

Abstract
Normal human retinal vascular development uses angiogenesis and vasculogenesis, both of which are interrupted in the vaso-obliteration phase of retinopathy of prematurity (ROP). Canine oxygen-induced retinopathy (OIR) closely resembles human ROP. Canine retinal endothelial cells (ECs) and angioblasts were used to model OIR and characterize the effects of hyperoxia on angiogenesis and vasculogenesis. Cell cycle analysis showed that hyperoxia reduced the number of G1 phase cells and showed increased arrest in S phase for both cell types. Migration of ECs was significantly inhibited in hyperoxia (P < 0.01). Hyperoxia disrupted the cytoskeleton of angioblasts but not ECs after 2 days. Differentiation of angioblasts into ECs (determined by acetylated low-density lipoprotein uptake) was evaluated after basic fibroblast growth factor treatment. Differentiation of angioblasts into pericytes was determined by smooth muscle actin expression after treatment with platelet-derived growth factor. Differentiation into ECs was significantly inhibited by hyperoxia (P < 0.0001). The percentage of CXCR4+ cells (a marker for retinal vascular precursors) increased in both treatment groups after hyperoxia. These data show novel mechanisms of hyperoxia-induced disruption of vascular development.
Keywords: angioblasts, endothelial cells, hyperoxia, retina, vasculogenesis, angiogenesis, CXCR4
INTRODUCTION
Two critical processes in normal vascular development are vasculogenesis and angiogenesis. Initially, vasculogenesis begins with clustering (or aggregation) of mesenchymal progenitors called angioblasts. In human and canine retinal vascular development, angioblasts are present in avascular retina. The angioblasts aggregate and differentiate to form the primary or superficial retinal vasculature (McLeod et al., 1987; Hughes et al., 2000; Chan-Ling et al., 2004). In vitro, retinal angioblasts can differentiate into endothelial cells or pericytes (Lutty et al., 2006). Alternately, during angiogenesis, the endothelial cells sprout from pre-existing vessels by degrading vascular basement membrane, and then the endothelial cells migrate and proliferate to form new vessels. This is the process by which the deep capillary plexus of retina develops in most species (McLeod et al., 1987, 1996; Hughes et al., 2000). Oxygen levels play key roles in vascular development, but relatively little work has been done to elucidate the effects of hyperoxia on vascular development (Maltepe and Simon, 1998). Disruption of appropriate oxygen levels creating either hyperoxia or hypoxia, can lead to vaso-obliteration and abnormal neovascularization, respectively, in the developing retina (Penn et al., 1992; Smith et al., 1994; McLeod et al., 1998).
One of the leading causes of blindness during childhood, retinopathy of prematurity (ROP), is a result of exposure to hyperoxic conditions. Developing retinal blood vessels in premature infants are particularly susceptible to increased oxygen levels. This finding is in contrast to adult retinal vasculatures, which are in fact, not affected by hyperoxia (Patz, 1984; Gu et al., 2002). Premature infants are reared in an oxygen-enriched environment after birth. The exposure to hyperoxia during this period is suspected to cause developing retinal blood vessels to stop growing and retinal capillaries drop out in a manner similar to vaso-obliteration in the canine oxygen-induced retinopathy (OIR) model. The OIR model shows severe vaso-obliteration where 60% of retinal capillaries are eliminated (McLeod et al., 1996).
Vaso-obliteration is difficult to detect in the premature infant because ocular fundus examinations for ROP begin 4 – 6 weeks after birth, long after vaso-obliteration is observable (Subhani et al., 2001). This finding is because the premature infant’s optical medium may not be clear before that time. However, a recent report shows undeveloped retinal capillary beds in the case of extremely low birth weight infants with ROP (Schulenburg and Tsanaktsidis, 2004), a common feature of vaso-obliteration. Animal models of OIR reveal obvious vaso-obliteration during hyperoxia exposure, suggesting that it happens in ROP (McLeod et al., 1996).
Relative hypoxia after hyperoxia-induced vaso-obliteration stimulates pro-angiogenic factor synthesis. This results in retinal neovascularization that grows into the vitreous body and forms a fibrovascular membrane, known as the vasoproliferative stage in ROP and OIR. Retinal detachment can occur through fibrovascular membrane traction, thus, leading to blindness (Recchia and Capone, 2004; International Committee for the Classification of Retinopathy of Prematurity, 2005).
An important target for future therapies is vaso-obliteration, where normal vascular development is prevented by unknown mechanisms. Several studies have focused on characterizing the mechanisms of hypoxia induction of retinal neovascularization (Wilkinson-Berka et al., 2004; Neu et al., 2006). While research in this area is important and has broad applications, at this late stage, the human retinal vasculature is irreversibly damaged. Hyperoxia-induced apoptosis of endothelial cells has been one of the putative mechanisms by which vaso-obliteration is thought to occur in the different animal and in vitro models of OIR (Alon et al., 1995; Gu et al., 2003; Kermorvant-Duchemin et al., 2005). Studies have also shown that hyperoxia inhibits endothelial cell proliferation (D’Amore and Sweet, 1987) and impairs the expression of the cell survival factor vascular endothelial growth factor (VEGF; Alon et al., 1995).
Oxidative stress is another putative, but elusive, factor that can lead to endothelial cell apoptosis (Gu et al., 2003), resulting in vaso-obliteration. However, there are key factors that have not been accounted for in this and other studies. The developing human and canine retinal vasculatures form initially by vasculogenesis and later by angiogenesis (McLeod et al., 1987; Lutty et al., 2000; Chan-Ling et al., 2004; McLeod et al., 2006). Both angiogenesis and vasculogenesis are ongoing at the time premature infants are exposed to hyperoxia. Therefore, the effects of hyperoxia on both of these processes must be understood.
