

Abstract
Zn(II) is used in nature as a biocatalyst in hundreds of enzymes and the structure and dynamics of its catalytic activity is the subject of considerable interest. Many of the Zn(II)-based enzymes are classified as hydrolytic enzymes, in which the Lewis acidic Zn(II) center facilitates proton transfer(s) to a Lewis base, from proton donors such as water or thiol. This report presents the results of a quantum computational study quantifying the dynamic relationship between the zinc coordination number (CN), its coordination geometry, and the thermodynamic driving force behind these proton transfers originating from a charge-neutral methylthiol ligand. Specifically, density functional theory (DFT) and second-order perturbation theory (MP2) calculations have been performed on a series of [(Imidazole)nZn–S(H)CH3]2+ and [(Imidazole)nZn–SCH3]+ complexes with the CN varied from one to six, n=0–5. As the number of imidazole ligands coordinated to zinc increases, the S–H proton dissociation energy also increases, (i.e. −S(H)CH3 becomes less acidic), and the Zn–S bond energy decreases. Furthermore, at a constant CN, the S–H proton dissociation energy decreases as the S–Zn–(ImH)n angles increase about their equilibrium position. The zinc-coordinated thiol can become more or less acidic depending upon the position of the coordinated imidazole ligands. The bonding and thermodynamic relationships discussed may apply to larger systems that utilize the [(His)3Zn(II)–L] complex as the catalytic site, including carbonic anhydrase, carboxypeptidase, β-lactamase, the tumor necrosis factor-α-converting enzyme, and the matrix metalloproteinases.
TOC Graphic

Introduction
Zinc is one of the most abundant and important metals in living systems, serving as an essential cofactor in thousands of proteins.1,2 Found in all six classes of enzymes, hydrolases being the most common, zinc is also involved in signaling and plays both structural and regulatory roles.3,4 The coordinating environment of zinc in proteins is dominated by ligation to nitrogen atoms of imidazole (ImH) from histidine (His, or H) side chains and sulfur atoms of thiol and thiolate from cysteine (Cys, or C) side chain amino acid residues, as revealed by numerous X-ray crystal and solution NMR structures.5–7 Of particular interest is the three-His coordination to zinc, [(His)3Zn(II)–L], with the L site occupied by a ligand or ligands that may (H2O, Cys, Glu,.) or may not (inhibitor) be native to the enzyme. The [(His)3Zn(II)–L] center acts as the catalytic “active” site in numerous enzymes, including carbonic anhydrase, β-lactamase, cytosine deaminase, matrix metalloproteinases, and the tumor necrosis factor-α-converting enzyme (TACE). As the center of catalysis, it seems important to understand the physical and chemical properties governing the stability of the first coordination sphere in the [(His)3Zn(II)–L] system. This report constitutes a step in that direction. Herein we present a detailed investigation of the interplay among coordination number (CN), molecular geometry, and both the bond strength and proton dissociation energy of the Zn–S(H)CH3 moiety for a series of [(Imidazole)nZn(II)–S(H)CH3] complexes. These systems are models for the [(His)3Zn(II)–Cys] coordination environment, which, among other things, is an important entity in the activation and inhibition processes of the matrix metalloproteinase (MMP) family of endopeptidases, as outlined below.
The MMPs comprise a family of 26 Zn(II)-dependent hydrolytic enzymes, which are involved in degrading and remodeling the macromolecular components of the extra-cellular matrix.8–10 With such breadth in their physiological roles, the MMPs have been implicated in a host of ailments, including cardiovascular disease, arthritis, cancer, and play a role in the development of neuropathic pain.11–17 In this regard, a widespread effort has been made over the last three decades to control and regulate the activities of these enzymes through selective, competitive inhibition. While little clinical success has been realized, selective MMP inhibition may still be attainable18,19 by, in part, exploiting structural relationships of the type reported herein.
The MMPs are members of the metzincin family of enzymes, which are distinguished by two highly conserved motifs, one containing three histidine residues that bind zinc at the catalytic site, and the second being the conserved methionine turn that sits beneath the active site zinc forming a hydrophobic floor.8,20,21 The signature zinc-binding motif of all MMPs reads HExGHxxGxxH in the catalytic domain, wherein the resting catalytic site consists of an approximately tetrahedral zinc center that is bonded to the protein through nitrogen atoms provided by the imidazole side chains of the three conserved histidines. In the inactive proMMP (Scheme 1A), the thiolate group of a cysteine residue within the propeptide coordinates Zn(II) and blocks substrate access, thereby causing latency.
Scheme 1.
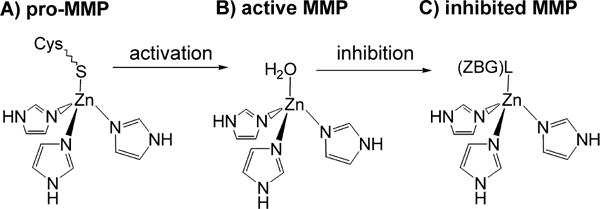
Depiction of the first coordination sphere of the catalytic zinc center of A) inactive proMMP, B) activated MMP, and C) inhibited MMP.
