

Abstract
Skeletal muscle terminal differentiation starts with the commitment of pluripotent mesodermal precursor cells to myoblasts. These cells have still the ability to proliferate or they can differentiate and fuse into multinucleated myotubes, which maturate further to form myofibers. Skeletal muscle terminal differentiation is orchestrated by the coordinated action of various transcription factors, in particular the members of the Muscle Regulatory Factors or MRFs (MyoD, Myogenin, Myf5, and MRF4), also called the myogenic bHLH transcription factors family. These factors cooperate with chromatin-remodeling complexes within elaborate transcriptional regulatory network to achieve skeletal myogenesis. In this, MyoD is considered the master myogenic transcription factor in triggering muscle terminal differentiation. This notion is strengthened by the ability of MyoD to convert non-muscle cells into skeletal muscle cells. Here we describe an approach used to identify MyoD protein partners in an exhaustive manner in order to elucidate the different factors involved in skeletal muscle terminal differentiation. The long-term aim is to understand the epigenetic mechanisms involved in the regulation of skeletal muscle genes, i.e., MyoD targets. MyoD partners are identified by using Tandem Affinity Purification (TAP-Tag) from a heterologous system coupled to mass spectrometry (MS) characterization, followed by validation of the role of relevant partners during skeletal muscle terminal differentiation. Aberrant forms of myogenic factors, or their aberrant regulation, are associated with a number of muscle disorders: congenital myasthenia, myotonic dystrophy, rhabdomyosarcoma and defects in muscle regeneration. As such, myogenic factors provide a pool of potential therapeutic targets in muscle disorders, both with regard to mechanisms that cause disease itself and regenerative mechanisms that can improve disease treatment. Thus, the detailed understanding of the intermolecular interactions and the genetic programs controlled by the myogenic factors is essential for the rational design of efficient therapies.
Keywords: Biochemistry, Issue 111, MyoD, TAP-Tag, Mass spectrometry, chromatin, transcription, muscle, HeLa-S3.
Introduction
Eukaryotic multi-cellular organisms are composed of different organs and tissues. Each functional tissue has a specific gene pattern expression, which is determined at each differentiation step. Cellular differentiation involves activation of specific genes, maintenance of their expression and, generally, silencing of a set of genes such those involved in cell proliferation. Skeletal muscle differentiation, or myogenesis, is thus a multi-step process, that begins with the determination of mesodermal stem cells into myoblasts, and then leads to the terminal differentiation of these myoblasts into first mono-nucleated, and then multi-nucleated, myotubes. Thus, myoblasts are "determined" cells, that are still able to proliferate, but they are committed to the skeletal muscle lineage, and thus can differentiate solely into skeletal muscle cells either during embryonic development or in adult muscle regeneration. The process of skeletal muscle terminal differentiation is orchestrated by a specific genetic program that begins with the permanent exit from the cell cycle of myoblast precursor cells that leads to a definitive silencing of proliferation associated genes, such as E2F target genes1. Indeed, during the process of terminal differentiation, myoblast proliferation arrest is a crucial step that precedes the expression of skeletal muscle specific genes and the fusion of myoblasts into myotubes2. Such a program permits adult muscle stem cells, also called satellite cells, to differentiate during the regeneration process following skeletal muscle injury.
Mammalian myogenesis is critically dependent on a family of myogenic basic helix-loop-helix (bHLH) transcription factors MyoD, Myf5, MRF4 and Myogenin, frequently referred to as the family of skeletal muscle determination factors or MRFs (Muscle Regulatory Factors)3. Each of them plays an essential role in specification and differentiation of skeletal muscle cells and has a specific expression pattern45-7. The activation of Myf5 and MyoD constitutes the determinative step that commits cells to the myogenic lineage, and subsequent expression of Myogenin triggers myogenesis with activation of skeletal muscle specific genes, such as MCK (Muscle Creatine Kinase). Myogenic bHLH transcription factors cooperate with members of the MEF2 family in the activation of muscle genes from previously silent loci8. They also stimulate skeletal muscle gene transcription as heterodimers with ubiquitous bHLH proteins, E12 and E47, known as E proteins, which bind so-called E-boxes in various gene-regulatory regions8. Twist, Id (inhibitor of differentiation) and other factors negatively regulate this process, by competing with MyoD for E proteins binding8.
MyoD is considered as the major player in triggering muscle terminal differentiation9 since it has the capacity to induce a myogenic determination/differentiation (trans-differentiation) program in many fully differentiated non-muscle systems10-13. Indeed, forced expression of MyoD induces the trans-differentiation of different cellular types even those derived from another embryologic origin12. For example, MyoD can convert hepatocytes, fibroblasts, melanocytes, neuroblasts, and adipocytes into muscle-like cells. The trans-differentiative action of MyoD involves an abnormal activation of the myogenic genetic program (notably its target genes) in a non-muscular environment, concomitant to the silencing of the original genetic program (notably, proliferation genes).
In proliferating myoblasts, MyoD is expressed but is unable to activate its target genes even when it binds to their promoters14-16. Therefore, the requirement for MyoD to be continuously expressed in undifferentiated myoblasts remains quite elusive. MyoD could repress its target genes due to recruitment of repressive chromatin-modifying enzymes in proliferating myoblasts prior to loading of activating chromatin remodeling enzymes14,17. For example, in proliferating myoblasts, MyoD is associated with transcriptional co-repressor KAP-1, histone deacetylases (HDACs) and repressive lysine methyltransferases (KMTs), including histone H3 lysine 9 or H3K9 and H3K27 KMTs, and actively suppress its target genes expression by establishing a locally repressive chromatin structure14,17. Importantly, a recent report indicated that MyoD is itself directly methylated by the H3K9 KMT G9a resulting in inhibition of its transactivating activity16.
The epigenetic mechanisms involved in this trans-differentiation of non-muscle cells by MyoD are largely unknown. Notably, some cell lines are resistant to MyoD-induced trans-differentiation. Thus, in HeLa cells, MyoD is either inactive or even might function as repressor rather than activator of transcription due to lack of expression of the BAF60C subunit of the chromatin remodeling complex SWI/SNF18. This model can thus be of choice to better characterize the mechanisms of MyoD-induced gene repression. It is also suitable to assay the ability of MyoD to induce repressive chromatin environment at its target loci with its associated partners and therefore uncover the MyoD-dependent repressive mechanisms in proliferating myoblasts to fine-tune terminal differentiation.
Here we describe the protocol for the identification of MyoD partners by using Tandem Affinity Purification (TAP-Tag) coupled to mass spectrometry (MS) characterization. The use of HeLa-S3 cells stably expressing Flag-HA-MyoD permitted to get enough material to purify the MyoD complexes from fractionated nuclear extracts. The identification of MyoD partners in the heterologous system was followed by validation in a relevant system.
Protocol
1. Preparation of HeLa-S3 Nuclear Salt-extractable and Chromatin-bound Fractions
- Collection of the Cells
- Grow HeLa-S3 Flag-HA-MyoD and HeLa-S3 Flag-HA (control cell line) in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal calf serum, 100 U/ml penicillin and 100 µg/ml streptomycin (growth medium) at 37 °C and 5% CO2 in a humid incubator. Note: HeLa-S3 cells stably expressing Flag-HA-MyoD and HeLa-S3 cells transduced with the empty vector (HeLa-S3 Flag-HA) could be either established using protocol described in:19,20, or provided by the authors.
- Amplify cells up to 10 fully confluent 150 mm dishes of each cell line (HeLa-S3 Flag-HA-MyoD and HeLa-S3 Flag-HA cells).