We have recently established an in vitro canine angioblast differentiation assay (Lutty et al., 2006). This model permits the evaluation of hyperoxia on differentiation in the presence of essential vascular development growth factors, basic fibroblast growth factor (bFGF) and platelet-derived growth factor-BB (PDGF-BB). Recent studies show that the CXCR4/platelet-derived growth factor (PDGF)system plays a critical role in vascular development (Salvucci et al., 2002; McLeod et al., 2006) and CXCR4 is expressed by retinal angioblasts (Lutty et al., 2006). CXCR4 is expressed in endothelial precursor cells and is a receptor of SDF-1. SDF-1 is a chemotactic factor for CXCR4+ cells, thereby serving as a potential endothelial and precursor homing factor in vascular development. Therefore, any perturbation of the CXCR4/SDF-1 system could have adverse effects on normal vascular development. However, the effect of hyperoxia on CXCR4 expression and endothelial differentiation has not been investigated. The working hypothesis for this study was that hyperoxia disrupts retinal vascular development in canine OIR in vitro. This disruption includes the proliferation, migration, and apoptosis induction of canine retinal endothelial cells, and differentiation of canine retinal angioblasts. As hypothesized, hyperoxia induced apoptosis in both endothelial cells and angioblasts, in a time-dependent manner. Endothelial cell migration was inhibited by hyperoxia. The proliferation of both cell types was also inhibited. Cell cycle analysis suggests that the inhibition may be carried out through different mechanisms. Differentiation of angioblasts into endothelial cells (by means of bFGF treatment and determined by acetylated low-density lipoprotein [AcLDL] uptake) was significantly inhibited by hyperoxia. Of interest, the percentage of CXCR4+ cells increased in both treatment groups. These data indicate that differentiation of angioblasts is disrupted in hyperoxia. Additionally, hyperoxia exposure leads to significant disruption of the angioblast cytoskeleton, which could contribute to their inability to migrate and proliferate.
RESULTS
Effects of Hyperoxia on Endothelial Cell and Angioblast Proliferation
We have previously characterized the severe vaso-obliteration that occurs in the canine model of OIR after 100% oxygen exposure for 4 days after birth. The levels of oxygen used in the following in vitro experiments are based on those used in the in vivo canine OIR model (McLeod et al., 1996, 1998). Hyperoxia (95%O2/5% CO2) and normoxia (21% O2/5% CO2) exposed endothelial cells and angioblasts were counted after 24, 48, and 72 hr. In normoxia, both endothelial cells and angioblasts proliferated significantly with time (P < 0.01). The growth rate of angioblasts was lower than that of endothelial cells in normoxia. Of interest, angioblast numbers were significantly decreased by hyperoxia with time (Fig. 1B), whereas endothelial cell numbers did not decrease significantly from the 24-hr count (Fig. 1A).
Fig. 1.

Cell counts, apoptosis, and necrosis after normoxia or hyperoxia exposure. A,B: Endothelial cells (A) and angioblasts (B) were counted after 24, 48, and 72 hr of normoxia (dashed lines) and hyperoxia (solid lines) exposure. Bars indicate SD. *P < 0.01 when normoxia is compared with hyperoxia; **P < 0.01 when hyperoxia values are compared with the 24-hr normoxia values. C: Induction of apoptosis and necrosis in endothelial cells by hyperoxia. Flow cytometry was used to determine the percentage of apoptotic (filled symbols) and necrotic endothelial cells (open symbols) with Annexin V and propidium iodide, respectively. The normoxic controls are shown with solid lines, and the hyperoxia-treated cells are represented by dashed lines. Bars indicate SD.
Mode of Cell Death in Hyperoxia
Endothelial cell apoptosis and necrosis were analyzed after 4, 24, 48, 72, and 96 hr after hyperoxia and normoxia exposure. Approximately 5% of endothelial cells were apoptotic after 4 hr of hyperoxia and normoxia. The amount of apoptosis was increased by 24 hr through 96 hr of hyperoxia and normoxia exposure. The rate of apoptosis in hyperoxia-exposed endothelial cells was consistently higher than that of normoxic cells. Of interest, the level of induced apoptosis peaked at 24 hr and then did not increase over the course of the experiment. This finding indicates that the apoptotic event is early and only affects a minority of the endothelial cell population. The level of necrosis increased after 24 hr, suggesting that the necrotic cells are likely to be late stage apoptotic cells (Fig. 1C).
Effect of Oxygen on Cell Cycle Distribution
Endothelial cell exposure to hyperoxia for 24 hr caused a marked accumulation of cells in the S phase and a reduction of cells in the G1- and G2/M phases. Apoptotic cells in hyperoxia were increased approximately threefold compared with normoxia-exposed cells (Fig. 2A), agreeing with the Annexin V staining data (Fig. 1C). The effect of hyperoxia on the angioblast cell cycle was different than observed in endothelial cells. The angioblasts were markedly accumulated in the Sand G2/M phases, whereas cells in G1 phase were reduced to less than 5% (Fig. 2B). The number of apoptotic angioblasts increased to approximately 5% when treated with hyperoxia for 24 hr (Fig. 2B).
Fig. 2.
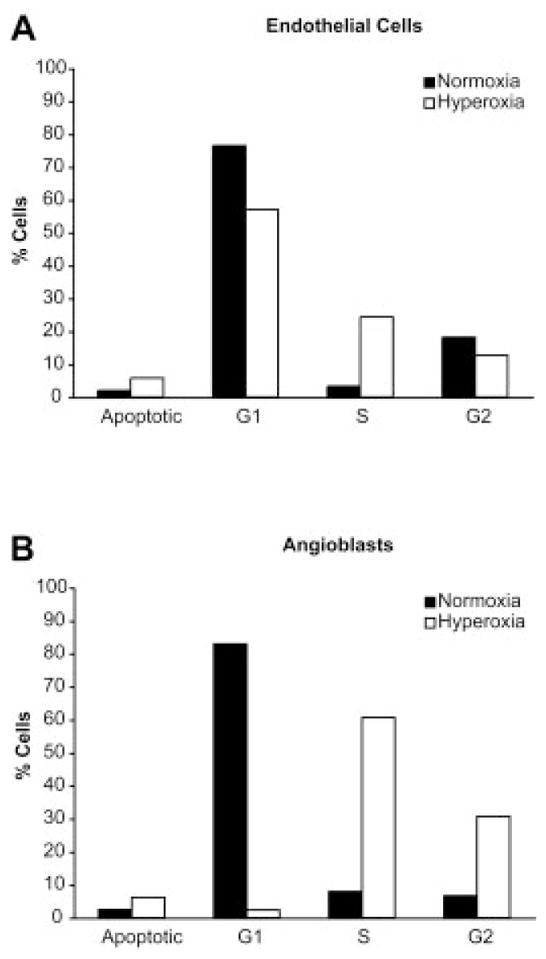
Cell cycle alterations in hyperoxia. A,B: The DNA content histograms for normoxia and hyperoxia-treated endothelial cells and angioblasts are shown in A and B, respectively. The percentages of cells in the various phases of the cell cycle and apoptosis are shown, where the normoxic values are indicated by solid bars and hyperoxic values by open bars.
