engineering of apomixis. However, as the corresponding cells are deeply embedded within the floral tissues, they are very difficult to access for experimental analyses, including cell-type specific transcriptomics. To overcome this limitation, sections of individual cells can be isolated by laser-assisted microdissection (LAM). While LAM in combination with transcriptional profiling allows the identification of genes and pathways active in any cell type with high specificity, establishing a suitable protocol can be challenging. Specifically, the quality of RNA obtained after LAM can be compromised, especially when small, single cells are targeted. To circumvent this problem, we have established a workflow for LAM that reproducibly results in high RNA quality that is well suitable for transcriptomics, as exemplified here by the isolation of cells of the female germline in apomictic Boechera. In this protocol, procedures are described for tissue preparation and LAM, also with regard to RNA extraction and quality control.
Keywords: Plant Biology, Issue 111, Boechera, cell type-specific, female germline, gametophyte, laser-assisted microdissection, plant reproduction, RNA quality, transcriptome
Introduction
In transcriptional studies done at the tissue level, the transcriptomes of highly specialized but rare cell types are often masked by the more abundant surrounding cells. An example for such highly specialized cell types are the cells of the female reproductive lineage (germline) in plants. The female germline is specified within developing ovules, the precursors of seeds inside the gynoecium of the flower 1,2. The megaspore mother cell (MMC) is the first cell of the female germline. It undergoes meiosis to form a tetrad of reduced megaspores. Typically, only one of these megaspores survives and divides mitotically without cytokinesis, i.e., in a syncytium. These mitoses are followed by cellularization to form the mature gametophyte, which typically consists of four cell types: three antipodals, two synergid cells, the egg, and the central cell. The egg and central cells are the female gametes that get fertilized by two sperm cells during double fertilization to give rise to the embryo and endosperm of the developing seed 1,2. In the sexual model system Arabidopsis thaliana, only ~ 50 seeds develop per flower while about 50 - 80 seeds develop per flower in the closely related genus Boechera. Thus, the female germline consists of only a few highly specialized cell types, making it an excellent model to study developmental processes, such as cell specification and differentiation.
Moreover, insights into the gene regulatory processes governing plant reproduction can be of applied value. In plants, both sexual and asexual reproduction through seeds (apomixis) can occur. While sexual reproduction generates genetic diversity in a population, apomixis leads to the formation of clonal offspring that is genetically identical to the mother plant. Therefore, apomixis has great potential for applications in agriculture and seed production, as even complex maternal genotypes can be maintained unaltered over several generations 3,4,5. Because apomixis does not naturally occur in any major crop species, the engineering of apomixis in crops is of great interest 3,4,5. However, this long-term goal is difficult to achieve because the underlying genetic and molecular basis of apomixis is not understood in sufficient detail 6.
To gain insights into the transcriptional basis governing apomictic reproduction, cell type-specific transcriptional profiling using laser-assisted microdissection (LAM) and next generation sequencing (NGS) represents a very powerful approach 7,8. LAM has first been established for animal and biomedical research. In the past few years LAM has also been applied to plant biology 6,9,10. In contrast to other methods allowing profiling of individual cell and tissue types, LAM does not require the generation of marker lines 6,9,10. Therefore, it can be applied to any cell or tissue type without prior molecular knowledge. Another advantage of LAM is that it can be applied to any cell type as long as the cell can be recognized in dry sections based on position and/or structural features. LAM has the additional advantage that fixed tissues are used, which prevents changes of the transcriptional profile during processing.
The tissue of interest, e.g., floral tissue, is fixed in a non-crosslinking fixative prior to embedding in paraffin wax. Embedding in paraffin wax can be done manually, following established protocols 9,11. However, the use of an automated tissue processor for dehydration and infiltration with the wax generally results in higher reproducibility in terms of the conservation of RNA quality and tissue morphology. The alternative strategy of embedding tissues in resin has also been successfully used for cell type-specific analyses by LAM 8. However, the use of an automated tissue processor for embedding in wax is very time efficient, as many samples can be processed at once requiring a minimum of hands-on time. While typically no significant loss of RNA quality occurs during fixation and embedding, the preparation of thin sections with the microtome and, in particular, the mounting on the frameslides used for LAM remains a critical step for preservation of RNA quality. This has previously been noted and the use of a tape transfer system has been described to result in better RNA quality at this step 12. However, this adds an additional time-consuming step during preparation of the slides and also requires special equipment. The optimized protocol described below reproducibly produces RNA that is of sufficient quality for transcriptional profiling with GeneCHIPs and Next Generation Sequencing (NGS) approaches 7,11,13,14. In addition, with the laser microdissection microscope used, a high purity of the isolated cell types is routinely produced 7,11,13,14.
The genus Boechera is an excellent model system for studying the key steps of apomictic reproduction. In Boechera, a variety of different sexual and apomictic accessions have been identified and can be used for comparative analyses 15,16,17. In a comparison of cell type-specific transcriptomes of cells from the female germline from sexual Arabidopsis and apomictic Boechera, we identified genes and pathways that are differentially expressed, thereby identifying new aspects of the regulatory processes governing apomixis 7. In addition, this study verified the suitability of LAM for cell type-specific transcriptional analyses of small and rare cell types. We have already used this protocol for the analysis of different cell types in a variety of plant species, but species- and tissue-specific modifications to the protocol may be required in certain cases.