Upon activation, the cysteine-thiolate ligand is replaced by a water molecule, producing the proteolytically active [(His)3Zn(OH2)]2+ complex, as shown in Scheme 1B. Several lines of evidence suggest that the extent of ligand protonation is crucial. For example, in the proposed MMP mechanism the Zn–OH2 moiety plays a central role, losing both hydrogen atoms, as protons, to the hydrolysis products.22,23 Reports also argue for a critical protonation step of the Zn–S moiety, and possibly a change in CN, as a prerequisite for activation of proMMPs.24,25 When the activated enzyme is rendered inactive by competitive inhibition, whether through binding of a tissue inhibitor of metalloproteinase (TIMP) or an exogenous inhibitor, the H2O ligand is replaced by the inhibitor’s zinc binding group (ZBG), the functional group that directly coordinates to the catalytic Zn(II) ion, as illustrated in Scheme 1C. Proton transfer events may also occur to varying extents upon inhibitor binding; protic site(s) on the ZBG can remain protonated, serve as a hydrogen bond donor to a nearby amino acid, or donate a proton essentially completely to a stronger base/weaker acid in the active site cleft.
These putative steps involving proton transfer reactions between the Zn–L moiety and the protein environment are likely crucial to the processes of MMP activation, catalysis, and inhibition. Examination of the many MMPs in the RCSB protein data bank reveals an array of experimentally determined active site geometries. For instance, X-ray crystal and NMR solution structures of MMPs have shown a variety of 4- and 5-coordinate [(His)3Zn(II)–L] geometries that include tetrahedral, trigonal-bipyramidal, and square-pyramidal, demonstrating that the zinc active site of the MMPs is both coordinatively and geometrically flexible. To illustrate, Figure 1 shows the first coordination shell of Zn(II) centers that are from three X-ray crystal structures of MMPs, with selected bonding parameters listed in Table 1. Figure 1 (TBP) shows a complex of MMP-3 with a synthetic inhibitor (PDB code: 1BIW), wherein the inhibitor is coordinated through a bidentate hydroxamic acid ZBG, yielding a pentacoordinate Zn(II) center.26 The zinc geometry in Figure 1(TBP) is best described as trigonal-bipyramidal with the NImH–Zn–NImH angles slightly distorted from the idealized 90.0° and 120.0° (see Table 1). Figure 1 (SQP) shows a complex of MMP-1 with a synthetic inhibitor (PDB code: 1HFC), also having a bidentate hydroxamic acid ZBG.27 However, in this case the geometry about the pentacoordinate zinc is best described as a square-based pyramid. Last, Figure 1 (Tet.) shows the active site of proMMP-3 (PDB code: 1SLM), in which zinc is 4-coordinate and tetrahedral with NImH–Zn–NImH angles only slightly less than the idealized 109.5°.28 The variability in these experimentally determined [(ImH)3Zn(II)–L] geometries, despite the high degree of sequence and structure homologies of the MMP catalytic domain, clearly show that the active sites of the MMPs are not rigid.
Figure 1.

First coordination spheres of the catalytic zinc centers of three MMPs. Coordinates taken from published X-ray crystal structures; color scheme is CPK. TBP: trigonal bipyramidal, MMP-3, chelated by synthetic hydroxamic acid based inhibitor (PDB code: 1BIW.) SQP: square pyramidal, MMP-1, chelated by synthetic hydroxamic acid based inhibitor (PDB code: 1HFC.) Tet.: tetrahedral, cysteine thiolate from the pro domain of proMMP-3 (PDB code: 1SLM.)
Table 1.
Selected bonding parameters from first coordination shells of catalytic Zn(II) centers taken from MMP X-ray crystal structures shown in Figure 1. Bond lengths in Angstroms and angles in degrees.
| MMP (PDB code) | MMP-3 (1BIW) | MMP-1 (1HFC) | MMP-3 (1SLM) |
|---|---|---|---|
| Coord. Geometry | TBP | SQP | Tet. |
| Zn–NImH (Å) | 2.20 | 2.08 | 1.93 |
| 2.12 | 2.02 | 1.87 | |
| 2.11 | 1.88 | 1.80 | |
|
| |||
| mean Zn–NImH (Å) | 2.14 | 1.99 | 1.87 |
|
| |||
| ∠NImHZnNImH(°) | 107.1 | 104.0 | 105.6 |
| 101.4 | 100.4 | 105.1 | |
| 92.2 | 96.6 | 104.0 | |
|
| |||
| mean ∠NImHZnNImH(°) | 100.2 | 100.3 | 104.9 |
The extent to which changes in the [(His)3Zn(II)–L] coordination environment are a factor in Zn–L bond cleavage or proton transfer events that occur upon activation, catalysis, and inhibition is unknown. Experimentally, the various protic Zn–L species and their interactions are difficult to examine in the enzyme, since electron density maps from crystal structures are, in general, not sufficiently defined to locate hydrogen atoms.29,30 However, small-molecule model complexes of the active sites, their structures, and relative energies, are tractable by computational methods. Thus, we herein report the results of a series of quantum chemical calculations carried out to investigate the driving forces for both Zn–S bond dissociation and S–H deprotonation of Zn(II)-coordinated methylthiol, S(H)CH3. Using density function theory and second-order perturbation theory, calculations are carried out on a series of [(ImH)nZn(II)–S(H)CH3] and [(ImH)nZn(II)–SCH3], n=0–5, complexes. Particular attention is given to the geometry and geometry changes within the four-coordinate [(ImH)3Zn–S(H)CH3]2+, and its conjugate base, as models for the pro- or thiol inhibited-MMPs. The results of the calculations reveal that the coordination environment about the zinc center has a significant impact on the driving force for both bond dissociation and proton transfer processes involving the Zn(II)–S(H)CH3 moiety.
