

Abstract
We describe detailed methods to measure thymidine (dThd) and deoxyuridine (dUrd) concentrations and thymidine phosphorylase (TP) activity in biological samples. These protocols allow the detection of TP dysfunction in patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). Since the identification of mutations in TϒMP, the gene encoding TP, as the cause of MNGIE (Nishino et al. Science 283:689–692, 1999), the assessment of TP dysfunction has become the best screening method to rule out or confirm MNGIE in patients. TϒMP sequencing, to find the causative mutations, is only needed when TP dysfunction is detected. dThd and dUrd are measured by resolving these compounds with high-performance liquid chromatography (HPLC) followed by the spectrophotometric monitoring of the eluate absorbance at 267 nm (HPLC-UV). TP activity can be measured by an endpoint determination of the thymine formed after 1 h incubation of the buffy coat homogenate in the presence of a large excess of its substrate dThd, either spectrophotometrically or by HPLC-UV.
Keywords: Thymidine, Deoxyuridine, Thymidine phosphorylase, Mitochondrial neurogastrointestinal encephalomyopathy, MNGIE, HPLC, Biochemical diagnosis
1. Introduction
The assessment of thymidine phosphorylase (TP) function is the reference method to diagnose mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) (1) in patients with clinical features suggestive of this disorder. TP initiates the catabolism of thymidine (dThd) and deoxyuridine (dUrd) in humans (2) by catalyzing the phosphorolysis of these nucleosides to deoxyribose phosphate and the corresponding bases (Fig. 1). In MNGIE patients, TP dysfunction caused by TϒMP gene mutations results in the systemic accumulation of these nucleosides (3, 4) and increased urinary excretion of dThd and dUrd (5, 6).
Fig. 1.
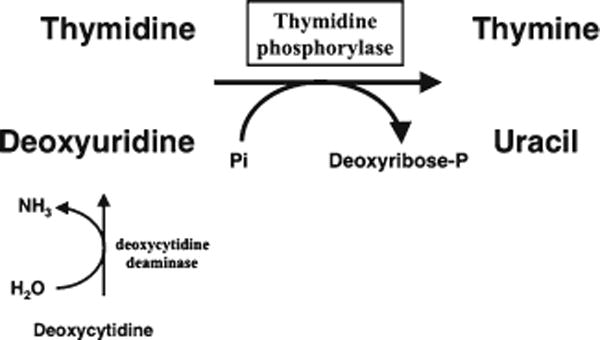
Role of TP in the catabolism of pyrimidine deoxyribonucleosides in humans. Thymidine and deoxyuridine are the TP substrates; indirectly, TP also contributes to the degradation of the third pyrimidine deoxyribonucleoside, deoxycytidine, because it is first converted in deoxyuridine via deamination.
Analyzing plasma dThd and dUrd concentrations is the easiest way to test for TP dysfunction. Screening of urine is also useful, but precautions should be taken to prevent bacterial growth in the sample; otherwise, in vitro catabolism of dThd and dUrd via bacterial TP can lead to underestimation of urinary excretion. For both plasma and urine samples, dThd and dUrd concentrations can be easily assessed by high-performance liquid chromatography coupled to ultraviolet spectrophotometric detection (HPLC-UV) or to tandem mass spectrometry (HPLC-MS/MS). UV detection is less sensitive and selective than MS/MS but simpler to set up and more accessible to many nonspecialized laboratories that may not have access to HPLC-MS/MS. In addition, the sensitivity of the UV detection (typically 0.05 μM in our hands (7)) is sufficient to detect and accurately quantify dThd and dUrd levels typically found in MNGIE patients, including those with residual TP activity (late-onset MNGIE patients (8, 9)), in contrast to plasma and urine levels that are undetectable by UV in healthy subjects and TϒMP mutation carriers.
In our experience, there is complete concordance between nucleoside accumulation and reduced TP activity in buffy coat. Therefore, testing only nucleoside levels is generally sufficient for diagnostic purposes. Nevertheless, we prefer to measure TP activity in buffy coat in all cases, including those with undetectable dThd and dUrd plasma concentrations, because several late-onset MNGIE, with moderately elevated plasma levels of nucleosides as low as 0.4 μM, have been reported (8, 9). In such cases, TP activity can be around 15–20% of normal in contrast to most MNGIE cases with little or no detectable TP activity. If blood samples are not kept in cold and immediately centrifuged to separate the plasma from the cellular fraction, residual TP activity in vitro can artifactually reduce dThd and dUrd to levels near or below the detection limit, thus masking the moderate nucleoside accumulation in late-onset MNGIE patients.