Inhibition of Endothelial Cell Migration by Hyperoxia
The effect of hyperoxia on endothelial cell migration was evaluated with an in vitro wound healing assay in the presence of Mitomycin C. Mitomycin C was used to inhibit proliferation of endothelial cells, allowing a more accurate assessment of migration (Fig. 3). From 0 to 180 μm from the wound line (bottom of the photomicrographs), there were no significant differences in the number of cells in either normoxia or hyperoxia groups. Very few cells migrated further than 720 μm. Most of the hyperoxia-exposed cells migrated less than 180 μm. The major differences between normoxia and hyperoxia could be seen between 180 and 720 μm from the wound line where significantly more normoxic cells were present (P < 0.01; Fig. 3).
Fig. 3.

Inhibition of endothelial cell migration by hyperoxia. The inset photos show cells that have migrated beyond the initial scrape line (bottom of the image) after 24 hr of normoxia (left photo) and hyperoxia (right photo) exposure. Mitomycin C was used to inhibit proliferation of endothelial cells before scraping. The graph depicts nuclei counts at various migration intervals. Bars indicate SEM values. *P < 0.01.
Disruption of the Angioblast Cytoskeleton by Hyperoxia
To clarify the effect of hyperoxia on the angioblast and endothelial cytoskeletons, cells were incubated in hyperoxia for 0, 1, 2, 3, 4, and 5 days. The cells were then labeled with Phalloidin, a marker for polymeric actin. Additionally, angioblasts were immunostained with anti–smooth muscle actin (SMA) antibody. A day before incubation in hyperoxia, endothelial cells were wounded to identify differences between quiescent and dividing/migrating cells. Under normoxic conditions, both angioblasts and endothelial cells had organized cytoskeletal structures as visualized by Phalloidin/SMA and Phalloidin staining, respectively (Fig. 4A,D). The angioblast cytoskeleton was degraded in cells after the second day of hyperoxia exposure. Disorganized or degraded actin was observed as nonfilamentous or aggregated actin (Fig. 4B,C,F). The percentage of cells with an intact angioblast cytoskeleton was significantly decreased after only 2 days, but a dramatic decline in organized angioblast cytoskeleton occurred after 3 days of hyperoxia treatment. On the other hand, the endothelial cell cytoskeleton was not significantly affected during 3 days of hyperoxia exposure. The dividing endothelial cell cytoskeleton was significantly affected after 4 days of hyperoxia, but the percentage of affected cells never increased above 20% (Fig. 4G).
Fig. 4.

Disruption of angioblast and endothelial cell cytoskeletons by hyperoxia. A–F: Photomicrographs were taken of angioblasts (A–C) and endothelial cells (D–F) exposed to 0 (A,D), 3 (B,E), and 5 (C,F) days of hyperoxia. These cells were then labeled with Phalloidin (green), and angioblasts were also labeled with anti-smooth muscle actin (red, SMA). The cells were counterstained with 4′,6-diamidine-2-phenylidole-dihydrochloride (DAPI, blue). G: The percentage of cells with an intact cytoskeleton, as shown in upper panels, was determined. The SEM is indicated by bars. *P < 0.05 to normoxic angioblasts, and **P < 0.05 when compared with either normoxic/hyperoxic quiescent or normoxic dividing/migrating endothelial cells. All angioblast counts were significantly less (P < 0.05) than endothelial counts after the first day.
Hyperoxia Inhibits Angioblast Differentiation
Our previous in vitro differentiation study showed that canine angioblasts differentiate into endothelial cells (AcLDL+) or pericytes (SMA+) after 8 days stimulation with bFGF or PDGF-BB, respectively (Lutty et al., 2006). In the current study, angioblasts were incubated in normoxia and hyperoxia for 8 days with complete medium supplemented with bFGF or PDGF-BB to determine the effects of hyperoxia on angioblast differentiation. As described earlier (Fig. 1), angioblasts have a relatively slow rate of proliferation, but the number of bFGF-treated angioblasts increased approximately threefold compared with PDGF-treated angioblasts in normoxia. In hyperoxia, however, bFGF-treated cells significantly decreased in number compared with cells in normoxia (P < 0.0001; Fig. 5A–C).
Fig. 5.

Effect of hyperoxia on basic fibroblast growth factor (bFGF) -treated angioblast differentiation and CXCR4 expression. A,B: Upper panels (A,B) show acetylated low-density lipoprotein (AcLDL) uptake by bFGF-treated angioblasts after normoxia (A) and hyperoxia (B) exposure. AcLDL uptake was observed as punctate staining in the cytoplasm (A,B). C: The graph shows the number of cells per 10 mm2. D,E: Lower panels show CXCR4 immunostaining in bFGF-treated angioblasts after normoxia (D) and hyperoxia (E) exposure. F: The graph shows the number of cells per 10 mm2. G: The percentages of AcLDL+ and CXCR4+ cells are shown. Bars in C, F, and G indicate SEM values. A,B,D,E: Nuclear counterstaining with DAPI is shown in blue, and AcLDL/CXCR4 is shown in red. NOX, normoxia; HOX, hyperoxia. *P < 0.05. Scale bar = 20 μm in A.
The number of cells with AcLDL incorporation, a known endothelial cell marker, made up approximately 86% of the population. On the other hand, hyperoxia treatment decreased the number of AcLDL+ cells by 20-fold when compared with normoxia (P < 0.0001). AcLDL incorporation was observed as punctate staining in the cytoplasm of the cells (Fig. 5A).