Computational Methods
All quantum mechanical calculations were performed using Gaussian 09 software (RevA.02),31 with most input files having been built using GaussView 5.0.8,32 all running on in-house MacPro and iMac computers. Density functional theory (DFT) calculations were performed using the M05-2X hybrid meta functional,33 incorporating an ultrafine integration grid. Full geometry optimizations and vibrational frequency calculations were performed to ensure true minima and obtain the zero-point vibrational energy and thermodynamic parameters, unless stated otherwise. Single-point ab initio calculations were performed using the frozen-core second order Møller-Plesset perturbation theory (MP2) method with a small number of calculations obtained using the frozen-core coupled-cluster theory with single and double excitations and a quasiperturbative treatment of connected triple excitations, CCSD(T), both at the M05-2X geometries. Calculations were performed in the gas-phase, and the all electron cc-pVDZ or cc-pVTZ basis sets were used on all atoms including zinc.34–36 The closed shell singlet electronic state was used in all calculations.
The reaction(s) to determine the Zn–S bond dissociation energy (BDE, or binding energy) and the S–H proton dissociation energy (PDE) are shown in equations (1) through (3) below. In these reactions the imidazole (ImH) variable n is set at 0 through 5.
| (1) |
| (2) |
| (3) |
Reaction (1) is used to calculate the Zn–S BDE for the binding of the charge neutral S(H)CH3 to the Zn(II)–ImH complexes, while (2) depicts anionic CH3S− binding. With the release of the proton in (3), it is sometimes referred to as the gas-phase acidity reaction. In all cases the BDE and PDE were determined by calculating the difference in energy of the products minus the reactants. The reported BDE and PDE values are at 0 K and include zero-point vibrational energies, therefore they are Do values (= ΔHo = ΔGo). Inclusion of thermal and entropic effects at 298K (ΔG298) are also reported at the M05-2X/cc-pVTZ level, under the particle-in-a-box, rigid rotor, and harmonic oscillator approximations.
As a test of our methods used, M05-2X/cc-pVTZ calculations yield a 298 K gas-phase Gibbs’ deprotonation energy, ΔG298, of 1480 kJ/mol (353.6 kcal/mol) for HSCH3, while single-point MP2/cc-pVTZ//M05-2X/cc-pVTZ results yield 1488 kJ/mol (355.6 kcal/mol), respectively. Both these values compare satisfactorily with the experimental value of 1476±9 kJ/mol (352.7±2.1 kcal/mol) for HSCH3.37 Single-point CCSD(T)/cc-pVTZ//M05-2X/cc-pVTZ results yield a value that is roughly 1.5% higher than experiment for ΔG298 at 1498 kJ/mol. Experimental bond lengths, binding energies, and deprotonation energies are not available for Zn(II) complexes like the ones investigated in this study. However, two recent reports by Amin, Truhlar, and co-workers, assessing the performance of theoretical methods on energies and geometric properties of Zn(II) complexes found that the M05-2X method performs very well.38,39 In fact, the M05-2X performed best, on average, for calculations of bond lengths, dipole moments, and Zn-ligand bond dissociation energies among 39 DFT methods studied, and they recommend the M05-2X functional for the accurate calculation of geometries, dipole moments, and energetics of Zn compounds. While a recent report by Weaver et. al., has evaluated numerous DFT methods for their accuracy in calculating heats of formation of small Zn(II) complexes, the M05-2X functional was not included in their study.40
Results and Discussion
1) Coordination Number Influence on Zn–S(H)CH3 Bond Energies
Foremost, both HSCH3 and CH3S− coordinate to Zn(II) through their sulfur atoms to yield stable gas-phase complexes for all six of the CNs studied. Figure 2 shows the optimized structures for [(ImH)1–5Zn(II)–S(H)CH3] and [(ImH)1–5Zn(II)–SCH3] at the M05-2X/cc-pVTZ level of theory. Selected parameters are also listed in Table 2, with complete coordinates included in the supplementary material. For comparison, Table 2 also includes parameters at the M05-2X/cc-pVDZ level, which differ only slightly from those using the larger basis set.
Figure 2.
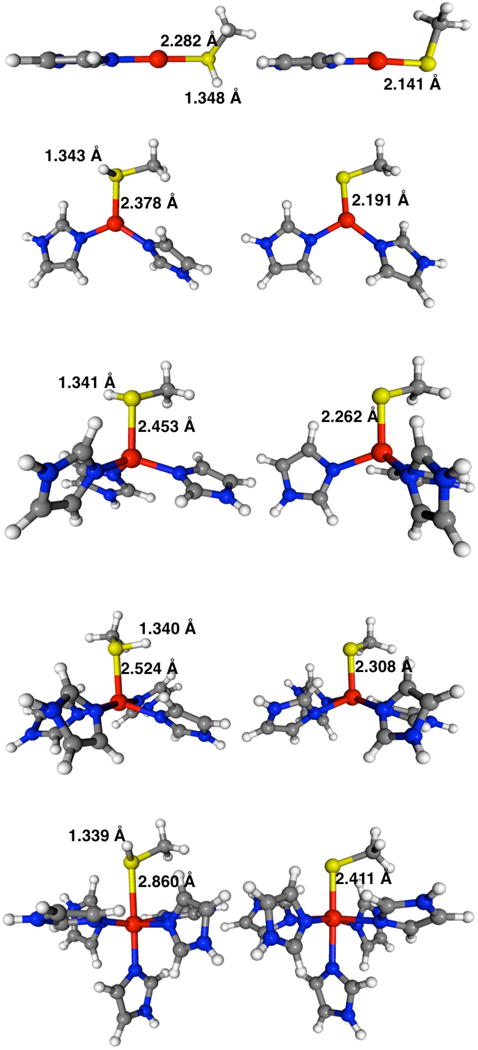
Geometry optimized structures for the [(ImH)nZn–S(H)CH3]2+ complexes on the left and their deprotonated [(ImH)nZn–SCH3]+ counterparts on the right, n = 1 through 5 from top to bottom. Selected parameters (in Angstroms) listed at the M05-2X/cc-pVTZ level of theory.