2. Materials
Prepare all solutions using HPLC grade water (e.g., obtained by deionization to a resistivity of 18.2 MΩ-cm).
2.1. Reagents
Hemolysis buffer: 10 mM NH4HCO3, 144 mM NH4Cl. Dissolve 0.791 g of NH4HCO3 (M = 79.06 g/mol) and 7.70 g of NH4Cl (M = 53.49 g/mol) in 1 L of water. Sterilize by filtering through a 0.22-μm membrane and keep it refrigerated for less than 6 months.
-
Eluent A: 20 mM potassium phosphate monobasic (KH2PO4), pH 5.6.
Dissolve 2.72 g of KH2PO4 (anhydrous salt, M = 136.09 g/mol) in approximately 0.9 L of water, adjust to pH 5.6 with a concentrated solution of KOH (e.g., 0.5 M), and add water up to 1 L. Filter eluent A through a 0.2-μm nylon or PVDF membrane before use.
Methanol, gradient grade (see Note 1).
Thymidine phosphorylase from Escherichia coli, recombinant (TP, SIGMA). Specific activity ~1,000 U/ml, the exact value varies according to lot.
Concentrated 70–72% (approximate 11.7 M) perchloric acid (PCA; H3ClO4).
0.55 M PCA: Dilute 11.7 M PCA 1:21 in HPLC grade water.
Phosphate buffered saline (PBS): 140 mM NaCl, 10 mM Na2HPO4, pH 7.4 (see Note 2). This reagent is only needed for the dilution of urine samples. Dissolve 8.2 g of NaCl (M = 58.5 g/mol) and 1.42 g of Na2HPO4 (M = 141.96 g/mol) in nearly 1 L of water. Adjust to pH 7.4 with HCl. Then, complete the volume to 1 L with water.
Lysis buffer: 50 mM Tris–HCl, pH 7.2, 1% (w/v) triton X-100, 2 mM phenylmethylsulfonyl fluoride (PMSF), 0.02% (v/v) 2-mercaptoethanol. Dissolve 0.606 g of Tris base (M = 121.14 g/mol) in 80 ml of water and adjust the pH to 7.2 with HCl. Add 1 g of triton X-100, 20 μl of 2-mercaptoethanol and 2 ml of stock PMSF solution (stock PMSF solution: 17.4 mg/ml PMSF in isopropanol). Add water to reach 100 ml final volume.
5× TP reaction buffer: 0.5 M Tris–arsenate, pH 6.5 (see Note 3). Dissolve 6.06 g of Tris base (M = 121.14 g/mol) and 15.6 g of disodium arsenate heptathydrate (Na2HAsO4 ·7H2O, M = 312.0) to nearly 100 ml of water, adjust to pH 6.5 with HCl, and complete to 100 ml with water. Store at room temperature.
167 mM Thymidine: Dissolve 0.404 g of thymidine (M = 242.2 g/mol) in 10 ml of water by sustained stirring, with slight heating to favor dissolution. Once completely dissolved, keep in frozen aliquots at −20°C until used. Upon thawing before its use, make sure that dThd eventually precipitated is completely redissolved (warming at 37°C and several vortexings will help to complete resolubilization).
0.3 M NaOH: Dissolve 1.2 g of NaOH (M = 40 g/mol) in 100 ml of water (exercise caution with this exothermic dissolution). Keep the solution in a well-closed plastic bottle at room temperature and discard when signs of carbonate appear in the surroundings of the cap.
2.2. Standards for HPLC-UV Quantification
-
To make the standard curve (see Note 4), prepare 10 mM aqueous solutions of:
dThd (M = 242.2 g/mol): dissolve 0.242 g of dThd in 100 ml of water.
dUrd (M = 228.2 g/mol): dissolve 0.228 g of dUrd in 100 ml of water.
Thymine (M = 126.1 g/mol): dissolve 0.126 g of thymine in 100 ml of water (see Note 5).
Prepare a 100 μM multistandard (100 μM dThd, 100 μM dUrd, 100 μM thymine) by diluting 1 ml of 10 mM dThd + 1 ml of 10 mM dUrd + 1 ml of 10 mM thymine up to 100 ml final volume with water).