- Collect the supernatant (contain non-adherent subpopulation) into 5 L spinner.
- Wash the adherent subpopulation with 10 ml of PBS per 150 mm dish, trypsinize cells for 1 min at room temperature (0.05% trypsin-EDTA solution, 2 ml for a 15 cm dish).
- Add 4 ml of the medium per 150 mm dish and collect the cells into 50 ml tubes.
- Combine the supernatant from step 1.1.3 (in the 5 L spinner) and the cells from step 1.1.5, add 500 ml of fresh pre-warmed growth medium for each cell line.
- Transfer cell suspension into 5 L spinner and grow cells for at least 60 hr at 37 °C and 5% CO2 in a humid incubator.
- Add 500 ml of fresh growth medium and grow cells for 24 hr.
- Add 1 L of fresh growth medium and grow cells for 24 hr.
- Take out 1 ml of the cell suspension to count the cells and check cell viability. Color 30 µl of cell suspension with 30 µl of 0.4% trypan blue and count alive (not blue) cells under microscope using hemocytometer.
- Keep the cell density at 2 - 6 x 105 cells/ml by adding fresh growth medium daily; do not exceed 1 x 106 cells/ml. The maximum volume for one 5 L spinner is 2.5 L. Note: For the following steps, proceed by batches of 2 - 2.5 L of culture at a density 1 x 106 cells/ml. Repeat steps 1.1.12 - 1.1.14 to collect about 20 L(≈ 20 g of dry pellet) of each cell line before proceeding to next step.
- Pellet cells by centrifugation at 500 x g for 5 min at 4 °C and discard the supernatant.
- Resuspend cells in 100 ml of ice-cold PBS by pipetting and centrifuge at 500 x g for 5 min at 4 °C to wash the pellet.
- Repeat the wash by resuspending the cells with 25 ml of ice-cold PBS, transferring the suspension into 50 ml tube and centrifuging at 500 x g for 5 min at 4 °C. Note: Cell pellets can be either used immediately or frozen in liquid nitrogen and stored at -80 °C for several days.
- Isolation of the Nuclei Note: Perform steps 1.2.1 - 1.4.3 in the cold room.
- Pre-chill buffers and homogenizers.
- If using frozen material, quickly thaw the cell pellets in water bath at 37 °C.
- Resuspend 20 g (≈ 20 ml) of cells in 20 ml (a volume equal to the size of the cell pellet) of hypotonic buffer A (Table 3) by using 10 ml of the buffer at first, then adding 5 ml, then adding another 5 ml. Wash the pipettes well with fresh buffer to get the maximum of cells.
- Transfer 40 ml cell suspension into the pre-chilled homogenizer with a tight-fitting pestle.
- Homogenize cells with 20 strokes in a homogenizer (20 in and out movements) and transfer cell suspension into 50 ml tube.
- Wash the homogenizer with 7 ml of sucrose buffer (1/3 of the initial cell volume, Table 3) to recover the maximum of cells. Transfer this suspension quickly into the 50 ml tube from step 1.2.5 to preserve the nuclei and limit the leakage.
- Stain a 30 µl aliquot with 30 µl of 0.4% trypan blue, analyze under the microscope to control the lysis efficiency (if the lysis is successful, all nuclei should be blue). If necessary, repeat homogenization step.
- Centrifuge for 7 min at 10,000 x g and 4 °C to pellet the nuclei. Save the supernatant as the cytoplasmic fraction.
- Preparation of Nuclear Salt-extractable (SE) Fraction
- Resuspend the nuclei pellet in 8 ml of sucrose buffer (a volume equal to the size of the nuclei pellet). Add drop by drop while mixing thoroughly on a vortex 8 ml of high salt buffer (1 nuclei pellet volume, Table 3) to get a final concentration of 300 mM NaCl. Incubate for 30 min on ice and mix every 5 min.
- Decrease NaCl concentration to 150 mM by adding 8 ml (1/3 of total volume) of sucrose buffer. Centrifuge for 10 min at 13,000 x g and 4 °C. Collect the supernatant (nuclear salt-extractable fraction, SE). Note: Either proceed to step 1.3.3 or leave the SE fraction on ice during preparation of chromatin-bound fraction and then ultra-centrifuge both fractions simultaneously.
- Ultra-centrifuge SE for 30 min at 85,000 x g at 4 °C. Take supernatant (≈ 26 ml).
- Freeze 100 µl aliquot in liquid nitrogen. Proceed to step 2 "Protein complex purification", using the rest of the extract. Note: The aliquot is to be used as an input control during step 5.1.1.
- Preparation of Chromatin-bound Fraction
- Resuspend the pellet (from step 1.3.5.) meticulously in 7 ml (a volume equal to the size of the pellet) of sucrose buffer.
- Add CaCl2 to a final concentration of 1 mM (28 µl of 0.5 M CaCl2 for 14 ml of suspension) to activate MNase (step 1.4.4) and mix.
- Pre-heat suspension for 1 min at 37 °C.
- Add 70 µl micrococcal nuclease (MNase) to get final concentration of 0.0025 U/µl (1/200 dilution from 0.5 U/µl stock) and mix.
- Incubate exactly 12 min at 37 °C. Mix every 4 min.
- Immediately place the reaction on ice.
- To stop MNase activity, add 112 µl of 0.5 M EDTA pH 8.0 (1/125 dilution) to a final concentration of 4 mM. Incubate for 5 min on ice.
- Sonicate 5 times for 1 min at high amplitude, between two sonications allow a break of 1 min (10 min in total).
- Ultracentrifuge for 30 min at 85,000 x g and 4 °C.
- Collect the supernatant (nucleosome enriched fraction, NE, ≈ 12 ml).
- Freeze 50 µl aliquot in liquid nitrogen. Proceed to step 2 "Protein complex purification", using the rest of the extract. Note: The aliquot is to be used as an input control during step 5.1.1.
2. Protein Complexes Purification
Note: Proceed in parallel extracts from each cell line (HeLa-S3 Flag-HA-MyoD and the control, HeLa-S3 Flag-HA).
- Flag- based Purification
- Pre-wash Flag resin. Use 600 µl of Flag resin (from the commercial 50% stock) for each experimental point (SE and NE fractions for each cell line).
- Transfer resin into 15 ml tube, resuspend in 13 ml of cold TEGN (Table 3), centrifuge for 2 min at 1,000 x g and remove supernatant. Repeat 5 times. After the last wash resuspend Flag resin into equal volume of TEGN (300 µl for each experimental point).
- For each experimental point, mix 600 µl of washed Flag resin and the corresponding protein extracts (in a 15 ml tube for the 12 ml NE and in a 50 ml tube for the 26 ml SE fractions, respectively). Incubate on a rotating wheel overnight at 4 °C.
- Centrifuge for 2 min at 1,000 x g at 4 °C, put aside the supernatant and keep at 4 °C until the result of the purification efficiency test (2.2).
- For the SE samples, resuspend the Flag resin in 4 ml of TEGN and transfer to a 15 ml tube. Repeat this step 3 times, each time transferring to the same 15 ml tube to avoid losing beads. Centrifuge 2 min at 1,000 x g and 4 °C and remove supernatant.
- Wash Flag resin 7 times in a 15 ml tube using 13 ml of TEGN and centrifuging 2 min at 1,000 x g and 4 °C.
- After the last wash, resuspend in 1 ml TEGN and transfer into a 1.5 ml low-binding tube. Centrifuge 2 min at 1,000 x g and 4 °C and remove the supernatant.