Table 2.
Selected bond lengths (Å) and partial atomic charges (a.u., atomic units) for [(ImH)nZn(II)–S(H)CH3] and [(ImH)nZn(II)–SCH3] systems at the M05-2X/cc-pVTZ level of theory. Last three columns contain M05-2X/cc-pVDZ values.
| Molecule/Ion | Bond Lengths (Å) | Mulliken Atomic Charges | cc-pVDZ Bond Lengths (Å) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Thiols | Zn–S | Zn–N(avg) | S–H | H | S | Zn | Zn–S | Zn–N(avg) | S–H |
| [S(H)CH3] | 1.3368 | 0.111 | −0.173 | 1.3503 | |||||
| [Zn–S(H)CH3]2+ | 2.308 | 1.3547 | 0.231 | 0.213 | 1.207 | 2.322 | 1.3670 | ||
| [(ImH)1Zn–S(H)CH3]2+ | 2.282 | 1.892 | 1.3477 | 0.213 | 0.092 | 0.983 | 2.289 | 1.895 | 1.3598 |
| [(ImH)2Zn–S(H)CH3]2+ | 2.378 | 1.958 | 1.3434 | 0.201 | −0.020 | 0.945 | 2.385 | 1.960 | 1.3557 |
| [(ImH)3Zn–S(H)CH3]2+ | 2.453 | 2.017 | 1.3406 | 0.178 | −0.046 | 0.882 | 2.454 | 2.020 | 1.3528 |
| [(ImH)4Zn–S(H)CH3]2+ | 2.524 | 2.133 | 1.3403 | 0.175 | −0.071 | 0.942 | 2.520 | 2.136 | 1.3525 |
| [(ImH)5Zn–S(H)CH3]2+ | 2.860 | 2.168 | 1.3387 | 0.169 | −0.147 | 0.996 | 2.837 | 2.171 | 1.3508 |
| Δ[(n=5)–(n=1)]* | 0.578 | 0.276 | −0.0090 | −0.044 | −0.239 | 0.548 | 0.276 | −0.0090 | |
| Thiolates | |||||||||
| [SCH3]1− | −0.851 | ||||||||
| [Zn–SCH3]+ | 2.179 | −0.068 | 0.879 | 2.190 | |||||
| [(ImH)1Zn–SCH3]+ | 2.141 | 1.935 | −0.224 | 0.776 | 2.145 | 1.938 | |||
| [(ImH)2Zn–SCH3]+ | 2.191 | 2.009 | −0.346 | 0.807 | 2.192 | 2.014 | |||
| [(ImH)3Zn–SCH3]+ | 2.262 | 2.070 | −0.411 | 0.775 | 2.269 | 2.070 | |||
| [(ImH)4Zn–SCH3]+ | 2.308 | 2.186 | −0.429 | 0.830 | 2.295 | 2.196 | |||
| [(ImH)5Zn–SCH3]+ | 2.411 | 2.246 | −0.474 | 0.890 | 2.403 | 2.251 | |||
| Δ[(n=5)–(n=1)]* | 0.270 | 0.311 | −0.250 | 0.258 | 0.313 | ||||
Differences between (ImH)5 & (ImH)1 systems.