Use water to serially dilute the 100 μM multistandard to obtain the following concentrations: 50, 25, 10, 8, 5, 2, 1, 0.5, 0.1 and 0.05 μM.
Once prepared, the aqueous multistandards can be frozen at −20°C in several aliquots and used as needed.
2.3. Other Materials and Equipment
HPLC apparatus. The procedure described here has been set for a HPLC apparatus with at least three independent eluent lines and mixer (see Note 6).
Vials appropriate for the injection device included in your HPLC apparatus. Vials with reduced dead volume are preferable in cases of limited volume of sample.
Column: The method, retention times, and overall runtimes described here have been setup and obtained with a column Alltima C18 NUC, 100 Å pore size; 5 μm particle size, 250 × 4.6 mm (Alltech). A different C18 column with similar length and particle size can be used. In this case, retention times and work pressures may change.
Spectrophotometer and quartz cuvettes (semimicro size). Although any spectrophotometer that can measure absorbance at wavelength 300 nm can be used, models with multisample capacity, enabling the simultaneous load of several cuvettes for reading, are preferable because they minimize the time of UV reading. A spectrophotometer is not needed if the final thymine measurement for TP activity determination is performed by HPLC-UV.
Nylon or PVDF 0.2 μm pore-size filter membranes, and filtering device for vacuum-driven filtration of aqueous solutions (eluent A and water) are used prior to HPLC (see Note 7).
3. Methods
3.1. Sample Collection and Preparation
Centrifuge 5–10 ml of anticoagulated blood in a 15-ml falcon tube at 1,500 × g for 10 min. Then separate the plasma and keep it −20°C until nucleoside analysis (see Note 8).
On the cell fraction (erythrocytes + buffy coat), add approximately two volumes of hemolysis buffer (e.g., 8 ml of buffer on 4 ml of cellular fraction) and shake vigorously 15 s, at room temperature. Then keep the tube on ice 30 min, followed by vigorous shaking.
Centrifuge at 1,500 × g for 10 min and discard the supernatant.
Resuspend the pellet in 10 ml of hemolysis buffer, shake vigorously, and maintain on ice 15 min.
Centrifuge at 1,500 × g for 10 min and discard the supernatant.
Resuspend the pellet in 2 or 3 ml of hemolysis buffer and transfer the suspension to two or three 1.5-ml microcentrifuge tubes, vortex 5 s, and keep for 10 min on ice.
Centrifuge at 20,000 × g for 10 min and discard the supernatant. Eliminate as much as possible the liquid red crown that covers the surface of the white buffy coat, to minimize hemoglobin contamination.
Keep the dry buffy coat at −80°C until TP activity analysis (see Note 9).
For the assessment in urine samples, collect fresh random urine and freeze immediately until the analysis (see Note 10).
3.2. dThd and dUrd Determination
Divide the sample (plasma or urine) (see Note 11) in two aliquots of equal volume (e.g., 200 μl each aliquot). If the sample is urine, because of the presence of many metabolites at high concentrations, 1:20 dilution with PBS will help avoiding interferences.
Add E. coli TP to one of the aliquots to a final catalytic concentration of approximately 10 U/ml (e.g., 200 μl sample + 2 μl of 1,000 U/ml TP). Vortex and incubate TP treated and untreated aliquots for 10 min at 37°C.
To deproteinize the sample, add concentrated perchloric acid (PCA, 11.7 M approximately) up to a final concentration of 0.5 M (e.g., 200 or 202 μl of incubated sample + 9 μl of concentrated PCA) to the TP treated and untreated aliquots. Vortex and keep on ice for 5 min to facilitate complete protein precipitation.
Centrifuge at 20,000 × g for 10 min. Discard the pellet and save the clear supernatant, which is now ready to be injected to the HPLC or kept at −20°C until analysis (see Note 12).
Thaw the following multistandards: 25, 10, 8, 5, 2, 1, 0.5, 0.1, and 0.05 μM. Once thawed, treat each multistandard with 11.7 M PCA in the same proportion as done for the samples. Because the multistandards will be injected more than once, you should use larger volumes, for example 500 μl of multi-standard + 22.5 μl of 11.7 M PCA.
-
Separation of nucleosides is carried out through HPLC with gradient elution and UV detection. The conditions of the separation are as follows:
- Column: Alltima C18 NUC (see Subheading 2).