The number of SMA+ cells was approximately 8 per 100 mm2. Of interest, hyperoxia did not reduce the number of SMA+, PDGF-treated cells (Fig. 6A–C). Even though the cells were positive for SMA, the staining pattern was different than typically observed. In the normoxia groups, SMA generally has a cable-like or striated appearance, but in the hyperoxia groups, the SMA appeared more granular and few cables were present (Fig. 6A,B). These observations suggest that the SMA is being disassembled in hyperoxia and may not be functional as observed in Figure 4A–C.
Fig. 6.

Effect of hyperoxia on platelet-derived growth factor (PDGF) -treated angioblast differentiation. A,B: Upper panels show smooth muscle actin (SMA) immunostaining in PDGF-treated angioblasts after normoxia (A) and hyperoxia (B) exposure. C: The graph shows the number of cells per 10 mm2. D,E: Lower panels show CXCR4 immunostaining in PDGF-treated angioblasts after normoxia (D) and hyperoxia (E) exposure. F: The number of nuclei and SMA+ or CXCR4+ cells per field of view (10 mm2) were also counted and are shown. G: The percentages of SMA+ and CXCR4+ cells are shown. Bars in C, F, and G indicate SEM values. A,B,D,E: Nuclear counterstaining with 4′,6-dia-midine-2-phenylidole-dihydrochloride (DAPI) is shown in blue. NOX, normoxia; HOX, hyperoxia. *P < 0.05. Scale bar = 20 μm in A.
The number of CXCR4+ cells per 100 mm2 was significantly decreased in the PDGF-treated (Fig. 6D–F), but not the bFGF-treated groups (Fig. 5D–F). On the contrary, the proportion of CXCR4+ cells was greatly increased in hyperoxia-exposed bFGF-treated cells (Fig. 5G) and only slightly increased in the PDGF-treated cells (Fig. 6G). The loss of AcLDL uptake by bFGF-treated angioblasts suggests that these cells may dedifferentiate after exposure to hyperoxia.
DISCUSSION
The present study demonstrated the effects of hyperoxia on retinal vascular development by evaluating the proliferation, migration, and death of endothelial cells and differentiation of angioblasts in vitro. Although this study uses 95% oxygen, premature infants are not presently reared in 95% oxygen. This finding is due to increased lung complications and severity of ROP at saturation of oxygen levels above 93% (hyperoxia). Furthermore, hyperoxia increases neonatal mortality (Saugstad, 2005; Vanderveen et al., 2006). However, there are situations in which high levels of inspired oxygen are needed to maintain saturation of oxygen for infant survival. Additionally, the oxygen levels in premature infant retinal tissues are not known.
As mentioned earlier, the in vitro levels of oxygen used in this study are based on the canine model of OIR, which does use 100% oxygen for 4 days. These conditions lead to canine disease that is quite similar to human ROP. Therefore, the present study is designed to shed light on the effects of a rapid and sustained increase in oxygen on the developing vasculature. Proliferation of both endothelial cells and angioblasts was inhibited by hyperoxia. Hyperoxia also inhibited the migration of endothelial cells. Hyperoxia was sufficient to induce apoptosis in endothelial cells immediately after hyperoxia exposure. However, the contribution of endothelial apoptosis to the decrease in endothelial cell numbers was much less than the decrease in cell number due to inhibition of proliferation (18% decrease vs. a 75% decrease). Differentiation of angioblasts is an essential step in vasculogenesis that was impaired by hyperoxia.
These data support the hypothesis that therapies for protecting angioblasts and endothelial cells should be investigated. Every effort is made, internationally, to limit the hyperoxic phase for many reasons including lung damage and ROP. However, it is very difficult, at best, to treat infants in hyperoxia. Therefore, additional therapeutic approaches need to be developed for the posthyperoxic infant. Ideally these therapies would stimulate normal differentiation of angioblasts to endothelial cells, so that the retina can regenerate a functional vasculature. These therapies could then be used in high-risk infants with careful consideration of other health concerns.
Apoptosis of retinal vascular endothelial cells has been considered as the major cause of hyperoxia-induced vaso-obliteration, but the peak period of endothelial cell death is still controversial. Alon et al. reported that abundant apoptotic endothelial cells were observed in rat retinas exposed to hyperoxia for 48 hr (Alon et al., 1995). Gu et al. showed increased expression of caspase-3 and rate of bovine retinal endothelial apoptosis after 48 hr hyperoxia exposure in vitro (Gu et al., 2003). However, Hughes et al demonstrated that a significant induction of endothelial apoptosis was observed only at early time points (10 or 24 hr) after hyperoxia exposure in postnatal rat (Hughes et al., 2000). In the current canine study, apoptosis of endothelial cells increased after 24 hr of hyperoxia exposure and it continued until 96 hr. Necrosis of endothelial cells increased after 96 hr of hyperoxia exposure (Fig. 1C). These data suggest that apoptosis from hyperoxia, in the canine model, is an early event in vaso-obliteration. The peak period of apoptosis in our endothelial cells was approximately 25% after 24 hr of hyperoxia exposure. Therefore, apoptosis of endothelial cells from hyperoxia may not be the major cause of vaso-obliteration. In fact, hyperoxia did not greatly decrease endothelial cell numbers, with respect to the 24-hr values (Fig. 1A), but the inhibition of cell cycle progression was prominent after 24 hr.
Hyperoxia inhibited endothelial cell and angioblast proliferation, as shown in Figures 1 and 2. Oxygen-induced DNA damage results in impairment of cell proliferation in several cell types (O’Reilly, 2001). Previous studies have also shown that oxygen levels greater than 90% inhibit cell proliferation in G1, S, and G2 phase of the cell cycle and caused chromosomal abnormalities (Helt et al., 2001; Ko et al., 2004). The data in Figure 3 demonstrate that the G1 phase of the cell cycle was reduced and the S phase was obviously increased in both endothelial cells and angioblasts, resulting in inhibition of cell proliferation. Of interest, the cell cycle blockade effects of hyperoxia were limited to only the S phase in the angioblasts, whereas the endothelial cells showed an accumulation in both the S and G2 phases. Therefore, hyperoxia has dissimilar effects on different cells, possibly depending on their differentiation state. These data show that the effect of hyperoxia on the cell cycle distribution is not the same in angioblasts as seen in endothelial cells. However, the mechanism is currently not known. The similarities and differences of hyperoxic effects on these cells indicate that, although the effects are somewhat related, there are different mechanisms involved.