The calculated Zn(II)–S BDEs of [(ImH)n Zn–S(H)CH3]2+ and [(ImH)nZn–SCH3]+, n=0–5, are listed in Table 3 at the M05-2X/cc-pVTZ level of theory, with MP2/cc-pVTZ//M05-2X/cc-pVTZ values listed for comparison. Both methylthiol and methylthiolate are bound very strongly to the bare Zn(II) ion with BDEs of 576 and 1785 kJ/mol, respectively. In fact, the Zn(II)–Sthiolate bond is much stronger than its Zn(II)–Sthiol counterpart, for all CNs. The binding energy of both methylthiolate and methylthiol decrease significantly with the addition of ImH ligands to the Zn(II) center. The Zn–Sthiolate BDEs decrease step-wise first to 1401 kJ/mol in [(ImH)Zn–SCH3]+ down to their minimum of 699 kJ/mol in the 6-coordinate, octahedral [(ImH)5Zn–SCH3]+ complex. The Zn–Sthiol BDEs drop to 385 kJ/mol in [(ImH)Zn–S(H)CH3]2+ down to their minimum of a very weak 14 kJ/mol in the 5-coordinate square-pyramidal [(ImH)4 Zn–S(H)CH3]2+ complex, in which HSCH3 occupies the apex position, see Figure 2. The Addison parameter (τ) can be used to quantify square-pyramidal/trigonal-bipyramidal geometries for five-coordinate complexes, with a value of 0.0 indicating a true square-pyramid and a value of 1.0 indicating a trigonal-bipyramid.41 In the case of [(ImH)4Zn–S(H)CH3]2+, τ = 0.45 indicating a structure closer to square-pyramid. A meager 3 kJ/mol higher in energy is a structure described as trigonal-bipyramidal, τ = 0.63, in which HSCH3 occupies an equatorial position (2 kJ/mol higher at MP2/cc-pVTZ//M05-2X/cc-pVTZ level). The Zn–S BDE of the 6-coordinate [(ImH)5Zn–S(H)CH3]2+, 38 kJ/mol, is low but noticeably greater than that of its 5-coordinate counterpart. The stable 5-coordinate [(ImH)4Zn–S(H)CH3]2+ complexes were difficult to locate computationally. Starting with an initial trigonal-bipyramidal geometry having HSCH3 in an axial position resulted in dissociation of the thiol ligand from zinc to form the unbound tetrahedral [(ImH)4Zn]2+ 4 and S(H)CH3 systems using multiple levels of theory: M05-2X/cc-pVDZ, M05-2X/cc-pVTZ, M05-2X/aug-cc-pVDZ, B3LYP/cc-pVDZ, BLYP/cc-pVDZ, or MP2/cc-pVDZ. Both square-pyramidal (τ = 0.30) and trigonal-bipyramidal (τ = 1.04) geometries were also located for the 5-coordinate thiolate complex, [(ImH)4Zn–SCH3]+, of which the square-pyramidal is again lower in energy, by 2 kJ/mol, with a considerable BDE of 740 kJ/mol. The low stability of the protonated [(ImH)4Zn–S(H)CH3]2+ to Zn–S dissociation may have implications regarding the activation process of the MMPs wherein the Zn–SCH3 bond must break. It has been reported that a CN change from four to five accompanies protonation of the coordinated sulfur atom of Cys upon Zn–S bond cleavage.25 In that study it is the carboxylate oxygen of the conserved Glutamic acid that binds to zinc. Our results are consistent with that finding; the calculations reveal a CN change from 4 to 5 would indeed result in a significantly weakened Zn–S bond. Surprisingly, while [(ImH)4Zn–S(H)CH3]2+ is stable at 0 K, inclusion of thermal and entropic effects show that it would undergo spontaneous thiol dissociation at 298 K, with ΔG298 = −37 kJ/mol (Table 3, values in parentheses). Thiol dissociation from the 6-coordinate [(ImH)5Zn–S(H)CH3]2+ complex would also be spontaneous with ΔG298 = −10 kJ/mol. Lastly, the Zn–S BDEs obtained using the MP2/cc-pVTZ//M05-2X/cc-pVTZ method are in good agreement with the M05-2X/cc-pVTZ values. The energy differences between the two methods are much less than their overall magnitudes, and the two methods yield identical trends.
Table 3.
Zn–S Bond Dissociation Energies (BDEs) and S–H Proton Dissociation Energies (PDEs), both Do values (=ΔHo), for [(ImH)nZn(II)–S(H)CH3] and [(ImH)nZn(II)–SCH3] systems at various levels of theory. All numbers in kJ/mol. (ΔG298 values listed in parentheses)
| Zn–S Bond Dissociation Energies (kJ/mol) | S–H Proton Dissociation Energies (kJ/mol) | |||||
|---|---|---|---|---|---|---|
| cc-pVTZ | cc-pVTZ | cc-pVDZ | ||||
| M05-2X | MP2//M05-2X | M05-2X | MP2//M05-2X | CCSD(T)//M05-2X | M05-2X | |
| Thiols | ||||||
| [S(H)CH3] | 1504 (1480) | 1512 | 1522 | 1533 | ||
| [Zn–S(H)CH3]2+ | 576 (547) | 545 | 296 (270) | 294 | 303 | 287 |
| [(ImH)1Zn–S(H)CH3]2+ | 385 (341) | 374 | 488 (461) | 487 | 499 | 482 |
| [(ImH)2Zn–S(H)CH3]2+ | 175 (133) | 165 | 618 (592) | 618 | 616 | |
| [(ImH)3Zn–S(H)CH3]2+ | 117 (73) | 113 | 721 (692) | 723 | 715 | |
| [(ImH)4Zn–S(H)CH3]2+ | 14 (−37) | 11 | 778 (749) | 779 | 784 | |
| [(ImH)5Zn–S(H)CH3]2+ | 38 (−10.) | 42 | 843 (820) | 846 | 848 | |
| Δ[(n=5)−(n=1)]* | 355 (359) | 359 | 366 | |||
| Thiolates | ||||||
| [Zn–SCH3]+ | 1785 (1756) | 1764 | ||||
| [(ImH)1Zn–SCH3]+ | 1401 (1360) | 1400 | ||||
| [(ImH)2Zn–SCH3]+ | 1061 (1021) | 1059 | ||||
| [(ImH)3Zn–SCH3]+ | 901 (861) | 902 | ||||
| [(ImH)4Zn–SCH3]+ | 740 (693) | 745 | ||||
| [(ImH)5Zn–SCH3]+ | 699 (649) | 708 | ||||
| Δ[(n=5)−(n=1)]* | −702 (−711) | −692 | ||||
Difference in energy between (ImH)5 & (ImH)1 systems.