- Column temperature: 30°C (see Note 13).
- Injection volume: 50 μl.
- Detection: UV absorbance at wavelength 267 nm.
- Eluent A: 20 mM KH2PO4, pH 5.6; eluent B: methanol gradient grade; eluent C: water.
For each run, the gradient elution should be programmed as follows:Time (min) Flow (ml/min) %A %B %C Comment
0 1.5 100 0 0 5 min isocratic segment
5 1.5 100 0 0 From 5 to 25 min, linear gradient from 0 to 17.4% MeOH 25 1.5 82.6 17.4 0
26 1.5 0 0 100 Washout of eluent A to avoid salt precipitation (see Note 14) 30 1.5 0 0 100
31 1.5 0 100 0 Washout of compounds still retained in the column 35 2.0 0 100 0 45 2.0 0 100 0 46 1.5 0 100 0
47 1.5 0 0 100 Washout of MeOH to avoid salt precipitation (see Note 14) 50 1.5 0 0 100
51 1.5 100 0 0 Re-equilibration of the column at 100% A for the next run 60 1.5 100 0 0
Generate a sample set for analysis. Inject, for every sample, both TP-treated and untreated aliquots. Intercalate the multistandards among the samples. To obtain reliable calibration curves, two or three injections of every multistandard are advisable for every run (see Note 15). Figure 2 shows two typical chromatograms of untreated and TP-treated plasma from a MNGIE patient. Retention times and resolution of the peaks will vary depending on the HPLC apparatus and the column used (see Note 16).
Integrate the areas of the standards’ peaks using the software of your HPLC apparatus. Generate each a calibration curve for dThd and dUrd by adjusting peak areas (ϒ axis) versus standards’ concentrations (X axis) to a linear regression with no offset (i.e., force the linear regression to pass by the x = 0, y = 0).
Calculate dThd and dUrd concentrations from the peak areas, using the calibration curves. Because TP from E. coli degrades dThd and dUrd, the results from any remaining peaks present in the TP-treated aliquots at the dThd and dUrd retention times should be subtracted from the untreated result, as only the TP-labile portions of these peaks are true dThd or dUrd (see Note 17).
Fig. 2.
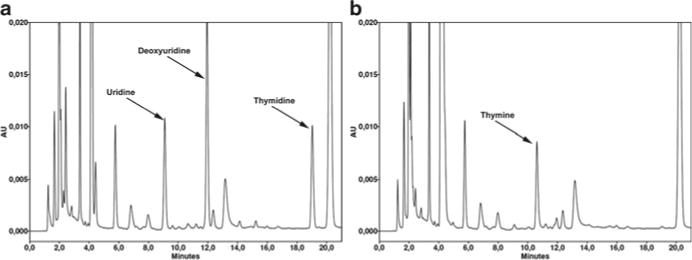
Chromatograms of plasma from a MNGIE patient showing thymidine and deoxyuridine peaks. Representative chromatograms obtained from plasma of a MNGIE patient (a), and the same plasma after the selective elimination of TP substrates by treatment with Escherichia coli TP (b). The peaks of dThd and dUrd (5.4 and 10.6 μM in this specific case) virtually disappear in the TP-treated aliquot, whereas the thymine observed in the panel b is the product of dThd phosphorolysis by TP. Uracil, derived from phosphorolyses of dUrd and uridine, is also present, but it elutes very early in the chromatogram and is not resolved well due to coeluting peaks. Note that the ribonucleoside uridine, which is normally found in plasma of healthy controls and MNGIE patients at similar concentrations, is also degraded by E. coli TP.
3.3. TP Activity Assay
Always work with samples on ice:
Resuspend the buffy coat obtained as indicated in the Subheading 3.1 in approximately 300 μl of lysis buffer and homogenize the pellet by passing the suspension through a 27 g × 1 in. needle several times.
After homogenization, centrifuge at 20,000 × g for 30 min at 4°C and save the supernatant (homogenate).
Determine the protein concentrations of the homogenates (see Note 18).
Dilute all the homogenates with lysis buffer up to a protein concentration of 1.35 mg/ml (diluted homogenate). At this concentration, the reaction mixture will contain 100 μg of protein (we will add 74 μl of this extract to a final volume of 100 μl of the reaction mixture). If the undiluted homogenate has a protein concentration lower than 1.35 mg/ml, the assay can be performed with undiluted homogenate, but this will result in a slight overestimation of the activity (see Note 19).