Protocol
Note: This protocol describes tissue preparation, laser assisted microdissection, and RNA extraction for transcriptional profiling. Always use gloves throughout all steps of the protocol. Study and consider the safety instructions for each chemical used. In particular, keep in mind that Xylol is harmful and can penetrate gloves and that methanol is toxic. For all instruments used, please refer to user manuals accordingly.
1. Removal of RNAse Activity from Glassware and Other Equipment
Prior to use, wrap any glassware in aluminum foil prior to baking for ~ 8 hr at 180 °C to ensure RNAse free quality.
Treat any metal surfaces (e.g., pair of tweezers) as well as the work bench and the heating plate with an appropriate RNAse decontamination solution. Afterwards, wash the surfaces with RNAse-free water to remove the decontamination solution. Subsequently, clean the surfaces further with 70% EtOH.
Use all plastic consumables (e.g., tubes) brand-new without any pretreatment. If available, use plastic consumables that are certified to be RNAse free.
2. Tissue Fixation
Prepare 10 ml of farmer's fixative (3:1; v/v ethanol:acetic acid) in a fresh 15 ml tube on ice. (Always prepare fresh Farmer's fixative before use). As an alternative, the non-crosslinking fixative ethanol:acetic acid (5:1; v/v) can be used 18.
- Use a fine pair of tweezers to collect buds at the developmental stage of interest. Immediately submerge buds in the ice-cold fixative. Use 10 ml of fixative for the fixation of ~ 20 buds or flowers from Arabidopsis or Boechera. Note: When using plant species with larger flowers it is recommended to dissect the flowers with a razor blade to generate smaller pieces prior to fixation.
- In order to obtain mature gametophytes, emasculate the 2 - 3 largest flower buds of the inflorescence 2 days prior to harvest.
- Fill the bottom of an exsiccator with ice. Open the tubes containing the samples, e.g., flowers, and place them on ice, either by directly putting them into the ice in a way that they cannot fall over or by using an appropriate holder. Vacuum infiltrate for 15 min prior to release of the vacuum.
- Repeat the vacuum infiltration once for 15 min.
Release the vacuum and store overnight (~ 12 hr) on ice. Cover the ice bucket with a lid or aluminum foil and store the bucket in a 4 °C room or refrigerator in order to prevent the ice from thawing. Note: Prolonging the fixation time is not recommended.
3. Tissue Embedding, Thin Sectioning, and Mounting on Slides for LAM
- Tissue embedding.
- Exchange the fixative to 70% ethanol prepared with pure ethanol and RNAse free water. Leave the samples on ice until further processing. Start embedding of the samples the same day. In case no tissue-processing machine is available, proceed to manual embedding as described elsewhere 9,11 and continue with step 3.1.11.
- Gently transfer the samples to tissue embedding cassettes with a pair of tweezers. Firmly close the cassettes. CRITICAL STEP: Avoid even partial drying of the sample during this step. Avoid squeezing of samples with the tweezers. Note: To label the cassettes a pencil should be used, ideally by writing on the inclined surface of the cassette.
- Store the embedding cassette loaded with the buds or flowers in 70% ethanol until all material is transferred to cassettes and the embedding machine can be loaded. For this purpose, use a glass staining trough filled with 70% ethanol.
- Remove the basket of the tissue processor from the retort of the machine and transfer the embedding cassettes to the basket with the inclined surfaces facing the top and in the same direction. CRITICAL STEP: Perform this quickly to prevent any drying of the tissues. If processing many embedding cassettes at once, place the basket into an appropriate RNAse free container filled with 70% ethanol prior to loading the samples.
- Put the lid on the basket and put the basket back into the retort. Close the retort. Note: The tissue processor automatically dehydrates the samples, changes the solvent to Xylol, and does the infiltration with melted paraffin wax. Typically, this is done overnight.
- Start the program of the tissue processor. Recommended standard settings are 1 hr 70% EtOH, three times 1 hr 90% EtOH, three times 1 hr 100% EtOH, two times 1 hr 100% Xylol, one time 1 hr 15 min 100% Xylol at room temperature, followed by infiltration with paraffin wax two times for 1 hr and one time for 3 hr at 56 °C 11. Note: To ensure that the blocking station is ready with the wax melted, switch the machine on several hours before start or use the timer function to select the approximate starting time. Note: In case the tissue cannot be processed immediately afterwards, longer incubation times in paraffin wax at 56 °C are possible instead of the 3 hr at 56 °C. Refer to the user manual of the instrument. Typically, the cassettes with tissues will automatically remain in melted wax until the "empty retort" command is approved. If the wax is not melted when starting the tissue processor, the retort is filled with 70% ethanol and the program remains on hold until ready to start.
- Empty the retort and immediately transfer the samples to a paraffin wax bath at 56 °C in a blocking station. Remove the lid from the basket and cover the paraffin bath of the blocking station with its lid.
- Pile as many fresh balancing trays on the surface of the blocking station heated to 56 °C as there are cassettes in the basket. Note: An ethanol-resistant permanent marker can be used to label the bottom of the balancing trays. Acetone-resistant permanent markers are not recommended.
- Using the blocking station, fill a pre-warmed fresh plastic balancing tray with melted paraffin wax at 56 °C. Lift the lid of the paraffin bath, pick up one cassette only at a time and transfer the samples from the cassettes to the liquid paraffin wax at 56 °C in the balancing tray using a pair of tweezers. CRITICAL STEP: Avoid any hardening of the wax surrounding the sample while handling the cassette by working quickly.