- Resuspend in 200 µl of Flag peptide solution at 4 mg/ml at pH 7.8 for each experimental point. Mix the beads and peptide. Add 200 µl of TEGN buffer and incubate at 4 °C for at least 4 hr or overnight to elute bound proteins.
- Centrifuge for 2 min at 1,000 x g at 4 °C and transfer the resin and supernatant (Flag eluate) to an empty spin column placed in a 2 ml tube.
- Centrifuge the column, collect the left-over supernatant in the 2 ml tube.
- Collect the Flag resin by adding TEGN buffer to the column and proceed to the second Flag elution with 200 µl of Flag peptide solution (4 mg/ml) overnight at 4 °C for each experimental point.
- Repeat steps 2.1.9 - 2.1.10 to collect second elution. Combine first and second eluates.
- Test of Purification Efficiency Note: During step 2.1.11 - 2.1.12, perform SDS-PAGE and silver staining (2.2.1 - 2.2.2).
- Take 15 µl of the eluates, add 5 µl of 4x loading buffer and 2 µl of 10x reducing agent, boil for 5 min at 96 °C and run a SDS-polyacrylamide 4 - 12% gel according to the manufacturer's instructions.
- Stain the gel using silver staining kit (follow the manufacturer's instructions). If there is clear differences in protein patterns between Flag-HA-MyoD and Flag-HA mock eluates, in particular the band of Flag-HA MyoD (around 50 kDa, Figure 1A), proceed to the next step.
- Hemagglutinin (HA)-based Purification
- Wash HA resin by transferring into 15 ml tube, resuspending in 13 ml of TEGN and centrifuging 2 min at 1,000 x g at 4 °C. Repeat washing step 5 times. After last wash resuspend beads in equal volume of TEGN buffer. Note: The volume must be adjusted according to the efficiency of Flag-based purification. Start with 300 µl from the commercial 50% stock for each experimental point.
- Transfer HA resin for each experimental point into a 1.5 ml low binding tube. Centrifuge 2 min at 1,000 x g at 4 °C, eliminate the maximum of the wash buffer (Table 3) and add the eluates from Flag-based purification to the HA resin.
- Incubate tubes on the rotating wheel overnight at 4 °C.
- Centrifuge for 2 min at 1,000 x g at 4 °C, put aside the supernatant and keep at 4 °C until the result of the purification efficiency test (2.3.12.).
- Resuspend the HA resin in 0.5 ml of TEGN, transfer to a new low binding 1.5 ml tube. Repeat resuspension once adding to the same 1.5 ml tube to avoid losing beads and centrifuge 2 min at 1,000 x g and 4 °C. Remove supernatant.
- Wash beads 8 times by using 1 ml of TEGN each time, centrifuging 2 min at 1,000 x g and 4 °C and removing supernatant (avoid losing beads). After the last wash, transfer the beads into 0.5 ml tubes.
- Remove as much supernatant as possible. Add 100 µl of HA free peptide solution (4 mg/ml) at pH 7.8 on beads to elute bound proteins. Incubate on the rotating wheel over night at 4 °C. Note: The amount of HA-peptide needs to be adjusted depending on the abundance of the complexes.
- Centrifuge for 2 min at 1,000 x g at 4 °C and transfer the resin and supernatant (HA eluate) to an empty spin column placed in a 2 ml tube.
- Centrifuge the column to collect the left-over supernatant in the 2 ml tube.
- Perform a second elution with 100 µl of HA peptide (4 mg/ml) for each experimental point. Incubate on the rotating wheel over night at 4 °C. Repeat step 2.3.8 - 2.3.9 to collect second elution.
- Combine first and second eluates.
- Test purification efficiency by SDS-PAGE and silver staining (see steps 2.2.) (Figure 1A).
- Concentrate the HA eluate to 30 µl using centrifugal filter units with a 10 kDa cut-off (nominal molecular weight limit, NMWL) according to the manufacturer's instructions.
- Divide the concentrated samples into two parts: 22.5 (3/4) and 7.5 (1/4) µl. Snap freeze 1/4 of samples in liquid nitrogen and store at -80 °C. Use the 3/4 part for step 3. Note: The 1/4 frozen part is to be used for the confirmation of mass spectrometry results by western blot (see step 5.1).
3. Mass Spectrometry Analysis
Note: The following steps should be discussed with the Mass Spectrometry facility that will perform the analysis.
Supplement 3/4 (22.5 µl) of the samples with 7.5 µl of 4x loading buffer and 3.3 µl of 10x reducing agent and boil for 5 min at 96 °C.
Load samples on a SDS-polyacrylamide 4 - 12% gel. Run gel at 150 V constant for 5 min to allow all the proteins to enter the gel without separation.
Wash the gel with water; fix for 20 - 30 min with fixative solution (10% acetic acid, 50% methanol, 40% ultra-clean water) then wash the fixed gel 5 times in ultra-clean water.
Cut the bands containing the proteins, store in 1 ml of water in a 1.5 ml tube and send to mass spectrometry facility.
4. Data Analysis
Note: This is a general guidance for the analysis. The exact steps will depend on the particular mode of the data given by MS facility used to perform the analysis (e.g., 21,22).
Compare the list of the identified proteins in "MyoD" eluates with the list obtained from corresponding negative control preparation. Exclude from the analysis the proteins found in both lists.
Remove proteins that have ≤2 total number of identified peptides from remaining list.
Access functional annotation and functional classification of the obtained list of proteins using DAVID Bioinformatics Resources (http://david.abcc.ncifcrf.gov/home.jsp) and classify the list of the proteins23,24.
(Optional) Remove from the list "hitchhikers", charged non-nuclear proteins with strong adventitious binding to negatively charged DNA25. These contain cytoskeletal, cytoplasmic, mitochondrial, membrane and receptor proteins (Protein folding, Cytoskeleton and Miscellaneous group proteins in Tables 1 and 2).
Compare the obtained lists of the MyoD interactors from SE and NE fractions to identify common proteins and proteins specific for each fraction (Figure 2).
5. Confirmation of Identified Interactors by Western Blot
- Confirmation in HeLa-S3 Flag-HA-MyoD and HeLa-S3 Flag-HA
- Thaw eluate samples obtained at the step 2.3.14 (7.5 µl) on ice. Add 12 µl of TEGN buffer, 10 µl of 4x loading buffer and 3 µl of 10x reducing agent. Boil samples for 5 min at 96 °C.
- Prepare input samples for the western blotting.
- Thaw aliquots from the steps 1.3.9 and 1.4.11 and measure protein concentration, for example using BCA kit (follow manufacturer's instructions).
- Use 15 µg of the protein per lane. Measure the appropriate amount of the extract and adjust volume of the sample up to 15 µl with TEGN buffer.
- Add 5 µl 4x loading buffer and 1.5 µl 10x reducing agent. Boil samples for 5 min at 96 °C.
- Perform a western blot analysis with antibodies of interest (specific for partner candidate identified during MS analysis) using 15 µl of eluate samples. Use input extracts as a control of endogenous protein levels (Figure 3A).
- Confirmation in the Relevant Cellular System
- Preparation of total nuclear extract of C2C12 mouse myoblasts.
- Grow C2C12 mouse myoblasts in DMEM medium supplemented with 15% fetal calf serum, 100 U/ml penicillin and 100 µg/ml streptomycin at 37 °C and 5% CO2 in a humid incubator. Never exceed 80% cell confluence to maintain cells in proliferative state.