Structurally, two trends stand out in the first coordination sphere bond length parameters starting with the [(ImH)1Zn(II)–L] complex. First, with the addition of 1 to 4 imidazole ligands, the Zn–S bond length (RZn–S) increases, as listed in Table 2 and illustrated in Figure 2. In the thiolate complexes RZn–S increases from 2.141 to 2.411 Å, a lengthening of +0.270Å, an even larger increase occurs in the thiol complexes (+0.578 Å) with RZn–S values extending from 2.282 to 2.860 Å. Second, accompanying the increase in RZn–S, a corresponding increase in the average Zn–N bond lengths (RZn–N(avg)) occurs with the progression in coordination number. RZn–N(avg) increases 0.310Å and 0.276Å for the thiolate and thiol complexes, respectively. Even the very weakly bound [(ImH)4Zn–S(H)CH3]2+ follows the step-wise pattern. In other words, for all [(ImH)1–5Zn(II)–L] complexes, as the CN increases, all bonds to zinc increase in length. This parallels the step-wise reduction in the methylthiolate binding strength and the almost step-wise reduction (excluding [(ImH)4Zn–S(H)CH3]2+ in the methylthiol binding strength.
2) Coordination Number Influence on H–SCH3 Deprotonation Energies
Accompanying the increase in zinc’s coordination number, changes in bonds not directly adjacent to the zinc atom are also observed. The primary example is the methylthiol S–H bond lengths and corresponding proton dissociation energies. The S–H proton dissociation energy (PDE) of the [(ImH)nZn–S(H)CH3]2+ complexes are listed in Table 3 at the M05-2X, MP2 and selective CCSD(T) levels of theory. The calculated PDE’s of methylthiol are significantly diminished from its native value when its sulfur atom is bonded to the zinc atom in any of the [(ImH)nZn–S(H)CH3]2+ complexes. Thus, predictably, methylthiol becomes more acidic by virtue of its association with the Lewis acidic Zn(II) ion. Furthermore, the fewer the ligands coordinated to zinc, the lower the PDE becomes. Hence, the lowest PDE occurs when methylthiol is bound to the bare Zn(II) ion at 296 kJ/mol, a striking reduction of 1208 kJ/mol from the unbound HSCH3 value of 1504 kJ/mol. MP2 and CCSD(T) methods calculate similar PDE reductions of 1218 and 1219 kJ/mol, respectively. The thiol PDE increases monotonically with CN. As the number of ImH ligands is increased from 1 to 5, the PDE trends upward to 488 kJ/mol for the linear [(ImH)Zn–S(H)CH3]2+, finishing at 843 kJ/mol in the 6-coordinate [(ImH)5Zn–S(H)CH3]2+. This upper value is still 661 kJ/mol (44%) less than the PDE of the unbound HSCH3. Even the very weakly bound [(ImH)4Zn–S(H)CH3]2+ follows the step-wise progression of proton dissociation energies. Although the Zn–S BDE is only 14 kJ/mol in [(ImH)4Zn–S(H)CH3]2+, the reduction in the S–H PDE is over 726 kJ/mol, or almost one-half the unbound value.
These absolute PDEs and their changes are similar (≤ 3% difference) to those calculated using the smaller cc-pVDZ basis set, the ab initio MP2/cc-pVTZ//M05-2X/cc-pVTZ, and high-level CCSD(T)/cc-pVTZ//M05-2X/cc-pVTZ methods. Furthermore, among the ligand protons in these complexes, removal of the S–H proton is the least endergonic. For example, in [(ImH)3Zn–S(H)CH3]2+ the imidazole N–H PDE is significantly higher,42,43 at 826 kJ/mol, than the S–H PDE of 715 kJ/mol (M05-2X/cc-pVDZ values for Do).
The most noticeable structural change in methylthiol upon binding to Zn2+ is its S–H bond length (RS–H), as seen in Table 2 and Figure 2. When HSCH3 binds to the bare Zn(II) ion RS–H increases in length from its initial value of 1.337 Å to 1.355 Å (+0.018 Å). With the addition of imidazole ligands, RS–H decreases, first to 1.348 Å in [(ImH)Zn–S(H)CH3]2+, reaching 1.339 Å in the 6-coordinate [(ImH)5Zn–S(H)CH3]2+. This decrease in RS–H of 0.9 pm with increasing numbers of ImH ligands is accompanied by the increase in methanethiol’s proton dissociation energy of +355 kJ/mol.
Atomic (Mulliken) charges were used to track shifts in electron density on the Zn, S and H atoms as a function of the number of Lewis basic ImH ligands coordinated to Zn2+. Although the charge on zinc does not follow a simple pattern, the charges on S and H do (Table 2). As the CN increases, an accumulation of negative charge density on S is observed for both the thiol and thiolate complexes. Likewise, in the protonated [(ImH)nZn–S(H)CH3]2+ complexes the positive charge on H also decreases as n increases. Hence, although the –S(H) unit becomes more negatively charged as zinc’s CN increases, the partial charge differences between S and H indicate that the ionic character in the S–H bond increases with n. Although the PDE involves energies of both the protonated and deprotonated complexes, the results show a clear correlation between positive charge on H and the ease with which it is removed as a proton. This result has implications regarding the strength of intermolecular forces (i.e. H-bonding) required to modulate the Zn–S bond strength for coordinated −S(H)R ligands, since the amount of positive electrostatic charge collection would modulate the proton interactions with negative charges/bases in the vicinity.