- Position all samples according to the needs using a preparation needle before hardening of the wax. Typically, several flowers are arranged in a balancing tray individually spaced about 1 cm in each direction.
- Repeat step 3.1.9 until all samples are transferred.
- Place the basket back to the tissue processor and clean the retort using an appropriate program (refer to user manual).
- Switch off tissue processor and blocking station. Continue with step 3.1.13.
- In case no tissue infiltration machine and no embedding station is available, use a heating oven to melt paraffin wax at 56 °C. On the bench, fill the melted wax in plastic balancing trays, transfer the samples to the wax, and use preparation needles to position the samples.
- Allow the paraffin wax to harden at room temperature for ~ 1 - 3 hr before storing the samples at 4 °C. Samples can subsequently be processed freshly or stored for up to several months.
- Preparation of thin sections and slides for LAM.
- On a light table illuminating the wax block from below, remove the paraffin wax with the samples from the surrounding plastic trays. Dissect out a squared wax block containing the tissue, e.g., a bud or flower, using an RNAse free razor blade. To this aim, always orient the samples towards the top of the wax block (also see Figure 1).
- Prepare wax blocks of all samples that should be used for LAM at a time.
- Mount the blocks to process embedding cassettes filled with hardened paraffin wax: melt a small piece or droplet of paraffin wax on the tip of an appropriate spatula using an Ethanol lamp and use the hot wax to fix the block on the support (with the embedded tissues on top).
- Allow the wax to harden for at least 20 min at 4 °C.
- Remove surplus wax surrounding the sample with a razor blade on the light table. Make sure to keep a square surface with parallel edges (see Figure 1).
- Set the heating plate to 42 °C.
- Safely mount the samples to the microtome and prepare 6 - 10 µm thick sections. Refer to the manufacturer's instructions for using the microtome. Note: While thinner sections will often give better structural resolution, larger amount of RNA of higher quality can be obtained with thicker sections. For the cells of Boechera mature gametophytes we use 7 µm as the default. Moving rather slowly with the samples over the microtome knife often results in better histology.
- Always transfer the paraffin ribbons at a length of approximately 10 - 15 cm into a plastic box with a black cardboard at the bottom before positioning them in LAM slides.
- If possible, pre-select the parts of the paraffin strips containing the cell- or tissue types of interest, e.g., ovules, under a dissecting-scope and discard the rest.
- Preparation of the slides for LAM.
- Place the required amount of metal-framed slides (typically 5 - 10) with the flat surface on top on the heating plate.
- Pipet ~ 1 - 2 ml of RNAse free water on the plastic part of the slide. Using a razor blade, cut the paraffin ribbons in short pieces of about 4 - 5 cm long enough to span the plastic windows. Use a pair of tweezers and a preparation needle to ideally place the paraffin strips parallel to each other on the plastic windows of the slides. Carefully lift the slide at one end to remove the water and place the slides back.
- Alternatively, place the heating plate under a chemical hood. Apply a small volume of methanol to the plastic part of the slide just sufficient to wet the surface (this can cause a slight corrugation of the slides). Place the paraffin strips on the slides. Note: For reasons of toxicity, avoid use of methanol if possible. However, the use of methanol at this step does improve the RNA quality in case the quality achieved by mounting on water is not sufficient. It is worthwhile testing these alternatives at the start of a new project, as the RNA quality achieved varies with cell type and species under investigation.
- Dry the slides overnight on the heating plate at 42 °C for ~ 12 hr. Cover the heating plate with a plastic lid that does not touch the slides. Ensure that the plastic lid does not prevent the exchange of air. To this aim the lid can be placed on pipet boxes or similar to be lifted.
- Alternatively, reduce the drying time to a minimum of two hours or until the tissue appears totally dry. The reduction of time from slicing to collection may improve RNA quality.
- Under a chemical hood, de-wax the slides 2 times for 10 min in Xylol using appropriate slide holders. Make sure to fully submerge the plastic part of each slide only but to allow removal of the slide by touching the broader end of the metal frame that is kept dry.
- Allow the slides to dry for ~ 10 min under the chemical hood prior to LAM. Note: Slides not immediately processed can be placed back on the heating plate at 42 °C with the tissue facing up for up to a few hours. This will prevent any humidity from compromising RNA quality until further processing.
4. Laser-assisted Microdissection (LAM)
Note: The procedure for LAM will vary with the instrument used. Certain details of this protocol are adapted to a specific instrument and the technology of collecting the samples on a cap making use of electrostatic forces. In addition, details might vary even between different versions of the software. Please refer to manufacturer's instructions and user handbooks for a detailed description and specific instructions on instrument and software.
Switch on the devices for LAM (computer, microscope, laser control box).
Start the program steering the microscope and laser.
Switch on the laser by pressing the corresponding button on the control box.
Place a cap (0.5 ml, without diffusor) into the cap holder and position it in the instrument.
Support the LAM slide with a glass slide for microscopy. Ensure that the flat surface of the slide with the tissue attached faces the glass slide.
Place the slide in the instrument with the glass slide underneath.
Select the 4X Objective. In the program, select to move the cap to the "up" position and proceed to make a scan of the slide.
Search through the slide and identify the structures of interest. Pinpoint and save their positions on the slide to be able to move back to the exact position for the cutting step.
Select the appropriate objective (e.g. 40X or 60X).
If needed, adjust laser speed, focus, and laser power, and also calibrate the stage movement by referring to the user manual of the instrument. Once an account is set up for the specific tissue of interest, the settings can be reused.