- Grow at least two 150 mm dishes of C2C12 for one point (one antibody) of immunoprecipitation (IP). Aspirate the medium, wash cells twice with 10 ml of PBS.
- Scrape the cells and collect into 15 ml tube. Centrifuge at 250 x g for 5 min at 4 °C. Discard supernatant.
- Estimate the volume of the cell pellet (= Vcell). Gently resuspend the pellet in 3 volumes Vcell of Hypotonic buffer B to disrupt cytoplasmic membrane (Table 3).
- Add 0.44 x Vcell volume of 10% NP-40 to reach final concentration of 1% (v/v). Gently invert the tube several times to mix.
- Add 0.89 x Vcell volume of SR (Table 3) to preserve the nuclei integrity. Gently invert the tube several times to homogenize the suspension, and centrifuge for 5 min at 2,000 x g to pellet nuclei.
- Remove the supernatant (cytosolic fraction). Estimate the volume of nuclei pellet (= Vnuc). Resuspend nuclei pellet in one volume Vnuc of sucrose buffer.
- Add drop by drop while mixing thoroughly on a vortex one volume Vnuc of high salt buffer to get a final concentration of 300 mM NaCl, and incubate for 30 min on ice. Mix every 5 min.
- Add one volume Vnuc of sucrose buffer (to decrease NaCl concentration to 150 mM) and CaCl2 (final concentration 1 mM). Mix.
- Pre-heat suspension for 1 min at 37 °C, add micrococcal nuclease to get final of concentration 0.0025 U/µl (1/200 from stock of 0.5 U/µl). Mix.
- Incubate exactly 12 min at 37 °C. Mix every 3 min.
- Immediately place reaction on ice and add EDTA (final concentration 4 mM) to stop MNase activity. Incubate for 5 min on ice.
- Sonicate 5 times for 0.5 min each at high amplitude. Between two sonications, allow a break of 0.5 min (5 min in total).
- Ultra-centrifuge for 30 min at 85,000 x g and 4 °C. Collect the supernatant (total nuclear extract).
- Measure protein concentration of the total nuclear extract, for example using BCA kit (follow manufacturer's instructions) and proceed immediately to step 5.2.2.
- Immunoprecipitation (IP) of endogenous MyoD.
- To pre-clear the extracts, wash 10 µl of protein G agarose beads for each IP point.
- Transfer the needed volume of protein G agarose beads into a 1.5 ml tube, resuspend in 1.4 ml of cold TEGN, centrifuge for 2 min at 1,800 x g and remove supernatant. Repeat 3 times.
- Mix pre-washed protein G agarose beads with the appropriate volume of total nuclear extract. Use 500 µg of total nuclear extract protein per IP point.
- Incubate on a rotating wheel at 4 °C for 2 hr to pre-clear the extracts.
- Centrifuge for 5 min at 1,800 x g at 4 °C. Keep supernatant.
- Mix 5 µg of MyoD Ab or 5 µg of rabbit IgG with 500 µg pre-cleared total nuclear extracts in 1.5 ml low binding tubes. Add TEGN buffer up to 1 ml. Incubate on a rotating wheel at 4 °C overnight. Note: Perform the following steps during step 5.2.2.3.
- Pre-wash 7.5 µl protein A/G stock beads solution per IP point.
- Transfer the needed volume of beads into a 1.5 ml tube, resuspend beads in 1 ml of TEGN and centrifuge at 1,800 x g at 4 °C for 2 min. Remove the supernatant. Repeat 3 times.
- Resuspend protein A/G beads in 1.5 ml blocking solution (Table 3) and incubate on a rotating wheel overnight at 4 °C.
- Centrifuge total nuclear extracts incubated with Abs (step 5.2.2.5) at 16,000 x g for 10 min at 4 °C. Keep supernatant.
- Centrifuge blocked protein A/G resin (step 5.2.2.7) at 1,800 x g for 2 min at 4 °C. Discard supernatant and resuspend in 1 ml of TEGN buffer. Re-centrifuge at 1,800 x g for 2 min at 4 °C to wash out blocking buffer.
- Resuspend blocked protein A/G resin in 1 ml of TEGN buffer and aliquot into new low protein-binding tubes to obtain 7.5 µl blocked protein A/G resin per IP point. Centrifuge at 1,800 x g at 4 °C for 2 min and remove as much as possible of the supernatant.
- Add the total nuclear extracts incubated with the Abs (or control IgG) to the A/G resin. Mix and incubate for 2 hr at room temperature (RT) on a rotating wheel.
- Centrifuge suspension 1,800 x g at RT for 2 min. Discard supernatant, resuspend in 1 ml of wash buffer and transfer suspension into new low binding tubes. Incubate at RT on a rotating wheel for 5 min.
- Centrifuge suspension at 1,800 x g at RT for 2 min, resuspend in 1 ml of wash buffer and incubate at RT on a rotating wheel for 5 min. Repeat 4 times. For the last wash transfer to a new low binding tube.
- Remove maximum of the supernatant. Add 7 µl of wash buffer, 7 µl of 4x loading buffer and 3 µl of 10x reducing agent to the beads, mix and boil for 5 min at 96 °C.
- Perform a western blot analysis with antibodies of interest (specific for partner candidate for the immune precipitated protein). Use 1% (5 μg) of input extracts as a control of endogenous protein levels (Figure 3B).
Representative Results
To understand the regulation of MyoD activity, we undertook the exhaustive characterization of the MyoD complexes using biochemical purification, based on the immunopurification of a double tagged form of MyoD followed by mass spectrometry (MS). The use of HeLa-S3 cell line expressing Flag-HA-tagged MyoD and a control cell line expressing Flag-HA permits to get enough material to purify the MyoD complex by performing double-affinity purification of Flag-HA-MyoD.
We fractionated the cell extracts into cytoplasmic and nuclear fractions, then further fractionated the nuclear fraction to salt-extracted (nuclear salt-extractable, SE) and enriched for nucleosomes (nucleosome-enriched, NE) fractions. TAP-Tag purification from these separated nuclear fractions (Figure 1, Tables 1 and 2) permitted unravelling partners that have relatively low abundance when localized at one specific subnuclear compartment. Furthermore, such a strategy was exploited to uncover partners of unbound (SE) versus DNA-bound (NE) MyoD to get insights on MyoD activity regulation.
For TAP-Tag purification, the Flag-HA tandem epitope system was used. The small hydrophilic Flag and HA epitopes have minimal interference with protein function and are highly accessible for antibody-antigen interaction. Anti-Flag and anti-HA resin-based sequential immunopurification was performed, followed by elution of the immunopurified complexes using Flag and HA peptides. The eluted proteins were then run on an SDS-PAGE to allow all proteins to enter the gel. The pieces of gel containing all purified proteins were cut; the proteins were extracted, trypsin-digested and identified by mass spectrometry (MS).
As shown in Figure 2, MyoD complexes purified from NE fraction have higher enrichment in transcription factors and co-repressors. MS analysis unraveled a series of known partners of MyoD (such as Pbx, Id, E12/E47 (HTF4), BRG1 (SMCA4), MEIS1…) and new partners that were confirmed in the studies generated by this analysis (such as HP1, CBF, MBD3, BAF47 (SNF5/INI1) and all the other SWI/SNF complex subunits…) (Figure 2 and 3)26-28. This sheds light on DNA-bound MyoD partners and possible co-regulators that can establish a repressive-chromatin environment. For example, HP1 proteins, which were identified as MyoD partners by described methodology, are known to bind methylated H3K9 to maintain gene repression and heterochromatin structure. Indeed, HP1 inhibits MyoD transcriptional activity resulting in impaired MyoD target gene expression and muscle terminal differentiation26.