3) Molecular Geometry Influence on H–SCH3 Deprotonation Energies
Significant deprotonation energy differences have been established for the series of [(ImH)nZn–S(H)CH3]2+ complexes, with the PDE increasing as zinc’s coordination number increases. However, as the CN changes, so does its coordination geometry; linear for n=0&1, trigonal planar for n=2, tetrahedral for n=3, square-pyramidal for n=4, and octahedral at CN=6 (n=5). This begs the question of whether the change in PDE derives from (a) the change in CN, (b) the change in geometry, or (c) both CN and geometry changes? To test (b), the influence of geometry on the PDE, we need a systematic way to change the geometry while leaving the CN constant. Inspection of Figure 2 reveals that it is the angles about zinc that change the most with CN. Specifically, the S–Zn–Nn angles, ∠SZnNn, ranges from a minimum of 83° in the hexacoordinate [(ImH)5Zn–S(H)CH3]2+ structure to a maximum of 132° in the three-coordinate [(ImH)2Zn–SCH3]+ structure (omitting all angles near 180°). Therefore, to measure the influence of geometry on the PDE, we systematically modified the coordination geometry by incrementally adjusting the S–Zn–Nn angles and calculating the resulting PDE at constant CNs. This approach was applied to the 3 and 4-coordinate systems, [(ImH)2Zn–S(H)CH3]2+ and [(ImH)3Zn–S(H)CH3]2+, primarily because their symmetry-equivalent bond angles make it straight forward to systematically modify their geometries. Additionally, the 4-coordinate [(ImH)3Zn(II)–S(H)CH3] is structurally similar to [(His)3Zn(II)–L], which is an important catalytic center in many enzymes.
In the series of constrained geometry calculations, the two/three symmetry-related S–Zn–(ImH)n angles in [(ImH)2,3Zn–S(H)CH3]2+ were incrementally varied approximately ±20° about the idealized trigonal planar (120.0°) and tetrahedral (109.5°) angles; all other internal coordinates (bond lengths & angles) were allowed to vary during the geometry optimization process. Energies were obtained for both the thiol and thiolate complexes with the PDE calculated from the difference in energy between the optimized structures. The results of these constrained optimizations are used to systematically gauge the impact that geometry alone has on the PDEs/acidities. These constrained angle calculations were carried out with the cc-pVDZ basis set, which we have demonstrated performs on par with the larger cc-pVTZ basis set for PDE calculations in this study. As no vibrational frequency calculations were performed, no zero-point energies are included in these results. Therefore these PDE values are Des.
Results of the constrained angle calculations are arranged in Table 4, while Figure 3 shows 8 of these structures, 4 for each of the protonated and deprotonated complexes at the constrained ∠SZnN2,3 of 95° and 125°. The trend in PDEs is clearly apparent in Figure 4, which reveals the cause and effect relationship between the ∠SZnNn and PDE. This plot unambiguously demonstrates that as ∠SZnNn increases the PDE decreases for both [(ImH)2Zn–S(H)CH3]2+ and [(ImH)3Zn–S(H)CH3]2+; in other words, the complexes become more acidic. These results imply an intrinsic relationship between coordination geometry and deprotonation energy.
Table 4.
Selected bond lengths, partial charges, proton dissociation energies (De), and relative energies for [(ImH)2Zn–S(H)CH3]2+ (top set) and [(ImH)3Zn–S(H)CH3]2+ (bottom set) as a function of S–Zn–Nn angles. Values from M05-2X/cc-pVDZ calculations.
| Bond angles | Bond lengths (Å) | Partial charges (a.u.) | Energies (kJ/mol) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| (S–H) PDE | Relativec | ||||||||
| Zn–S | S–H | Zn–(N)na | Zn | H | S | (ImH)nb | Thiol (Thiolate) | ||
|
|
|||||||||
| ∠SZn(ImH)2 | Zn–(ImH)2 | (ImH)2 | |||||||
| 95° | 2.565 | 1.3536 | 1.943 | 0.760 | 0.168 | −0.062 | 0.921 | 680 | 36.7 (75.5) |
| 105° | 2.460 | 1.3545 | 1.948 | 0.727 | 0.175 | −0.009 | 0.882 | 662 | 10.0 (30.7) |
| 115° | 2.398 | 1.3554 | 1.957 | 0.697 | 0.182 | 0.034 | 0.856 | 651 | 0.6 (10.1) |
| 120° | 2.377 | 1.3556 | 1.963 | 0.689 | 0.185 | 0.050 | 0.840 | 647 | 0.5 (6.1) |
| 125° | 2.358 | 1.3560 | 1.972 | 0.685 | 0.188 | 0.060 | 0.829 | 642 | 3.5 (4.2) |
| 135° | 2.327 | 1.3563 | 2.009 | 0.671 | 0.193 | 0.080 | 0.804 | 627 | 28.8 (14.5) |
| Δ(135°−95°) | −0.238 | 0.0027 | 0.066 | −0.089 | 0.025 | 0.142 | −0.117 | −53 | |
| fully optd | 2.385 | 1.3557 | 1.960 | 0.699 | 0.186 | 0.035 | 0.842 | 641 | 0.0 (0.0) |
|
| |||||||||
| ∠SZn(ImH)3 | Zn–(ImH)3 | (ImH)3 | |||||||
| 85° | 2.857 | 1.3513 | 1.997 | 0.699 | 0.129 | −0.097 | 1.125 | 813 | 57.8 (129.4) |
| 95° | 2.592 | 1.3522 | 2.006 | 0.626 | 0.143 | −0.041 | 1.106 | 770 | 18.9 (47.3) |
| 105° | 2.473 | 1.3527 | 2.016 | 0.570 | 0.155 | 0.004 | 1.085 | 750 | 1.6 (9.9) |
| 110° | 2.436 | 1.3533 | 2.024 | 0.548 | 0.161 | 0.021 | 1.074 | 741 | 1.4 (0.9) |
| 115° | 2.409 | 1.3536 | 2.036 | 0.543 | 0.167 | 0.030 | 1.062 | 737 | 7.0 (3.2) |
| 125° | 2.361 | 1.3546 | 2.084 | 0.548 | 0.179 | 0.055 | 0.997 | 722 | 49.2 (30.4) |
| Δ(125°−85°) | −0.496 | 0.0033 | 0.087 | −0.151 | 0.050 | 0.152 | −0.128 | −91 | |
| fully optd | 2.454 | 1.3528 | 2.020 | 0.567 | 0.163 | −0.005 | 1.079 | 741 | 0.0 (0.0) |
average Zn–Nn distance,
total charge on the zinc coordinated imidazole ligands,
Energy of the constrained thiol and (thiolate) relative to their fully optimized (ImH)2 and (ImH)3 complexes.