Both angiogenesis and vasculogenesis are required for retinal vascular development, and both processes are affected by hyperoxia. The effect of oxygen on the differentiation of angioblasts in retinal vascular development is poorly understood. Our hypothesis was that hyperoxia inhibits key signaling events in the process of angio-blast differentiation. Recently, we demonstrated that, under certain conditions, the default phenotype for angioblasts is the pericyte and, additionally, angioblasts can differentiate into endothelial cells with the addition of bFGF (Lutty et al., 2006).
In this study, we cultured angio-blasts in the presence of 10% fetal bovine serum (FBS), as opposed to 1% Nuserum used in the past study, to better approximate the cellular milieu and to maintain media consistency through the diverse experiments conducted. The presence of angiogenic growth factors in the serum may have reduced the ability of these angioblasts to differentiate into pericytes as reported earlier (Lutty et al., 2006). These same factors may have contributed to the ease of angioblast differentiation into endothelial cells. bFGF has been shown to be important for the induction of angioblast growth and differentiation in many tissues (Bikfalvi et al., 1997; Poole et al., 2001). Therefore, the effect of bFGF and hyperoxia on angioblast proliferation and differentiation were assessed in the present study.
After bFGF treatment, the number of angioblasts was increased compared with cultures without bFGF (Figs. 5, 6). Hyperoxia inhibited angioblast differentiation into endothelial cells (AcLDL+ ), but not pericytes (SMA+). We also evaluated the expression of CXCR4, a receptor involved in homing of precursors during development. Our hypothesis was that hyperoxia would depress the expression of this homing receptor. If this were true, the implication would be that the reduced capacity of angioblasts to home to the correct site may result in inhibited vascular development and repair. The data showed a decrease in the number of CXCR4+ angioblasts that were treated with PDGF-BB. Of interest, the number of CXCR4+, bFGF-treated angioblasts remained the same. In both cases, the proportion of CXCR4+ cells increased, especially in the bFGF-treated cells. CXCR4/SDF-1 is thought to play an important role in retinal vascular development, but this role is not mechanistically defined (McLeod et al., 2006). So, while these results are intriguing, we can only postulate that the inhibition of CXCR4 in PDGF-BB differentiated angioblasts and apparent dedifferentiation of endothelial lineage may have a detrimental effect on the progression of vasculogenesis during hyperoxia. In turn, this dedifferentiation may contribute to vaso-obliteration.
Proliferation of angioblasts and endothelial cells was significantly inhibited by hyperoxia (Fig. 1A,B). Additionally, migration of endothelial cells was inhibited by hyperoxia (Fig. 3). Therefore, we speculated that the angioblast and endothelial cell cytoskeleton was being disassembled in hyperoxia and may not be functional. Therefore, a quantitative time course study of changes occurring in the cytoskeleton organization was undertaken using Phalloidin, a marker for polymeric actin, in hyperoxia-exposed angioblasts and endothelial cells. In addition, angioblasts were stained with anti-SMA antibody to visualize the smooth muscle actin cables. These data demonstrated that the angioblast cytoskeleton was significantly degraded after hyperoxia exposure for only 2 days, when compared with angioblasts in normoxic conditions, and were further degraded after 3 days of hyperoxia exposure. Interestingly, the cytoskeleton of endothelial cells was not nearly as susceptible to the damaging effects of hyperoxia as the angioblast cytoskeleton.
It is possible that different cells could have different intracellular oxygen levels with the same extracellular oxygen level, thereby causing different effects in the respective cells. Another hypothesis is that the effects seen from hyperoxia exposure are due to different molecular mechanisms in the two cell types examined. A final alternative may be that the antioxidant capacity differs between cells and between differentiation states. We speculate that the disruption in angioblast cytoskeleton could impair angioblast migration and mitosis. Interestingly, similar phenomena have also been observed in pericytes after hyperoxia exposure in the feline ROP model (Chan-Ling, T., personal communication).
This in vitro study evaluates critical features of vascular development in hyperoxia that need to be validated in the canine OIR model. The current study demonstrated that the inhibition of endothelial cell/angioblast proliferation by cell cycle perturbation may play a role in vaso-obliteration, an early stage of OIR. This role is likely to be important in the formation of the deep capillary network by angiogenesis. Migration, a critical feature of angiogenesis and vasculogenesis, was also inhibited by hyperoxia. Vasculogenesis is responsible for the initial formation of the retinal vasculature. This process relies on the ability of angioblasts to home and differentiate into both endothelial cells and pericytes. We are the first to show that the differentiation from angioblast into endothelial cell is overwhelmingly impaired by hyperoxia, while the differentiation of angioblast to pericyte is not greatly effected. Together, these data show that critical facets in vascular development are significantly disrupted by hyperoxia.
EXPERIMENTAL PROCEDURES
Cell Culture
Primary canine retinal endothelial cells and angioblasts were isolated and cultured as previously described (Lutty et al., 2006). Canine retinal endothelial cells and angioblasts were maintained in Dulbecco’s modified eagle medium (DMEM)/Ham’s F12 medium containing 10% FBS and 1% penicillin–streptomycin–fungizone (PSF) at 37°C in a 5% CO2 environment. Endothelial cells were passaged by washing with phosphate buffered saline (PBS) followed by trypsin digestion (0.25% trypsin), followed by washing with complete medium before plating. Endothelial cells passaged less than 10 times were used in these experiments. Angioblasts were passed by scraping and re-plating using a cell scraper in medium. Angioblasts were used before the third passage. In all experiments, except where noted, endothelial cells and angioblasts were plated in 24-well plates at a density of 20,000 cells per well. All cell culture reagents were purchased from Invitrogen (Carlsbad, CA). Each experiment was repeated at least three times unless otherwise noted.
Cell Proliferation Studies
Endothelial cells and angioblasts were plated in triplicate in 24-well plates and incubated for 2 days under normal conditions before treatment in hyperoxia (95% O2/5% CO2) or normoxia (21% O2/5% CO2) conditions. The cells were harvested at 24, 48, and 72 hr after incubation. The cells were harvested by trypsin digest followed by counting with an improved Neubauer hemocytometer.