Further fractionation of the SE and NE MyoD complexes on glycerol gradient (as described in29) uncovered the MyoD sub-complexes in the two subnuclear compartments (Figure 1B). In particular, SE MyoD is distributed in three sub-complexes, while the chromatin-bound MyoD belongs mainly to one complex.
Some of the TAP-tag/MS revealed interactors were confirmed by western blot on MyoD complexes. These include the transcription factor CBF, EBB, MTG8R and the SWI/SNF subunit BAF47 (SNF5) (Figure 3A, left) and HP1 proteins (CBX1 and CBX3) (Figure 3A, right). Importantly, since HeLa cells are not muscle cells and do not express MyoD, it is necessary to confirm interactions between newly identified interactors and MyoD in myoblasts (Figure 3 B-D). Notably, for such validation, the total nuclear extracts (without separation on SE and NE) are usually sufficient, which permits reduction of the amount of myoblasts used for sample preparation. The in vitro interaction assays as in26,27, help to further validated these findings. Finally, the functional meanings of these interactions in muscle cells should be further addressed as in26-28.
Taken together, presented data show a global view of ubiquitously expressed MyoD partners and pave the way to further functional studies in a more relevant muscle model.
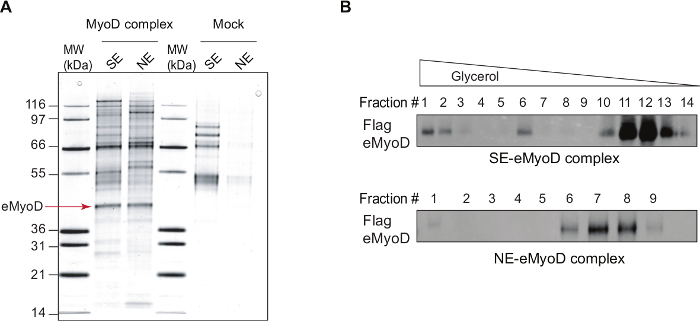 Figure 1: MyoD Complexes Isolated by Tandem Affinity Purification. (A) A silver staining of the double affinity-purified eMyoD complexes isolated from nuclear salt-extractable (SE) or nucleosome-enriched (NE) nuclear fractions of HeLa-S3 cell lines stably expressing Flag-HA-MyoD (MyoD complex) and control cell line (Mock). MW, molecular weight marker in kilo dalton (kDa). Arrow indicates Flag-HA-MyoD (eMyoD). This research was originally published in37. Copyright The American Society for Biochemistry and Molecular Biology. This figure has been modified from26: the lanes with mock purifications and eMyoD complex isolated from SE nuclear fractions are now shown. (B) Double affinity-purified eMyoD complexes as in (A) were fractionated on glycerol gradient ranging from 20% to 41%. Fractions were manually collected, concentrated and analyzed by western blot (WB) using anti-Flag antibodies. Note the presence of several eMyoD-containing sub-complexes in nuclear salt-extractable (SE) fraction. Please click here to view a larger version of this figure.
Figure 1: MyoD Complexes Isolated by Tandem Affinity Purification. (A) A silver staining of the double affinity-purified eMyoD complexes isolated from nuclear salt-extractable (SE) or nucleosome-enriched (NE) nuclear fractions of HeLa-S3 cell lines stably expressing Flag-HA-MyoD (MyoD complex) and control cell line (Mock). MW, molecular weight marker in kilo dalton (kDa). Arrow indicates Flag-HA-MyoD (eMyoD). This research was originally published in37. Copyright The American Society for Biochemistry and Molecular Biology. This figure has been modified from26: the lanes with mock purifications and eMyoD complex isolated from SE nuclear fractions are now shown. (B) Double affinity-purified eMyoD complexes as in (A) were fractionated on glycerol gradient ranging from 20% to 41%. Fractions were manually collected, concentrated and analyzed by western blot (WB) using anti-Flag antibodies. Note the presence of several eMyoD-containing sub-complexes in nuclear salt-extractable (SE) fraction. Please click here to view a larger version of this figure.
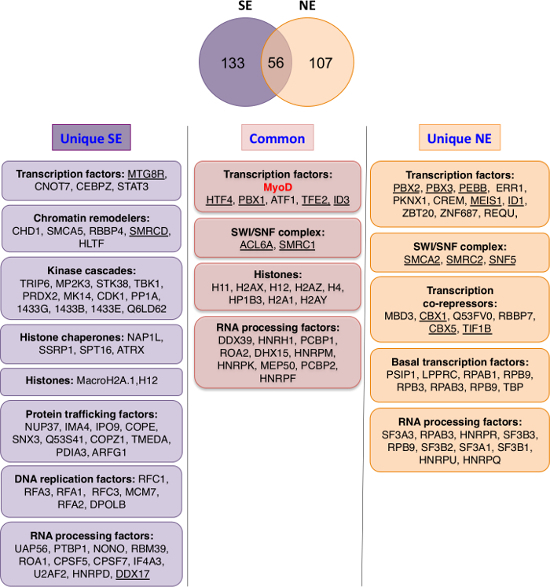 Figure 2: Comparison of Chromatin-bound (nucleosome enriched, NE) versus Nuclear Soluble (nuclear salt extractable, SE) MyoD Partners. Top: Venn diagram showing overlap between eMyoD interactors isolated from nuclear salt-extractable (SE) and nucleosome-enriched (NE) fraction. The ribosomal proteins, translation-initiation factors, DNA repair factors and the tubulin isoforms were excluded from the analysis as they are present in various different data-sets obtained by TAP and considered as non-specific. Bottom: The MyoD interactors found in both SE and NE fractions (common) or specific for one of the fractions (unique) were divided in groups based on their functional annotations. Cytoskeleton-related and other miscellaneous proteins are not depicted. The underlined proteins are MyoD interactors validated either in HeLa and/or in myoblasts by co-immunoprecipitation. Please click here to view a larger version of this figure.
Figure 2: Comparison of Chromatin-bound (nucleosome enriched, NE) versus Nuclear Soluble (nuclear salt extractable, SE) MyoD Partners. Top: Venn diagram showing overlap between eMyoD interactors isolated from nuclear salt-extractable (SE) and nucleosome-enriched (NE) fraction. The ribosomal proteins, translation-initiation factors, DNA repair factors and the tubulin isoforms were excluded from the analysis as they are present in various different data-sets obtained by TAP and considered as non-specific. Bottom: The MyoD interactors found in both SE and NE fractions (common) or specific for one of the fractions (unique) were divided in groups based on their functional annotations. Cytoskeleton-related and other miscellaneous proteins are not depicted. The underlined proteins are MyoD interactors validated either in HeLa and/or in myoblasts by co-immunoprecipitation. Please click here to view a larger version of this figure.