- Verify the laser position by a single shot by pressing the "laser shot" button of the program, ideally in a part of the membrane without tissue.
- If required, calibrate the laser position by following the manufacturer's instructions for the instrument.
Verify the calibration of the objective by moving an obvious structure to all four corners of the software window on the monitor, keeping the right mouse button pressed. Check that the position of the hand with respect to the obvious structure is the same in all four corners. Otherwise calibrate the objectives following the software manual.
With the cap in the 'down' position, use the 'hand-pen' tool to mark the borders of the cell type or tissue of interest, e.g., the egg cell. Dissect the cell of interest with the laser by pressing 'cut'. Note: Often, sectioning needs to be repeated if the border around the cell was not entirely dissected at once. In this case, it is recommended to set the software to automatically do two repeats of the section as a standard.
Lift the cap and move to the next saved position with the structure of interest. Repeat step 4.13 of the procedure to dissect the next cell sections until all cells of interest from one slide are collected. Make sure the cap is positioned in a way that the new section sticks to an empty position on the cap. Note: It is recommended to isolate larger pieces of tissues (e.g., sections of whole flowers or parts of it) from each slide after the isolation of single cell sections on a fresh cap. These can be used for RNA quality control.
Remove the slide and continue with the next slide by repeating steps 4.4 - 4.9 and 4.13 to 4.14.
Repeat step 4.15 and related steps until the cell types of interest from all slides have been isolated. Note: It is not recommended to harvest for longer than ~ 4 - 5 hr on the same cap.
- Visually inspect the cap surface after LAM to exclude unspecific contaminations of the sample. While the non-dissected tissues are protected with the foil, dust can stick to the surface due to electrostatic adhesion.
- To visually inspect the cap, remove the slide from the instrument. Place the cap holder in the 'down' position and inspect the surface with the 4X objective.
- In case small pieces of dust are visible, remove them with a small (2 µl) pipette tip. Alternatively, mount the slide back on the instrument, place the cap on a free part of the slide (no tissues), and completely destroy the contamination using the laser.
Close caps and freeze the dry sections at -80 °C until further use. If required, samples can be stored for up to several weeks. However, proceeding quickly at this step is recommended.
5. RNA Isolation and Quality Control
Note: RNA can be isolated by any method suitable for small amounts of RNA. In this protocol the use of an RNA isolation kit specified for small amounts of RNA is described. Follow the manufacturer's instructions.
- Use the tubes with the sample attached to the cap either directly or after removal from -80 °C.
- Open the tubes and carefully pipet 11 µl of extraction buffer to the wall of each tube using filter tips. Change tips between samples.
- Close the lid and shake the extraction buffer down to the cap of the tube.
- Place the tubes with the cap down in a heating oven preheated to 42 °C. Incubate for 30 min following the manufacturer's instructions.
- Follow the manufacturer's instructions for column preparation and collecting the extract at the bottom of the tubes.
- If material from more than one cap needs to be combined into one sample, proceed with steps 5.1.1 - 5.1.2 individually for each tube/cap.
- Afterwards, pool the extracts for one sample in one tube and measure the amount of pooled extract with a pipette prior to addition of an equivalent amount of EtOH (see manufacturer's instructions).
- Ensure that the amount of EtOH added before loading the column is equal to the volume of the pooled extract.
- After RNA precipitation with EtOH, proceed quickly to the loading of the columns following manufacturer's instructions. Continue the protocol with the 100 x g centrifugation step.
- After the contents of each collecting tube have been passed through the column, perform a 16,000 x g centrifugation step for 30 sec to remove the flow-through following manufacturer's instructions.
- Continue with RNA extraction and purification following manufacturer's instructions.
- After loading of the elution buffer to the column allow the column to incubate for 1 min. Continue to load the tubes into the centrifuge following the instructions in the manufacturer's instructions. Note: An additional centrifugation step for 1 min at 100 x g prior to elution as indicated in the protocol can increase the efficiency of elution.
Extract RNA from the controls (larger tissue sections) for RNA quality control following steps 5.1 to 5.2.7 of this protocol.
For RNA quality control load 1 µl undiluted total RNA of each control sample on the Bioanalyzer using an RNA Pico Chip following the manufacturer's instructions. Note: The extracted RNA can be used for linear amplification and transcriptome analysis using microarrays or RNA-Seq 7,11,13,14. Ideally, the quality control should indicate an RNA Integrity Number (RIN) of at least 7. A number of suppliers provide suitable kits for amplification, suitable examples are given in 9.
Representative Results
Sample Preparation and LAM are done in Consecutive Steps
A number of consecutive steps are required to prepare RNA for transcriptional analysis from selected cell types by LAM (Figure 1). This starts with the harvest of the flowers and immediate fixation to ensure that the RNA population remains unchanged after harvest. The tissue is embedded, sectioned, and mounted on slides. This allows the isolation of the cells by LAM and the pooling of cell type-specific sections on one or several caps (Figure 1). The material can be frozen afterwards or directly processed by RNA extraction. Apart from the advantage of LAM to be applicable for cell type-specific analyses in both model and non-model species, the method remains rather time-consuming. At least one week of working time, sometimes more, is required for the steps from tissue harvest to RNA extraction (Figure 1). It needs to be taken into account that often sections harvested over several days of LAM are pooled in one biological sample and RNA extraction. For Boechera egg cells, the use of at least ~ 200 sections per sample is highly recommended, with up to ~ 100 sections isolated per day.