Apoptosis and Necrosis Measurements
Endothelial cells and angioblasts were plated in triplicate in 24-well plates and incubated for 2 days under normal conditions before exposure to normoxia or hyperoxia for up to 96 hr. At harvest time, the cells were rinsed once with PBS, trypsinized, and washed once with complete medium. The supernatant was aspirated from the cell pellet and the pellet resuspended in 100 μl Annexin-binding buffer (5 μl Annexin V conjugate, 10 mM HEPES, 140 mM NaCl, and 2.5 mM CaCl2, pH 7.4) with 50 μg/ml propidium iodide (Invitrogen). The cells were stained for 15 min at room temperature. After propidium iodide staining, the Annexin V staining solution (400 μl of annexin-binding buffer with 5 μl Annexin V–fluorescein isothiocyanate (FITC) conjugate [Invitrogen]) was added to the cells, and the samples were kept on ice until analyzed. The stained cells were then analyzed as soon as possible on a BDS FACScan, and the data analysis done with WinMDI. Annexin V–positive and propidium iodide–negative cells were considered to be apoptotic, whereas necrotic cells were any cells positive for propidium iodide.
DNA Content Analysis
Endothelial cells and angioblasts were plated in 24-well plates with DMEM/F12 medium containing 10% FBS and 1% PSF in 37°C, 5% CO2 environment. After exposure to hyperoxia or normoxia for 24 hr at 37°C, endothelial cells and angioblasts were trypsinized and washed with complete medium. The cells were then centrifuged at 600 × g for 5 min, and the supernatant was aspirated. The cell pellet was then resuspended in cold 70% ethanol for 24 hr. The fixed cells were then centrifuged for 5 min at 600 × g, and the supernatant was aspirated. The cell pellet was then re-suspended in 50 μl of phosphocitrate buffer (192 parts 0.2 M Na2HPO4, 8 parts 0.1 M citric acid, pH 7.8) as described previously (Gong et al., 1994) and incubated at room temperature for 20 min. After centrifugation for 5 min at 600 × g, the supernatant was aspirated, and the cells were stained with 50 mg/ml propidium iodide in the presence of 50 mg/ml RNase at a final concentration of 1 × 106 cells/ml. Three separate wells from each group were combined, the cells were analyzed on a BDS FACScan, and the data analysis was done with WinMDI.
Endothelial Cell Migration Assay
Endothelial cells were plated in six-well plates at a density of 50,000 cells per well and incubated until confluent. A confluent monolayer of cells was wounded with a razor blade and washed once with complete medium to remove loose cells (Lutty et al., 1998). Wounded cells were incubated for 24 hr in normoxia or hyperoxia. The cells were then fixed with 2% paraformaldehyde in Tris buffered saline (TBS) for 10 min. Cells were incubated with Hoechst 33342 (10 mg/L stock, diluted 1:20,000, Invitrogen) for 10 min and then washed with TBS. Mitomycin C (Sigma-Aldrich, St. Louis, MO) was used to inhibit endothelial cell proliferation at 0.5 mg/ml in DMEM containing 10% FBS (Schleef and Birdwell, 1982). Cells were treated with Mitomycin C for 30 min before wounding to prevent proliferation. The cells were mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA) and imaged with a confocal microscope (Zeiss 510META, Wilmer Imaging Core) using a ×5 objective lens. The photographs were acquired every 180 μm from wound line. Fifteen photographs were taken from each wounded culture. The number of nuclei per 180 μm was counted with ImageJ (National Institute of Health, NIH Version v1.36b).
Cytoskeleton Staining
Angioblasts and endothelial cells were plated on hyaluronic acid (HA)/fibronectin (FN) coated and noncoated four-well chamber slides (Nunc International Corp, Naperville, IL) respectively, at 25k cells per well in 1 ml of DMEM/F12 containing 10% FBS and 1% PSF. A day before incubation of hyperoxia, endothelial cells were wounded with a razor blade as described above. After 0, 1, 2, 3, 4, 5 days of hyperoxia exposure, angioblasts were labeled with Phalloidin and immunostained with anti-SMA antibody. Endothelial cells were only labeled with Phalloidin, because they are always negative for SMA staining. The cells were fixed, permeabilized as described below, and then they were incubated with FITC-conjugated Phalloidin (Sigma) for 30 min. After Phalloidin staining, angioblasts were labeled with anti-SMA antibody and then goat anti-mouse IgG conjugated with Cy3 as described below. All cells were counterstained with 4′,6-diamidine-2-phenylidole-dihydrochloride (DAPI). Photomicrographs for figures were taken by confocal microscopy (Zeiss 510META) using a ×40 objective lens. Cells with disrupted cytoskeletons were identified by a lack of clear actin cables and punctate and/or diffuse staining patterns. Cells with intact cytoskeletons were identified by clear actin cables and 3 fields of view (at ×30) in each of the three wells counted (total 12 fields at each time point were analyzed statistically).
Angioblast Differentiation Assay
Angioblasts were plated on 0.1 mg/ml of HA (Sigma-Aldrich) and 0.1 mg/ml of FN (GIBCO Life Technologies, Rockville, MD) coated 24-well plates with 1 ml of DMEM/F12 medium containing 10% FBS and 1% PSF, then incubated in 5% CO2 at 37°C (Lutty et al., 2006). The angioblasts were plated and incubated for 2 days. The angioblasts were then incubated with either 25 ng/ml of recombinant human bFGF (R&D Systems, Minneapolis, MN) or 50 ng/ml recombinant human PDGF-BB (R&D Systems) and incubated in normoxia or hyperoxia for 8 days. The cells were washed and fed fresh medium, and the growth factor replaced every other day. After 8 days of incubation, the cells were evaluated by immunocytochemistry and DiI-labeled AcLDL uptake as described below.