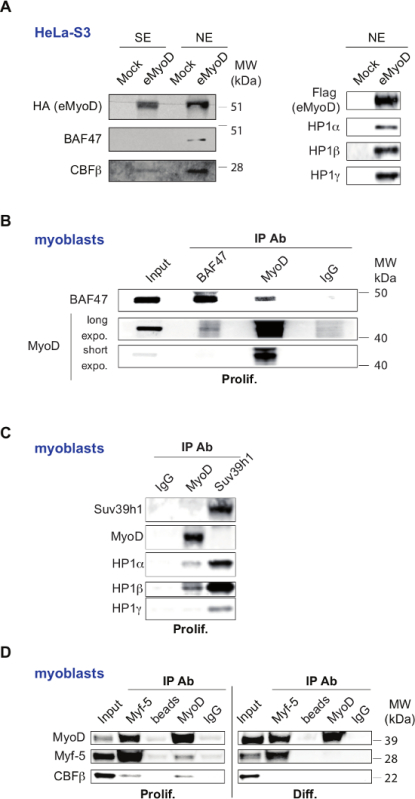 Figure 3: Validation of Selected Set of MyoD Interactors. (A) Validation of MyoD interactors, identified by mass spectrometry, in HeLa-S3 cell line stably expressing Flag-HA-MyoD (eMyoD). Left panel: Western blot analysis of double affinity-purified MyoD complexes isolated from nuclear salt-extractable (SE) or nucleosome-enriched (NE) fractions of HeLa-S3 cell line stably expressing eMyoD or control cell line (Mock) with the indicated antibodies. MW, molecular weight marker. Right panel: Western blot analysis of double affinity-purified MyoD complexes isolated from nuclear nucleosome-enriched fractions of HeLa-S3 cell line stably expressing eMyoD or control cell line (Mock) with indicated antibodies. This panel was originally published in The Journal of Biological Chemistry. Yahi H, Fritsch L, Philipot O, Guasconi V, Souidi M, Robin P, Polesskaya A, Losson R, Harel-Bellan A, Ait-Si-Ali S. J Biol Chem. 2008 Aug 29;283(35):23692-700. doi: 10.1074/jbc.M802647200. Epub 2008 Jul 2. Copyright The American Society for Biochemistry and Molecular Biology. This figure has been modified from26: police type and size was changed and the text was rotated to unify the labeling within the figure. MyoD was labeled as eMyoD to avoid the confusion with endogenous IP presented in the other panels. (B-D) Validation of MyoD interactors in C2C12 mouse myoblasts. (B) Nuclear total extracts from proliferating C2C12 myoblasts were used for immunoprecipitation (IP) with antibodies raised against BAF47 (SNF5) or MyoD, or with control IgG. The resulting precipitates were analyzed by WB with the indicated antibodies. For anti-MyoD antibodies longer (long expo.) and shorter (short expo.) exposure times are shown. Input extracts were loaded to show endogenous protein levels. This panel has been published in28 under the Creative Commons Attribution (CC BY) licence (http://creativecommons.org/licenses/by/4.0/). This figure has been modified from28: police type and size was changed and the test was rotated to unify the labeling within the figure. (C) Nuclear total extracts from proliferating C2C12 myoblasts were used for immunoprecipitation with antibodies raised against MyoD, Suv39h1 (positive control), or control IgG (negative control). The resulting precipitates were then subjected to WB with the indicated antibodies. This panel was originally published in The Journal of Biological Chemistry. Yahi H, Fritsch L, Philipot O, Guasconi V, Souidi M, Robin P, Polesskaya A, Losson R, Harel-Bellan A, Ait-Si-Ali S. J Biol Chem. 2008 Aug 29;283(35):23692-700. doi: 10.1074/jbc.M802647200. Epub 2008 Jul 2. Copyright The American Society for Biochemistry and Molecular Biology. This figure has been modified from26: police type and size was changed and the text was rotated to unify the labeling within the figure. (D) Nuclear total extracts from proliferating (prolif.) and differentiating C2C12 myoblasts (48 hr, indicated as Diff.) were used for immunoprecipitation (IP) with antibodies raised against MyoD and Myf5, or with normal rabbit IgG and with empty beads as negative control. The resulting precipitates were analyzed by WB with the indicated antibodies. Input extracts were loaded to show endogenous protein levels. This panel has been published in27 under the Creative Commons Attribution (CC BY) licence (http://creativecommons.org/licenses/by/4.0/). This figure has been modified from27: police type and size was changed and the text was rotated to unify the labeling within the figure. The panels with the results obtained from proliferating and differentiating C2C12 are separated in two as opposed to the original figure. Please click here to view a larger version of this figure.
Figure 3: Validation of Selected Set of MyoD Interactors. (A) Validation of MyoD interactors, identified by mass spectrometry, in HeLa-S3 cell line stably expressing Flag-HA-MyoD (eMyoD). Left panel: Western blot analysis of double affinity-purified MyoD complexes isolated from nuclear salt-extractable (SE) or nucleosome-enriched (NE) fractions of HeLa-S3 cell line stably expressing eMyoD or control cell line (Mock) with the indicated antibodies. MW, molecular weight marker. Right panel: Western blot analysis of double affinity-purified MyoD complexes isolated from nuclear nucleosome-enriched fractions of HeLa-S3 cell line stably expressing eMyoD or control cell line (Mock) with indicated antibodies. This panel was originally published in The Journal of Biological Chemistry. Yahi H, Fritsch L, Philipot O, Guasconi V, Souidi M, Robin P, Polesskaya A, Losson R, Harel-Bellan A, Ait-Si-Ali S. J Biol Chem. 2008 Aug 29;283(35):23692-700. doi: 10.1074/jbc.M802647200. Epub 2008 Jul 2. Copyright The American Society for Biochemistry and Molecular Biology. This figure has been modified from26: police type and size was changed and the text was rotated to unify the labeling within the figure. MyoD was labeled as eMyoD to avoid the confusion with endogenous IP presented in the other panels. (B-D) Validation of MyoD interactors in C2C12 mouse myoblasts. (B) Nuclear total extracts from proliferating C2C12 myoblasts were used for immunoprecipitation (IP) with antibodies raised against BAF47 (SNF5) or MyoD, or with control IgG. The resulting precipitates were analyzed by WB with the indicated antibodies. For anti-MyoD antibodies longer (long expo.) and shorter (short expo.) exposure times are shown. Input extracts were loaded to show endogenous protein levels. This panel has been published in28 under the Creative Commons Attribution (CC BY) licence (http://creativecommons.org/licenses/by/4.0/). This figure has been modified from28: police type and size was changed and the test was rotated to unify the labeling within the figure. (C) Nuclear total extracts from proliferating C2C12 myoblasts were used for immunoprecipitation with antibodies raised against MyoD, Suv39h1 (positive control), or control IgG (negative control). The resulting precipitates were then subjected to WB with the indicated antibodies. This panel was originally published in The Journal of Biological Chemistry. Yahi H, Fritsch L, Philipot O, Guasconi V, Souidi M, Robin P, Polesskaya A, Losson R, Harel-Bellan A, Ait-Si-Ali S. J Biol Chem. 2008 Aug 29;283(35):23692-700. doi: 10.1074/jbc.M802647200. Epub 2008 Jul 2. Copyright The American Society for Biochemistry and Molecular Biology. This figure has been modified from26: police type and size was changed and the text was rotated to unify the labeling within the figure. (D) Nuclear total extracts from proliferating (prolif.) and differentiating C2C12 myoblasts (48 hr, indicated as Diff.) were used for immunoprecipitation (IP) with antibodies raised against MyoD and Myf5, or with normal rabbit IgG and with empty beads as negative control. The resulting precipitates were analyzed by WB with the indicated antibodies. Input extracts were loaded to show endogenous protein levels. This panel has been published in27 under the Creative Commons Attribution (CC BY) licence (http://creativecommons.org/licenses/by/4.0/). This figure has been modified from27: police type and size was changed and the text was rotated to unify the labeling within the figure. The panels with the results obtained from proliferating and differentiating C2C12 are separated in two as opposed to the original figure. Please click here to view a larger version of this figure.