- For every sample, make two mixtures:
Reaction mixture (μl) Blank mixture (μl)
5× TP reaction buffera 20 20
Diluted homogenate (1.35 mg protein/ml) 74 74
167 mM thymidine 6 –
Final volume 100 94
aFinal concentrations in the reaction mixture: 0.1 M Tris–arsenate; 10 mM thymidine; 1 mg protein/ml. Incubate both blank and reaction mixtures for 1 h at 37°C. At the end of this incubation, the reaction must be stopped and the thymine concentration measured, either spectrophotometrically or by HPLC-UV. From here on, the protocol varies according to methods of detection of thymine.
- Spectrophotometric determination of thymine:
- After incubation for 1 h at 37°C, terminate the reaction by the addition of 1 ml of 0.3 M NaOH. Then, add 6 μl of 167 mM thymidine to all the blank mixtures and vortex (see Note 20).
- Measure the absorbance of the reaction and blank mixtures at 300 nm, subtract the blank result from that obtained for the reaction mixture and calculate from this difference the amount of thymine formed, based on the 3.4 × 103 L−1 mol cm−1 difference in molar absorptivity at 300 nm between dThd and thymine at alkaline pH.
- Express enzyme activity as nanomoles of thymine formed per hour and mg of protein.
- HPLC-UV determination of thymine:
- Stop the reaction by adding 1 ml of 0.55 M perchloric acid and vortex.
- Add 6 μl of 167 mM thymidine to all the blank mixtures and vortex (see Note 20).
- Keep all mixtures on ice for 5 min.
- Centrifuge at 20,000 × g for 10 min. Save the supernatant, which is now ready to be injected in the HPLC apparatus.
- Use the HPLC method and conditions described for dThd and dUrd determination to measure the thymine formed in the reaction and blank mixtures. Alternatively, the runtime can be shortened to around 15 min or less because the thymine in this supernatant can be readily separated and quantified using the following isocratic elution eluent A 90% and eluent B 10%. Flow 1.5 ml/min. In these conditions, the retention times for thymine and thymidine are around 4.5 and 9 min, respectively.
- Quantify the thymine formed from the areas of the peaks using a calibration curve done with aqueous standards. The range of concentrations of your calibration curve should cover concentrations between 0.5 and 100 μM.
- Calculate TP activity from the thymine formed in the reaction, taking in account that the blank must be subtracted (see Note 20). Express the results referred to protein (nmol of thymine formed/h/mg protein).
Acknowledgments
Dr. Martí is supported by a grant from the Spanish Instituto de Salud Carlos III (PS09/01591). Dr. López is supported by grants from the Marie Curie International Reintegration Grant Programme (COQMITMEL-266691) of the Seventh European Community Framework Programme, from Ministerio de Ciencia e Innovación, Spain (SAF2009-08315) and from the Consejería de Economía, Innovación y Ciencia, Junta de Andalucía (P10-CTS-6133). Dr. Hirano is supported by NIH grants R01 HD056103 (cofunded by NICHD and the NIH Office of Dietary Supplements), R01 HD057543, RC1 NS070232; MDA grant 115567; and the Marriott Mitochondrial Disorder Clinical Research Fund.
Footnotes
As a general rule, all eluents and other liquids to be passed through the column and the HPLC apparatus should be sterile and particle free to prevent damage to the column and system. Therefore, water and aqueous solutions must be filtered through a 0.2 μm filter prior to use, using a suitable vacuum-driven filter unit and hydrophilic nylon or PVDF membranes. HPLC grade organic solvents (e.g., methanol) do not need to be filtered because they are free of particles and do not support microbial growing.
Methanol can be purchased from various suppliers, but it is important to use HPLC “gradient grade” methanol. This will minimize the changes in the UV absorbance due to the increasing methanol proportion in the mobile phase for the HPLC separation.
Although the most common and physiological formulation of PBS includes potassium cation, for the purposes of this protocol, both formulations of PBS are acceptable. We have described here a formulation with sodium salts only. Moreover, it is also possible to purchase inexpensive preformulated PBS tablets to be dissolved in an appropriate volume of water to obtain PBS ready to use.
The final concentration in the TP reaction mixture is 0.1 M Tris–arsenate pH 6.5.