Isolation of the Cell Types of Interest Depends on their Visibility in Dry Sections
Isolation of the cell type-specific sections is the most time-consuming step of the protocol. So far, no automation of this step has been developed due to the complex nature of the samples. The identification of the cells of interest using a software tool is not feasible because the dry sections have low contrast and the section plane varies between the samples due to the fact that ovules are oriented at different angles within the tissue (Figure 2). Nevertheless, the unique morphology of the mature female gametophyte with the synergids, egg cell, and the central cell (antipodal cells degenerate before fertilization) allows the unambiguous identification of the egg cells by the researcher (Figure 2B, C; Figure 3). Indeed, in Boechera divaricarpa, the egg cell often shrinks a bit during preparation and thus separates from the surrounding tissues, a feature advantageous for the isolation of egg cell populations at high purity (Figure 2B, C; Figure 3).
Visual Inspection of the Cap after LAM is Recommended
Visual inspection of the cap surface after LAM allows the identification of any possible contaminations of the sample, e.g., by dust. In addition, it is helpful to ensure that the sections stick to the cap, as occasionally sections get lost during the procedure. Typically, many selected sections, e.g., egg cells, can be seen on one cap after several hours of LAM.
The Established Workflow Reproducibly Results in Good RNA Quality
From cell type-specific LAM isolations from female gametophytes, total RNA amounts are only in the low ng range. This limiting amount of RNA makes it necessary to use a representative control isolated from larger tissues of each slide as an approximation for RNA quality control after LAM. This is demonstrated by a comparison of cell type-specific RNA from B. divaricarpa egg cells as compared to controls from larger tissue areas isolated from the same slides, indicating similar RNA quality (Figure 4A, B). Using this method, a good to high RNA quality with RIN numbers ≥ 7 is reproducibly obtained. The RNA quality achieved was suitable for cell type-specific RNA-Seq, leading to the identification of 236 genes expressed in the apomictic egg cell but not in the apomictic central cell, synergid cell, or apomictic initial cell, nor in the cells or the mature sexual gametophyte of Arabidopsis thaliana (Figure 5) 7.
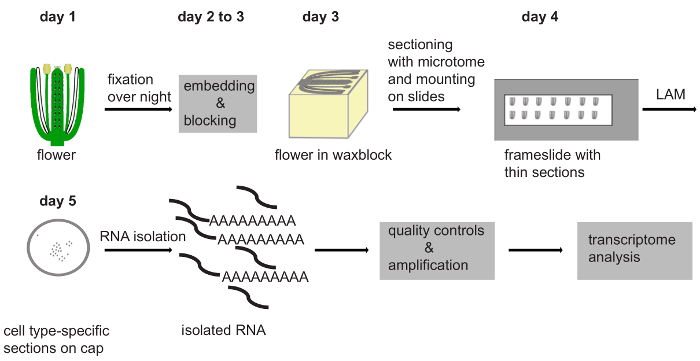 Figure 1: Scheme of the Steps of the Protocol, Indicating the Minimal Time Required per Step. Fixation of the flowers or tissues of interest is done overnight. The next day, embedding is started, which runs over night, resulting in embedded material at day 3 of the protocol. At day 3 microtome sections starting from embedded samples in wax blocks can be generated. The ribbons of the thin paraffin sections are mounted on slides and left to dry overnight. One to several days of LAM will be required to obtain enough material for one biological replicate. After RNA isolation and quality control, library preparation for RNA-Seq can be performed to prepare transcriptome analysis. Please click here to view a larger version of this figure.
Figure 1: Scheme of the Steps of the Protocol, Indicating the Minimal Time Required per Step. Fixation of the flowers or tissues of interest is done overnight. The next day, embedding is started, which runs over night, resulting in embedded material at day 3 of the protocol. At day 3 microtome sections starting from embedded samples in wax blocks can be generated. The ribbons of the thin paraffin sections are mounted on slides and left to dry overnight. One to several days of LAM will be required to obtain enough material for one biological replicate. After RNA isolation and quality control, library preparation for RNA-Seq can be performed to prepare transcriptome analysis. Please click here to view a larger version of this figure.
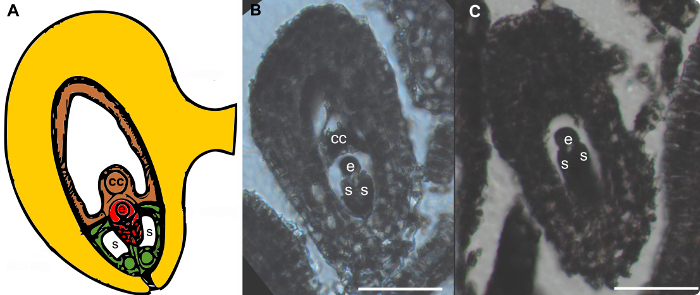 Figure 2: Egg Cells are Clearly Identifiable in Thin Sections, Due to the Characteristic Morphology of the Female Gametophyte. (A) Schematic drawing of the mature female gametophyte (Polygonum type). (B - C) Thin sections through ovules harboring female gametophytes in Boechera divaricarpa. Egg cells are clearly visible. Due to the morphology of the female gametophyte often not all cell types (egg cell: e, synergids: s, central cell: cc) are visible in a single section. Scale bars = 50 µm. Please click here to view a larger version of this figure.