Immunocytochemistry
The angioblasts were washed once for 5 min with TBS and fixed with 2% paraformaldehyde in TBS for 10 min at room temperature. After fixation, the cells were washed with TBS, permeabilized with 0.25% Triton in TBS, and then washed again with TBS. Blocking was carried out with 2% goat serum in TBS for 20 min at room temperature followed by TBS washing. The cells were incubated for 2 hr at room temperature with the following antibodies: mouse anti-SMA (DakoCytomation, Carpinteria, CA) and rabbit anti-CXCR4 (Novus Biologicals, Littleton, CO), diluted 1:1,000 and 1:2,500 in TBS with 1% bovine serum albumin, respectively. After washing with TBS, the cells were then incubated for 30 min at room temperature with the secondary antibodies: FITC-conjugated goat anti-mouse IgG (Jackson Immuno Research Lab, West Grove, PA) and Alexa fluor 647 goat anti-rabbit IgG (Invitrogen) diluted 1:500 in TBS with 1% bovine serum albumin. The cells were washed with TBS and mounted with Vectashield mounting medium with DAPI (Vector Laboratories).
DiI-labeled AcLDL Incorporation
It has been shown that vascular endothelial cells take up AcLDL by means of scavenger receptors on the cellular membrane (Voyta et al., 1984). Therefore, AcLDL was used to confirm the identity of endothelial cells in this study. Briefly, the cells were washed with DMEM/F12 and then incubated with 10 μg/ml of AcLDL (Biomedical Technologies, Inc., Stoughton, MA) in DMEM/F12 containing 10% FBS for 4 hr in normoxia or hyperoxia in the dark. After incubation, cells were washed three times with PBS, and then cells were washed with TBS before being mounted with Vectashield mounting medium with DAPI (Vector Laboratories).
Cell Counting and Confocal Microscopy
Photomicrographs were taken with a confocal microscope (Zeiss 510META, Carl Zeiss Microimaging, Thornwood, NY) with ×10 objective lens. Excitation wavelengths of 405, 488, 532, and 633 nm were used to excite DAPI, FITC, AcLDL, and Alexa fluor 647, respectively. Bandpass emission filters (460 – 480, 505–520, 560 – 600 nm) were used to capture DAPI, FITC, and AcLDL, respectively; whereas, a 670-nm-long pass filter was used to image Alexa fluor 647 stained cells. To determine the number of SMA+/CXCR4+ and AcLDL+ cells from four fields of view, each 10 mm2, were counted by two observers. A total of 12 fields and 20 fields from SMA/CXCR4-immunostained cells and AcLDL-treated cells, respectively, in each time point were analyzed for statistical significance.
Statistical Analysis
Unpaired two-tailed Student’s t-test was used to compare the scores from normoxia-treated cells with those from hyperoxia-treated cells. A P value of <0.05 was considered significant.
Acknowledgments
This work was supported by the Research to Prevent Blindness (Wilmer), National Eye Institute Grant R01EY09357 (G.L.), EY 01765 (Wilmer), the Johns Hopkins Hematology Training Grant T32HL007525 (T.P.), and Overseas Research Scholarship, Fukuoka University School of Medicine Alumni, Eboshikai (K.U.).
References
- Alon T, Hemo I, Itin A, Pe’er J, Stone J, Keshet E. Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat Med. 1995;1:1024–1028. doi: 10.1038/nm1095-1024. [DOI] [PubMed] [Google Scholar]
- Bikfalvi A, Klein S, Pintucci G, Rifkin DB. Biological roles of fibroblast growth factor-2. Endocr Rev. 1997;18:26–45. doi: 10.1210/edrv.18.1.0292. [DOI] [PubMed] [Google Scholar]
- Chan-Ling T, McLeod DS, Hughes S, Baxter L, Chu Y, Hasegawa T, Lutty GA. Astrocyte-endothelial cell relationships during human retinal vascular development. Invest Ophthalmol Vis Sci. 2004;45:2020–2032. doi: 10.1167/iovs.03-1169. [DOI] [PubMed] [Google Scholar]
- D’Amore PA, Sweet E. Effects of hyperoxia on microvascular cells in vitro. In Vitro Cell Dev Biol. 1987;23:123–128. doi: 10.1007/BF02623592. [DOI] [PubMed] [Google Scholar]
- Gong J, Traganos F, Darzynkiewicz Z. Staurosporine blocks cell progression through G1 between the cyclin D and cyclin E restriction points. Cancer Res. 1994;54:3136–3139. [PubMed] [Google Scholar]
- Gu X, Samuel S, El-Shabrawey M, Caldwell RB, Bartoli M, Marcus DM, Brooks SE. Effects of sustained hyperoxia on revascularization in experimental retinopathy of prematurity. Invest Ophthalmol Vis Sci. 2002;43:496–502. [PubMed] [Google Scholar]
- Gu X, El-Remessy AB, Brooks SE, Al-Shabrawey M, Tsai NT, Caldwell RB. Hyperoxia induces retinal vascular endothelial cell apoptosis through formation of peroxynitrite. Am J Physiol Cell Physiol. 2003;285:C546–C554. doi: 10.1152/ajpcell.00424.2002. [DOI] [PubMed] [Google Scholar]
- Helt CE, Rancourt RC, Staversky RJ, O’Reilly MA. p53-dependent induction of p21(Cip1/WAF1/Sdi1) protects against oxygen-induced toxicity. Toxicol Sci. 2001;63:214–222. doi: 10.1093/toxsci/63.2.214. [DOI] [PubMed] [Google Scholar]
- Hughes S, Yang H, Chan-Ling T. Vascularization of the human fetal retina: roles of vasculogenesis and angiogenesis. Invest Ophthalmol Vis Sci. 2000;41:1217–1228. [PubMed] [Google Scholar]
- International Committee for the Classification of Retinopathy of Prematurity. The International Classification of Retinopathy of Prematurity revisited. Arch Ophthalmol. 2005;123:991–999. doi: 10.1001/archopht.123.7.991. [DOI] [PubMed] [Google Scholar]