We detail here the procedure to prepare a multistandard of dThd, dUrd (for the quantification of these two compounds), and thymine (for the TP activity determination). Additional UV-absorbing compounds can be resolved using the HPLC procedure described here, so that more compounds can be included in the multistandard if needed for other purposes. In our hands, the following compounds are well resolved: cytosine, uric acid, cytidine, hypoxanthine, xanthine, uridine, thymine, dUrd, inosine, guanosine, dThd, tryptophan, and adenosine.
The dissolution of thymine in water to 10 mM is slow, but it will be easily accelerated by moderate heating or alkalinization with NaOH.
Under the conditions described here, the HPLC system must be able to bear pressures slightly above 4,000 pounds per square inches (psi). Alternatively, the column may be set to a higher temperature than that described in this protocol (described in Subheading 3.2 step 6), or the flow may be reduced. In this latter case, the gradient will have to be reformulated and the runtime for each injection increased.
Blood can be collected in tubes with any anticoagulant because they do not affect the plasma nucleoside concentration (unless the anticoagulant addition involves significant volume addition, which will have a dilution effect). Although the influence of the anticoagulant on the buffy coat TP activity has been only marginally studied (4), in our experience, standard anticoagulants (e.g., EDTA, heparin, and acid citrate) do not interfere with TP assessment.
Separating the buffy coat from each patient into two or three aliquots will provide one for TP assessment and the other(s) for additional studies (for example, DNA extraction for TϒMP sequencing in cases of TP deficiency).
The determination of the nucleosides in urine can be useful for diagnostic purposes, but it presents several pitfalls (variability, microbial contamination, sample collection problems, etc.) that commonly confound biochemical determinations with urine samples. A common way to normalize the results and correct from sample-related variability is to refer the results to milligram of creatinine. In addition, great care should be paid to avoid bacterial contamination, especially in 24-h urine samples that may be stored for several hours, often in poorly controlled conditions. Because of these potential problems, we recommend use of plasma rather than urine to test for dThd and dUrd overload in candidate MNGIE patients, but if urine should be finally tested, we recommend using freshly collected urine (less prone to be contaminated) that is frozen immediately until analysis, and refer the results to milligram of creatinine.
Although this protocol refers to plasma and urine samples, this method can be used for other biological fluids or tissue samples of interest (10).
Although it is safer to store the supernatant frozen if the sample will not be analyzed in the following days, we have observed that dThd, dUrd, as well as many other nucleosides are very stable in acidified supernatant, and their concentrations remain unchanged after more than 1 week at 4°C.
In our experience, the pressure of the HPLC system reaches maximum values of 4,000 or some higher when the column is set at 30°C. If the conditions described here lead to system pressure levels above those allowed by the HPLC apparatus, the best alternative is setting the column temperature above 30°C. Increasing the temperature of the column will reduce significantly the pressure, and the resolution of the peaks will be only slightly modified. Temperatures as high as 50 or 60°C can be tried, but the manufacturer instructions on the column specifications should always be checked to ensure the maximum temperature tolerated by the column.
The contact of high percentages of organic phases with phosphate buffers may result in salt precipitation, which may damage the column and the HPLC apparatus. To avoid this potential problem, we recommend including washout with water at the points indicated in the program, but these steps can be eliminated if you determine empirically that no salt precipitation occurs under your conditions. This would shorten the time needed for each injection.
In our experience, slopes of the calibration curves obtained for different runs are very reproducible. Typical slope variations are usually below 10%.
To find out the exact retention times of each compound in your conditions (e.g., dThd, dUrd, and thymine, as well as additional compounds of interest), separate injections of standards containing one compound only may be needed. Once the retention times for every compound have been established, standards of single compounds are not generally needed because the retention times change very little over different runs.
In our experience, one or several TP-resistant small peaks usually coelute or elute close to dUrd. TP-resistant peaks coeluting with dThd are rarer but sometimes happen (e.g., circulating UV-absorbing drugs). Care should be taken to avoid false-positives due to coeluting peaks.
The protein concentration of the homogenate can be determined with the Bradford method (11) by using bovine serum albumin for the preparation of the calibration curve, or with the method routinely used in your laboratory. It is imperative to use a reliable method for the determination of the protein concentration because biases or inaccuracies in this determination largely affect the final result, as the activity is expressed as nmoles of thymine/h/mg protein.