Table 1:List of Proteins Identified by MS Analysis in Double Affinity-purified eMyoD Complexes Isolated from Nuclear Salt-extractable Fraction after the Subtraction of the Background Proteins. (Proteins identified by MS in eluates from control cell line were considered as non-specific background.) The data represent the sum of four independent purifications. Please click here to download this file.
Table 2:List of Proteins Identified by MS Analysis in Double Affinity-purified eMyoD Complexes Isolated from Nucleosome-enriched Fraction after the Subtraction of the Background Proteins. The data represent the sum of three independent purifications. Please click here to download this file.
Table 3. Buffer Compositions. Please click here to download this file.
Discussion
The presented method permits exhaustive identification of partners of a transcription factor, MyoD. It revealed MyoD partners in a heterologous system resistant to MyoD-induced differentiation, namely HeLa-S3 cells. Thus, by definition, identified MyoD partners are ubiquitously expressed. These include general and sequence-specific transcription factors, chromatin-modifying enzymes, RNA processing proteins, kinases (Tables 1 and 2). Since HeLa-S3 cells are resistant to MyoD-induced trans-differentiation, MyoD activity is mainly repressive and the identified partners are likely to be MyoD co-repressors. This is highlighted by the presence of Id proteins (Tables 1 and 2, Figure 2), known to inhibit MyoD transactivation ability8. Importantly, such repressive state corresponds to the MyoD status in proliferating myoblasts. Indeed, we confirmed that MyoD interactions with CBFβ, a MyoD partner identified by the described method, can be detected in proliferating C2C12 myoblasts, but are lost when cells underwent differentiation (see Figure 3D). Nevertheless, it is important to note that one of the main limitations of the presented methodology is the lack of MyoD co-activators identification. Consistently, using this method none of the known MyoD co-activators, such as histone acetyltransferases32 were identified.
Purification of MyoD complexes either from the soluble fraction (nuclear salt extractable, SE) of the nucleus or the chromatin-enriched fraction (nucleosome enriched, NE) (Figure 1A) serves at least two major functions. Firstly, such fractionation increases the representation of the non-stoichiometric partners that would be masked if MyoD purification performed from total nuclear extracts. Secondly, this permits the separation of two functional subpopulations of MyoD: pre-deposited/evicted (SE) and chromatin-bound (NE). Among interactors specific for DNA-unbound MyoD, many kinases, transcription factors, trafficking proteins as well as some chromatin remodelers were identified (Figure 2). When fully validated, such a network could provide an insight into the mode of activity regulation of DNA-unbound MyoD. Additional search for MyoD post-translational modifications in these two nuclear compartments by mass spectrometry, could further unravel MyoD activity regulation. Finally, fractionation of soluble and chromatin-bound MyoD complexes on a glycerol gradient revealed that while the chromatin-bound MyoD is mainly contained in one complex, DNA-unbound MyoD is distributed in three sub-complexes (Figure 1B). Characterization of these sub-complexes by either MS and/or western blot against protein candidates should elucidate further the mode of MyoD regulation.
As stressed above, HeLa cells do not naturally express the muscle transcription factor MyoD. It was thus important to confirm the found interactions in a skeletal muscle model (Figure 3). Many reports confirmed in relevant models such functional interactions between MyoD and some of the partners identified in the presented TAP-tag assay. This is for example the case for prohibitin33, DDX1734 , Meis135, CBF27, HP126, and SWI/SNF chromatin-remodeling complex36,28,30.
Note that it is possible to perform such TAP-tag purification from low amount of cells, such as from myoblasts, when combined to sensitive MS. Indeed, a recent paper described MyoD partners characterization after inducible expression of Flag-tagged MyoD in myoblasts30. Since stable and continuous MyoD overexpression in myoblasts is deleterious, an alternative approach would be adding the Flag-HA tags into the endogenous MyoD allele(s) in myoblasts by using genome-editing methods, such as CRISPR-Cas931. Notably, addition of the tag(s) could potentially alter protein function and/or association with binding partners, therefore the place of the tag (N- or C-terminus) should be chosen with caution. A functional assay of the fusion protein in relevant system must be performed prior to TAP-tag purification to ensure the tagged protein is functional. Immunoprecipitation of endogenous proteins avoids these problems, however, it relies on the availability of a specific and high affinity antibody, which is rarely available.
Another added value of the described approach is the possibility to identify post-translational modifications (PTMs) of the purified protein itself and of its abundant partners. Thereby, with this feature the TAP-tag purification is suitable to identify not only new interaction partners but also new enzymatic functions associated to the protein of interest and/or its partners. In the case of a chromatin-binding protein (i.e., transcription factors, enzymes), this method is thus adapted for identification of the associated "histone code". Indeed, the amino-terminal histone tails, which are exposed on the nucleosome surface, are subject to multiple covalent PTMs. Histone PTMs confer a unique signature to the nucleosomes involved. A combination of different modifications on histone N-terminal tails can thus alter chromatin structure to allow gene expression regulation. Thus, characterizing such modifications associated to a given protein could provide insights into the roles and mechanisms of action of the studied chromatin-binding proteins22.
In summary, the presented methodology permits comprehensive identification of MyoD partners. The TAP-tag purification provides an alternative to other approaches such as GST pull-downs, yeast two-hybrid assays and phage display. Even if for practical reasons (production of large amount of nuclear extracts) we have to use a heterologous cellular system, we have been able to confirm the involvement of the identified MyoD partners in skeletal muscle differentiation. The obtained data shows that the MyoD myogenic factor seems to interact with a plethora of proteins ranging from transcriptional regulators to RNA binding proteins, suggesting the different mechanisms regulating the activity of a transcription factor.
In conclusion, the same methodological approach could be used to identify ubiquitously expressed partners of numerous nuclear factors that could be difficult to study in their specific cellular context.
Disclosures
The authors have nothing to disclose.
Acknowledgments
Work in the Ait-Si-Ali lab was supported by the Association Française contre les Myopathies Téléthon (AFM-Téléthon); Institut National du Cancer (INCa); Agence Nationale de la Recherche (ANR), Fondation Association pour la Recherche sur le Cancer (Fondation ARC); Groupement des Entreprises Françaises pour la Lutte contre le Cancer (GEFLUC); CNRS; Université Paris Diderot and the ''Who Am I?'' Laboratory of Excellence #ANR-11- LABX-0071 funded by the French Government through its ''Investments for the Future'' program operated by the ANR under grant #ANR-11-IDEX-0005-01. EB was supported by an INCa grant.