- Kermorvant-Duchemin E, Sennlaub F, Sirinyan M, Brault S, Andelfinger G, Kooli A, Germain S, Ong H, d’Orleans-Juste P, Gobeil F, Jr, Zhu T, Boisvert C, Hardy P, Jain K, Falck JR, Balazy M, Chemtob S. Trans-arachidonic acids generated during nitrative stress induce a thrombospondin-1-dependent microvascular degeneration. Nat Med. 2005;11:1339–1345. doi: 10.1038/nm1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko JC, Wang YT, Yang JL. Dual and opposing roles of ERK in regulating G(1) and S-G(2)/M delays in A549 cells caused by hyperoxia. Exp Cell Res. 2004;297:472–483. doi: 10.1016/j.yexcr.2004.03.033. [DOI] [PubMed] [Google Scholar]
- Lutty GA, Mathews MK, Merges C, McLeod DS. Adenosine stimulates canine retinal microvascular endothelial cell migration and tube formation. Curr Eye Res. 1998;17:594–607. [PubMed] [Google Scholar]
- Lutty GA, Merges C, McLeod DS. 5′ nucleotidase and adenosine during retinal vasculogenesis and oxygen-induced retinopathy. Invest Ophthalmol Vis Sci. 2000;41:218–229. [PubMed] [Google Scholar]
- Lutty GA, Merges C, Grebe R, Prow T, McLeod DS. Canine retinal angioblasts are multipotent. Exp Eye Res. 2006;83:183–193. doi: 10.1016/j.exer.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Maltepe E, Simon MC. Oxygen, genes, and development: an analysis of the role of hypoxic gene regulation during murine vascular development. J Mol Med. 1998;76:391–401. doi: 10.1007/s001090050231. [DOI] [PubMed] [Google Scholar]
- McLeod DS, Lutty GA, Wajer SD, Flower RW. Visualization of a developing vasculature. Microvasc Res. 1987;33:257–269. doi: 10.1016/0026-2862(87)90021-5. [DOI] [PubMed] [Google Scholar]
- McLeod DS, Brownstein R, Lutty GA. Vaso-obliteration in the canine model of oxygen-induced retinopathy. Invest Ophthalmol Vis Sci. 1996;37:300–311. [PubMed] [Google Scholar]
- McLeod DS, D’Anna SA, Lutty GA. Clinical and histopathologic features of canine oxygen-induced proliferative retinopathy. Invest Ophthalmol Vis Sci. 1998;39:1918–1932. [PubMed] [Google Scholar]
- McLeod DS, Hasegawa T, Prow T, Merges C, Lutty G. The initial fetal human retinal vasculature develops by vasculogenesis. Dev Dyn. 2006;235:3336–3347. doi: 10.1002/dvdy.20988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu J, Afzal A, Pan H, Gallego E, Li N, Li Calzi S, Caballero S, Spoerri PE, Shaw LC, Grant MB. The dipeptide Arg-Gln inhibits retinal neovascularization in the mouse model of oxygen-induced retinopathy. Invest Ophthalmol Vis Sci. 2006;47:3151–3155. doi: 10.1167/iovs.05-1473. [DOI] [PubMed] [Google Scholar]
- O’Reilly MA. DNA damage and cell cycle checkpoints in hyperoxic lung injury: braking to facilitate repair. Am J Physiol Lung Cell Mol Physiol. 2001;281:L291–L305. doi: 10.1152/ajplung.2001.281.2.L291. [DOI] [PubMed] [Google Scholar]
- Patz A. Current concepts of the effect of oxygen on the developing retina. Curr Eye Res. 1984;3:159–163. doi: 10.3109/02713688408997197. [DOI] [PubMed] [Google Scholar]
- Penn JS, Thum LA, Naash MI. Oxygen-induced retinopathy in the rat. Vitamins C and E as potential therapies. Invest Ophthalmol Vis Sci. 1992;33:1836–1845. [PubMed] [Google Scholar]
- Poole TJ, Finkelstein EB, Cox CM. The role of FGF and VEGF in angioblast induction and migration during vascular development. Dev Dyn. 2001;220:1–17. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1087>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Recchia FM, Capone A., Jr Contemporary understanding and management of retinopathy of prematurity. Retina. 2004;24:283–292. doi: 10.1097/00006982-200404000-00015. [DOI] [PubMed] [Google Scholar]
- Salvucci O, Yao L, Villalba S, Sajewicz A, Pittaluga S, Tosato G. Regulation of endothelial cell branching morphogenesis by endogenous chemokine stromal-derived factor-1. Blood. 2002;99:2703–2711. doi: 10.1182/blood.v99.8.2703. [DOI] [PubMed] [Google Scholar]
- Saugstad OD. Oxygen for newborns: how much is too much? J Perinatol. 2005;25(Suppl 2):S45–S49. doi: 10.1038/sj.jp.7211321. discussion S50. [DOI] [PubMed] [Google Scholar]
- Schleef RR, Birdwell CR. The effect of fibrin on endothelial cell migration in vitro. Tissue Cell. 1982;14:629–636. doi: 10.1016/0040-8166(82)90053-2. [DOI] [PubMed] [Google Scholar]
- Schulenburg WE, Tsanaktsidis G. Variations in the morphology of retinopathy of prematurity in extremely low birthweight infants. Br J Ophthalmol. 2004;88:1500–1503. doi: 10.1136/bjo.2004.044669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LE, Wesolowski E, McLellan A, Kostyk SK, D’Amato R, Sullivan R, D’Amore PA. Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci. 1994;35:101–111. [PubMed] [Google Scholar]
- Subhani M, Combs A, Weber P, Gerontis C, DeCristofaro JD. Screening guidelines for retinopathy of prematurity: the need for revision in extremely low birth weight infants. Pediatrics. 2001;107:656–659. doi: 10.1542/peds.107.4.656. [DOI] [PubMed] [Google Scholar]
- Vanderveen DK, Mansfield TA, Eichenwald EC. Lower oxygen saturation alarm limits decrease the severity of retinopathy of prematurity. J AAPOS. 2006;10:445–448. doi: 10.1016/j.jaapos.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Voyta JC, Via DP, Butterfield CE, Zetter BR. Identification and isolation of endothelial cells based on their increased uptake of acetylated-low density lipoprotein. J Cell Biol. 1984;99:2034–2040. doi: 10.1083/jcb.99.6.2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson-Berka JL, Babic S, De Gooyer T, Stitt AW, Jaworski K, Ong LG, Kelly DJ, Gilbert RE. Inhibition of platelet-derived growth factor promotes pericyte loss and angiogenesis in ischemic retinopathy. Am J Pathol. 2004;164:1263–1273. doi: 10.1016/s0002-9440(10)63214-2. [DOI] [PMC free article] [PubMed] [Google Scholar]