Figure 2: Egg Cells are Clearly Identifiable in Thin Sections, Due to the Characteristic Morphology of the Female Gametophyte. (A) Schematic drawing of the mature female gametophyte (Polygonum type). (B - C) Thin sections through ovules harboring female gametophytes in Boechera divaricarpa. Egg cells are clearly visible. Due to the morphology of the female gametophyte often not all cell types (egg cell: e, synergids: s, central cell: cc) are visible in a single section. Scale bars = 50 µm. Please click here to view a larger version of this figure.
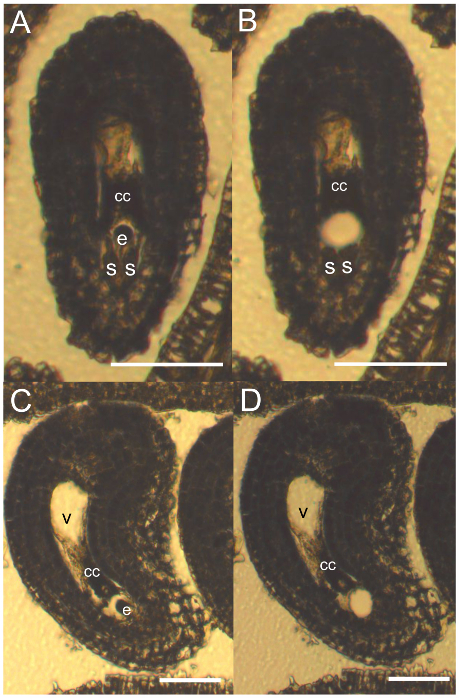 Figure 3. LAM of Egg Cells of Boechera divaricarpa. A and C. Section of the mature gametophyte of B. divaricarpa at 7 µm before and, B and D, after microdissection with the laser device (egg cell: e, synergids: s, central cell: cc). Scale bars = 50 µm. Please click here to view a larger version of this figure.
Figure 3. LAM of Egg Cells of Boechera divaricarpa. A and C. Section of the mature gametophyte of B. divaricarpa at 7 µm before and, B and D, after microdissection with the laser device (egg cell: e, synergids: s, central cell: cc). Scale bars = 50 µm. Please click here to view a larger version of this figure.
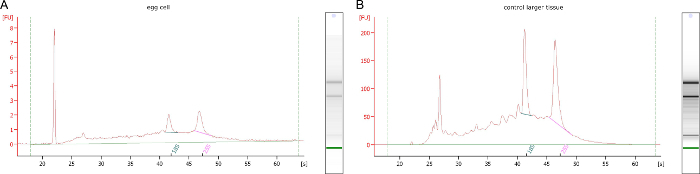 Figure 4. RNA Quality of Laser-Dissected Egg Cells and Control Sections. (A) RNA quality as analyzed from egg cell sections as compared to (B) larger tissue areas harvested from the same slides as controls. Please click here to view a larger version of this figure.
Figure 4. RNA Quality of Laser-Dissected Egg Cells and Control Sections. (A) RNA quality as analyzed from egg cell sections as compared to (B) larger tissue areas harvested from the same slides as controls. Please click here to view a larger version of this figure.
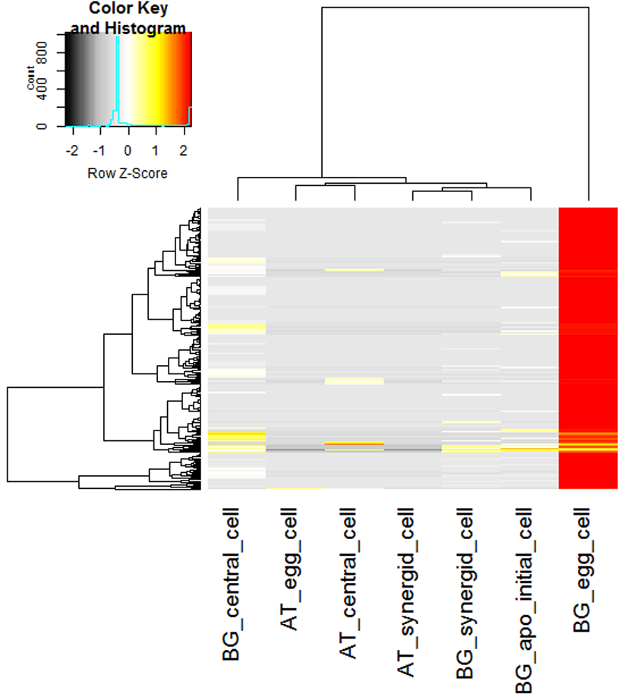 Figure 5. Identification of Genes Expressed Only in the Apomictic Egg Cell as Compared to the Cells of Apomictic and Sexual Mature Gametophytes or the Apomictic Initial Cell. Heatmap of 236 genes expressed in the apomictic egg cell but neither in the apomictic central cell, synergid cell, or apomictic initial cell of B. gunnisoniana nor the cells of the mature sexual gametophyte of A. thaliana7 Hierarchical clustering of samples and genes was based on Euclidean distance and hierarchical agglomerative clustering. Red denotes high expression and black low expression. Colors are scaled per row. apo: apomictic. Please click here to view a larger version of this figure.
Figure 5. Identification of Genes Expressed Only in the Apomictic Egg Cell as Compared to the Cells of Apomictic and Sexual Mature Gametophytes or the Apomictic Initial Cell. Heatmap of 236 genes expressed in the apomictic egg cell but neither in the apomictic central cell, synergid cell, or apomictic initial cell of B. gunnisoniana nor the cells of the mature sexual gametophyte of A. thaliana7 Hierarchical clustering of samples and genes was based on Euclidean distance and hierarchical agglomerative clustering. Red denotes high expression and black low expression. Colors are scaled per row. apo: apomictic. Please click here to view a larger version of this figure.