We describe here the assay for the enzyme reaction containing 100 μg of protein. Under these conditions, the reference values should be similar to those previously reported (4, 7), around 650–700 nmol of thymine/h/mg protein. The assay can be performed with smaller amounts of protein, but we have observed that the rate of thymine production is not strictly linear with the amount of protein, and smaller amounts of protein in the reaction mixture tend to result in slightly higher enzyme activities when expressed as nmoles of thymine formed per hour per mg of protein. We strongly recommend always working with the same amount of protein in the reaction mixture and to determine the reference values for healthy controls under these conditions.
In the blank mixture, the substrate thymidine should be added only at the end of the incubation, after TP protein contained in the homogenate has been inactivated. Therefore, any thymine detected in the blank is not produced by TP activity. We always observe very low but clearly measurable concentrations of thymine in the blank, as a result of the trace thymine contaminant in thymidine. We have estimated the thymine contamination of our thymidine preparations to be around 0.03%, but the substrate thymidine is added to the mixture in such a high concentration (10 mM) that this small contamination should be taken in account. It is negligible when TP activity is normal, but it may be significant when compared with the minute TP activities observed in MNGIE patients.
References
- 1.Nishino I, Spinazzola A, Hirano M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science. 1999;283:689–692. doi: 10.1126/science.283.5402.689. [DOI] [PubMed] [Google Scholar]
- 2.Focher F, Spadari S. Thymidine phosphorylase: a two-face Janus in anticancer chemotherapy. Curr Cancer Drug Targets. 2001;1:141–153. doi: 10.2174/1568009013334232. [DOI] [PubMed] [Google Scholar]
- 3.Marti R, Nishigaki Y, Hirano M. Elevated plasma deoxyuridine in patients with thymidine phosphorylase deficiency. Biochem Biophys Res Commun. 2003;303:14–18. doi: 10.1016/s0006-291x(03)00294-8. [DOI] [PubMed] [Google Scholar]
- 4.Marti R, Spinazzola A, Tadesse S, Nishino I, Nishigaki Y, Hirano M. Definitive diagnosis of mitochondrial neurogastrointestinal encephalomyopathy by biochemical assays. Clin Chem. 2004;50:120–124. doi: 10.1373/clinchem.2003.026179. [DOI] [PubMed] [Google Scholar]
- 5.la Marca G, Malvagia S, Casetta B, Pasquini E, Pela I, Hirano M, et al. Pre- and post-dialysis quantitative dosage of thymidine in urine and plasma of a MNGIE patient by using HPLC-ESI-MS/MS. J Mass Spectrom. 2006;41:586–592. doi: 10.1002/jms.1013. [DOI] [PubMed] [Google Scholar]
- 6.Schupbach WM, Vadday KM, Schaller A, Brekenfeld C, Kappeler L, Benoist JF, et al. Mitochondrial neurogastrointestinal encephalomyopathy in three siblings: clinical, genetic and neuroradiological features. J Neurol. 2007;254:146–153. doi: 10.1007/s00415-006-0255-3. [DOI] [PubMed] [Google Scholar]
- 7.Spinazzola A, Marti R, Nishino I, Andreu AL, Naini A, Tadesse S, et al. Altered thymidine metabolism due to defects of thymidine phosphorylase. J Biol Chem. 2002;277:4128–4133. doi: 10.1074/jbc.M111028200. [DOI] [PubMed] [Google Scholar]
- 8.Marti R, Verschuuren JJ, Buchman A, Hirano I, Tadesse S, van Kuilenburg AB, et al. Late-onset MNGIE due to partial loss of thymidine phosphorylase activity. Ann Neurol. 2005;58:649–652. doi: 10.1002/ana.20615. [DOI] [PubMed] [Google Scholar]
- 9.Massa R, Tessa A, Margollicci M, Micheli V, Romigi A, Tozzi G, et al. Late-onset MNGIE without peripheral neuropathy due to incomplete loss of thymidine phosphorylase activity. Neuromuscul Disord. 2009;19:837–840. doi: 10.1016/j.nmd.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 10.Valentino ML, Marti R, Tadesse S, Lopez LC, Manes JL, Lyzak J, et al. Thymidine and deoxyuridine accumulate in tissues of patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) FEBS Lett. 2007;581:3410–3414. doi: 10.1016/j.febslet.2007.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]