References
- Ait-Si-Ali S, et al. A Suv39h-dependent mechanism for silencing S-phase genes in differentiating but not in cycling cells. Embo J. 2004;23(3):605–615. doi: 10.1038/sj.emboj.7600074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckingham M. Skeletal muscle development and the role of the myogenic regulatory factors. Biochem Soc Trans. 1996;24(2):506–509. doi: 10.1042/bst0240506. [DOI] [PubMed] [Google Scholar]
- Arnold HH, Winter B. Muscle differentiation: more complexity to the network of myogenic regulators. Curr Opin Genet Dev. 1998;8(5):539–544. doi: 10.1016/s0959-437x(98)80008-7. [DOI] [PubMed] [Google Scholar]
- Buckingham M. Skeletal muscle formation in vertebrates. Curr Opin Genet Dev. 2001;11(4):440–448. doi: 10.1016/s0959-437x(00)00215-x. [DOI] [PubMed] [Google Scholar]
- Yun K, Wold B. Skeletal muscle determination and differentiation: story of a core regulatory network and its context. Curr Opin Cell Biol. 1996;8(6):877–889. doi: 10.1016/s0955-0674(96)80091-3. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Olson EN. Defining the regulatory networks for muscle development. Curr Opin Genet Dev. 1996;6(4):445–453. doi: 10.1016/s0959-437x(96)80066-9. [DOI] [PubMed] [Google Scholar]
- Arnold HH, Braun T. Targeted inactivation of myogenic factor genes reveals their role during mouse myogenesis: a review. Int J Dev Biol. 1996;40(1):345–353. [PubMed] [Google Scholar]
- Puri PL, Sartorelli V. Regulation of muscle regulatory factors by DNA-binding, interacting proteins, and post-transcriptional modifications. J Cell Physiol. 2000;185(2):155–173. doi: 10.1002/1097-4652(200011)185:2<155::AID-JCP1>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Tapscott SJ. The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development. 2005;132(12):2685–2695. doi: 10.1242/dev.01874. [DOI] [PubMed] [Google Scholar]
- Lassar AB, Paterson BM, Weintraub H. Transfection of a DNA locus that mediates the conversion of 10T1/2 fibroblasts to myoblasts. Cell. 1986;47(5):649–656. doi: 10.1016/0092-8674(86)90507-6. [DOI] [PubMed] [Google Scholar]
- Davis RL, Weintraub H, Lassar AB. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell. 1987;51:987–1000. doi: 10.1016/0092-8674(87)90585-x. [DOI] [PubMed] [Google Scholar]
- Tapscott SJ, et al. MyoD1: a nuclear phosphoprotein requiring a myc homology region to convert fibroblasts to myoblasts. Science. 1988;242:405–411. doi: 10.1126/science.3175662. [DOI] [PubMed] [Google Scholar]
- Weintraub H, et al. Activation of muscle-specific genes in pigment, nerve, fat, liver, and fibroblast cell lines by forced expression of MyoD. Proc Natl Acad Sci U S A. 1989;86(14):5434–5438. doi: 10.1073/pnas.86.14.5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mal A, Harter ML. MyoD is functionally linked to the silencing of a muscle-specific regulatory gene prior to skeletal myogenesis. Proc Natl Acad Sci U S A. 2003;100(4):1735–1739. doi: 10.1073/pnas.0437843100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkawa Y, Marfella CG, Imbalzano AN. Skeletal muscle specification by myogenin and Mef2D via the SWI/SNF ATPase Brg1. Embo J. 2006;25(3):490–501. doi: 10.1038/sj.emboj.7600943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling BM, et al. Lysine methyltransferase G9a methylates the transcription factor MyoD and regulates skeletal muscle differentiation. Proc Natl Acad Sci U S A. 2012;109(3):841–846. doi: 10.1073/pnas.1111628109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CL, McKinsey TA, Olson EN. Association of class II histone deacetylases with heterochromatin protein 1: potential role for histone methylation in control of muscle differentiation. Mol Cell Biol. 2002;22(20):7302–7312. doi: 10.1128/MCB.22.20.7302-7312.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forcales SV, et al. Signal-dependent incorporation of MyoD-BAF60c into Brg1-based SWI/SNF chromatin-remodelling complex. The EMBO journal. 2011;31(2):301–316. doi: 10.1038/emboj.2011.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatani Y, Ogryzko V. Immunoaffinity purification of mammalian protein complexes. Methods Enzymol. 2003;370:430–444. doi: 10.1016/S0076-6879(03)70037-8. [DOI] [PubMed] [Google Scholar]
- Ouararhni K, et al. The histone variant mH2A1.1 interferes with transcription by down-regulating PARP-1 enzymatic activity. Genes Dev. 2006;20(23):3324–3336. doi: 10.1101/gad.396106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsch L, et al. A subset of the histone H3 lysine 9 methyltransferases Suv39h1, G9a, GLP, and SETDB1 participate in a multimeric complex. Mol Cell. 2010;37(1):46–56. doi: 10.1016/j.molcel.2009.12.017. [DOI] [PubMed] [Google Scholar]
- Robin P, Fritsch L, Philipot O, Svinarchuk F, Ait-Si-Ali S. Post-translational modifications of histones H3 and H4 associated with the histone methyltransferases Suv39h1 and G9a. Genome Biol. 2007;8(12):R270. doi: 10.1186/gb-2007-8-12-r270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta S, et al. The protein composition of mitotic chromosomes determined using multiclassifier combinatorial proteomics. Cell. 2010;142(5):810–821. doi: 10.1016/j.cell.2010.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahi H, et al. Differential cooperation between heterochromatin protein HP1 isoforms and MyoD in myoblasts. J Biol Chem. 2008;283(35):23692–23700. doi: 10.1074/jbc.M802647200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philipot O, et al. The core binding factor CBF negatively regulates skeletal muscle terminal differentiation. PloS one. 2010;5(2) doi: 10.1371/journal.pone.0009425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joliot V, et al. The SWI/SNF subunit/tumor suppressor BAF47/INI1 is essential in cell cycle arrest upon skeletal muscle terminal differentiation. PloS one. 2014;9(10):e108858. doi: 10.1371/journal.pone.0108858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanese N. Small-scale density gradient sedimentation to separate and analyze multiprotein complexes. Methods. 1997;12(3):224–234. doi: 10.1006/meth.1997.0475. [DOI] [PubMed] [Google Scholar]
- Singh K, et al. A KAP1 phosphorylation switch controls MyoD function during skeletal muscle differentiation. Genes Dev. 2015;29(5):513–525. doi: 10.1101/gad.254532.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahi H, Philipot O, Guasconi V, Fritsch L, Ait-Si-Ali S. Chromatin modification and muscle differentiation. Expert Opin Ther Targets. 2006;10(6):923–934. doi: 10.1517/14728222.10.6.923. [DOI] [PubMed] [Google Scholar]
- Sun L, Liu L, Yang XJ, Wu Z. Akt binds prohibitin 2 and relieves its repression of MyoD and muscle differentiation. J Cell Sci. 117. 2004;117(Pt 14):3021–3029. doi: 10.1242/jcs.01142. [DOI] [PubMed] [Google Scholar]
- Caretti G, et al. The RNA helicases p68/p72 and the noncoding RNA SRA are coregulators of MyoD and skeletal muscle differentiation. Dev Cell. 2006;11(4):547–560. doi: 10.1016/j.devcel.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Knoepfler PS, et al. A conserved motif N-terminal to the DNA-binding domains of myogenic bHLH transcription factors mediates cooperative DNA binding with pbx-Meis1/Prep1. Nucleic Acids Res. 1999;27(18):3752–3761. doi: 10.1093/nar/27.18.3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albini S, Puri PL. SWI/SNF complexes, chromatin remodeling and skeletal myogenesis: it's time to exchange! Exp Cell Res. 2010;316(18):3073–3080. doi: 10.1016/j.yexcr.2010.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahi H, Fritsch L, Philipot O, Guasconi V, Souidi M, Robin P, Polesskaya A, Losson R, Harel-Bellan A, Ait-Si-Ali S. Differential Cooperation between Heterochromatin Protein HP1 Isoforms and MyoD in Myoblasts. J Biol Chem. 2008;283(35):23692–23700. doi: 10.1074/jbc.M802647200. [DOI] [PMC free article] [PubMed] [Google Scholar]