Discussion
The Protocol is Suitable for Different Cell and Tissue Types
LAM combined with transcriptome analyses by microarrays or RNA-Seq is a valuable tool to gain insights into the specific patterns of gene activity regulating developmental or physiological processes 7-11,13,14. However, the suitability of this method for any given cell type is critically dependent on structural issues. The cell needs to be clearly visible and unambiguously identifiable in the dry sections used for LAM. In Boechera divaricarpa, the morphology of the gametophyte allows an easy identification of the egg cell (Figure 2, Figure 3). In the end, the speed of isolation of a specific cell population also depends on the frequency at which the cell type can be found on one slide, as exchanging slides and scanning new slides are time-consuming steps. Given that dependent on the cell type, at least 200 - 250 cell sections should be pooled per sample, the suitability of the method for a certain study design depends largely on the time requirements for the LAM step.
In addition, depending on the morphology of the tissue, it might be advantageous in terms of structure to reduce the thickness of the sections to 6 µm. Similarly, thicker sections of ideally ≥ 8 µm typically result in higher RNA quantity and RNA of slightly higher quality from the same amount of tissue sections. In addition, the cells of interest should have at least a diameter of 8 - 10 µm to make isolation by LAM feasible.
In principle, the protocol described here can be adjusted to different cell and tissue types from distantly related species. However, the RNA quality can vary even between different cell types of the same species 7,10,13. This protocol has been adjusted based on small and critical cell types. Thus, it can now be applied to a broader range of samples without further adjustments. Slight modifications of the protocol might nevertheless be required, depending on the cell type and species under investigation. It is recommended to first use both alternative fixatives (see protocol 1.1) when setting up the method for a new species or cell type for a comparison of morphology and RNA quality.
In principal, the protocol can be adapted to and performed with different instruments suitable for laser microdissection 9. However, it needs to be noted that the instruments vary both in laser power and the minimal width of the laser beam that can be applied, as well as in the technology of sampling. Particularly for the isolation of small cells and tissue areas, lasers resulting in a narrow laser path are better suited. The laser beam of the instrument we use is rather fine (~ 1 µm broad) and allows dissection of small cells. The sampling technology of collecting the cells on a sticky cap surface by electrostatic adhesion is particularly advantageous for small cell types and the isolation of single cell sections. This minimizes the risk of sample loss by electrostatic effects.
The RNA Quality Obtained is Important for Further Transcriptional Analyses
One of the most critical points for successful RNA-Seq library preparation is RNA quality. While several providers of amplification kits optimized their technology for RNA of even lower integrity, the quality of the data obtained after transcriptome analysis by either microarrays or RNA-Seq typically increases with the quality of the input RNA. Using this established protocol, good and highly reproducible RNA quality for different developmental stages of the female reproductive tissues and the germline in Arabidopsis and Boechera was obtained (e.g., Figure 4). While the method is applicable also for more distantly related species (e.g., tomato, unpublished), it needs to be taken into account that small optimizations or modifications may be required depending on the species, tissue type, and even the laboratory environment as, for example, a high humidity might compromise RNA integrity during slide preparation and LAM.
A critical step during the protocol is the mounting of the sectioned wax stripes containing the samples to the slides. Here, in particular the contact to water is a critical step. While replacing water by EtOH does not result in any significant improvement of RNA integrity after isolation (unpublished), the replacement of water by methanol does. However, methanol should only be handled under the chemical hood and with great care. Depending on the sample type, as an alternative to the use of methanol, the drying times after mounting with water can be reduced to ~ 2 hr (see 3.3.3.1), resulting in equally good RNA quality. Performing a trial to test the RNA quality achieved by mounting either on water or methanol for the cell type and species of interest is recommended at the beginning of a new project.
LAM is a Powerful Technology for Transcriptional Analyses
In conclusion, we have successfully optimized and applied the combination of LAM and cell type-specific transcriptome analysis by microarrays and RNA-Seq to different cells of the sexual and apomictic germline lineage in Arabidopsis thaliana and in Boechera spp., thus allowing comparative transcriptional analyses (also see Figure 5) 7,11,13,14. The method described bears a number of advantages as compared to other technologies allowing cell type-specific transcriptional analysis. Importantly, depending on the recognizability of the cell types or tissues of interest in the section, it can be applied to rare cell types of both model and non-model species, such as the cells of the mature female gametophyte in Boechera spp., or it can even be used to isolate cellular domains, e.g., polar halves of cells 19. Similar applications would not be possible with other methods that rely on fluorescent activated sorting of cells or nuclei (FACS/FANS) 10.
A major drawback of the method is the time requirement. While the fixation and embedding does not require extended hands-on time and can simultaneously been done for a large amount of flowers, LAM as such is very time-consuming and, for a cell type-specific analysis, typically 3 days to 3 weeks of LAM (on average 5 hr per day at the laser microscope) needs to be planned. In this respect, it is helpful to do some pre-tests for the speed of isolation of the specific cell type targeted to properly design the study. In addition, even when using this optimized protocol, trials to test the RNA quality are highly recommended.
In summary, LAM is a powerful tool for the transcriptional analysis at the resolution of individual cell types or even subdomains of cells, which could not be achieved using alternative existing methods. In this respect, it bears tremendous potential to allow analyses, e.g., of developmental processes, with a very high temporal and spatial resolution. This was exemplified by the analyses of the transcriptomes of cells at all key steps of sexual and apomictic reproduction in Arabidopsis and Boechera7,11,13,14.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Timothy F. Sharbel (IPK Gatersleben) for providing Boechera divaricarpa seeds and Sharon Kessler (University of Oklahoma) for critical reading and proofreading. Work on cell type-specific transcriptome analyses to study gametophyte development and apomixis in UG´s laboratory is supported by the University of Zürich, by a fellowship of the "Deutsche Forschungsgemeinschaft" and the Marie Curie project IDEAGENA to AS, by grants from the "Staatssekretariat für Bildung und Forschung" in the framework of COST action FA0903 (to UG and AS) and the Swiss National Foundation (to UG).
References
- Maheshwari P. An Introduction to the Embryology of Angiosperms. New York: McGraw-Hill; 1950. [Google Scholar]
- Willemse MTM, Van Went JL. The female gametophyte. In: Johri BM, editor. Embryology of Angiosperms. Berlin: Springer-Verlag; 1984. pp. 159–196. [Google Scholar]
- Koltunow AM, Bicknell RA, Chaudhury AM. Apomixis: Molecular strategies for the generation of genetically identical seeds without fertilization. Plant Physiol. 1995;108:1345–1352. doi: 10.1104/pp.108.4.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vielle-Calzada JP, Crane C, Stelly DM. Apomixis - the asexual revolution. Science. 1996;274:1322–1323. [Google Scholar]
- Grossniklaus U, Koltunow A, van Lookeren Campagne M. A bright future for apomixis. Trends Plant Sci. 1998;3:415–416. [Google Scholar]
- Schmidt A, Schmid MW, Grossniklaus U. Plant germline formation: molecular insights define common concepts and illustrate developmental flexibility in apomictic and sexual reproduction. Development. 2015;142:229–241. doi: 10.1242/dev.102103. [DOI] [PubMed] [Google Scholar]
- Schmidt A, et al. Apomictic and sexual germline development differ with respect to cell cycle, transcriptional, hormonal and epigenetic regulation. PLOS GENET. 2014;10:e1004476. doi: 10.1371/journal.pgen.1004476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada T, et al. Enlarging cells initiating apomixis in Hieractium praealtum transition to an embryo sac program prior to entering mitosis. Plant Physiol. 2013;163:216–231. doi: 10.1104/pp.113.219485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuest SE, Grossniklaus U. Laser-assisted microdissection applied to floral tissues. Methods Mol Biol. 2014;1110:329–344. doi: 10.1007/978-1-4614-9408-9_19. [DOI] [PubMed] [Google Scholar]
- Wuest SE, Schmid MW, Grossniklaus U. Cell-specific expression profiling of rare cell types as exemplified by its impact on our understanding of female gametophyte development. Curr Opin Plant Biol. 2013;16(1):41–49. doi: 10.1016/j.pbi.2012.12.001. [DOI] [PubMed] [Google Scholar]
- Wuest SE, et al. Arabidopsis female gametophyte gene expression map reveals similarities between plant and animal gametes. Curr Biol. 2010;20:1–7. doi: 10.1016/j.cub.2010.01.051. [DOI] [PubMed] [Google Scholar]
- Cai S, Lashbrook CC. Laser capture microdissection of plant cells from tape-transferred paraffin sections promotes recovery of structurally intact RNA for global gene profiling. Plant J. 2006;48(4):628–637. doi: 10.1111/j.1365-313X.2006.02886.x. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Wuest SE, Vijverberg K, Baroux C, Kleen D, Grossniklaus U. Transcriptome analysis of the Arabidopsis megaspore mother cell uncovers the importance of RNA helicases for plant germ line development. PLOS BIOL. 2011;9:e1001155. doi: 10.1371/journal.pbio.1001155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid MW, Schmidt A, Klostermeier UC, Barann M, Rosenstiel P, Grossniklaus U. A powerful method for transcriptional profiling of specific cell types in eukaryotes: laser-assisted microdissection and RNA sequencing. PLOS ONE. 2012;7:e29685. doi: 10.1371/journal.pone.0029685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mau M, et al. Hybrid apomicts trapped in the ecological niches of their sexual ancestors. Proc Natl Acad Sci.U S A. 2015;112(18):2357–2365. doi: 10.1073/pnas.1423447112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aliyu OM, Seifert M, Corral JM, Fuchs J, Sharbel TF. Copy number variation in transcriptionally active regions of sexual and apomictic Boechera. demonstrates independently derived apomictic lineages. Plant Cell. 2013;25(10):3808–3823. doi: 10.1105/tpc.113.113860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corral JM, et al. A conserved apomixis-specific polymorphism is correlated with exclusive exonuclease expression in premeiotic ovules of apomictic boechera species. Plant Physiol. 2013;163(4):1660–1672. doi: 10.1104/pp.113.222430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeken R, Ache P, Kajahn I, Klinkenberg J, Bringmann G, Hedrich R. Identification of Arabidopsis thaliana. phloem RNAs provides a search criterion for phloem-based transcripts hidden in complex datasets of microarray experiments. Plant J. 2008;55:746–775. doi: 10.1111/j.1365-313X.2008.03555.x. [DOI] [PubMed] [Google Scholar]
- Schmid MW. Genome Scale Quantitative Biology in Arabidopsis thaliana. University of Zürich; 2015. pp. 40–104. PhD Thesis. [Google Scholar]