Fully opt numbers are from optimizations with no constraints, in which the average ∠SZn(ImH)2 = 117.6° and the average ∠SZn(ImH)3 = 108.0°, for the thiol complexes
Figure 3.
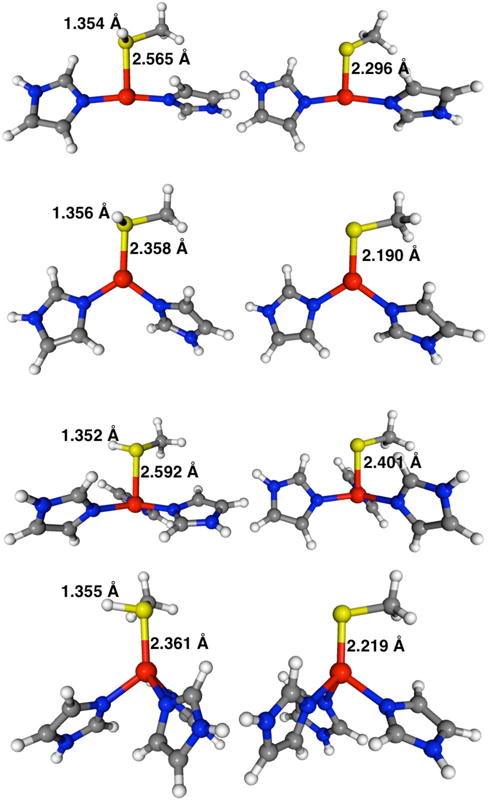
Constrained geometry optimized structures for [(ImH)2Zn–S(H)CH3]2+ and [(ImH)3Zn–S(H)CH3]2+ on the left and the deprotonated [(ImH)2Zn–SCH3]+ and [(ImH)3Zn–SCH3]+ complexes on the right, at S–Zn–(ImH)n angles of 95° (1st and 3rd set) and 125° (2nd and 4th set). Selected bonding parameters (in Angstroms) at the M05-2X/cc-pVDZ level of theory.
Figure 4.
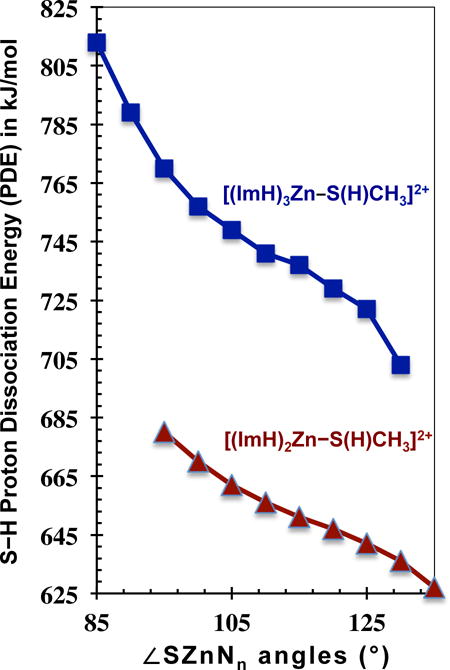
Proton dissociation energy (kJ/mol) plotted versus the S–Zn–(ImH)n angle for [(ImH)3Zn–S(H)CH3]2+, top square data, and [(ImH)2Zn–S(H)CH3]2+, bottom triangle data. Proton dissociation energies calculated at the M05-2X/cc-pVDZ level of theory. This figure represents the energy difference between the geometry-optimized deprotonated and geometry-optimized protonated complexes, with the only constraint being ∠SZn(ImH)2,3.
A close analysis of the PDE variations in Table 4 shows that the PDE decreases by 91 kJ/mol across the 40° increase in ∠SZn(ImH)3, and by 53 kJ/mol across the 40° increase in ∠SZn(ImH)2. The magnitudes of these PDE changes are considerable and, for [(ImH)3Zn–S(H)CH3]2+, equivalent to the strength of a weak covalent bond. The 91 kJ/mol is also of the same order as the PDE change from CN three to four, and is greater than the four to five or five to six changes (Table 3). While a 40° change in S–Zn–N2,3 angle is a large geometry change for a given CN, changes of a few degrees to either side of the lowest energy structure requires little energy. The last columns of Table 4 lists the relative energies upon ∠SZn(ImH)2,3 change. These relative energies are with respect to the fully optimized M05-2X/cc-pVDZ structures, in which the average ∠SZn(ImH)n are 117.6° for the trigonal planar [(ImH)2Zn–S(H)CH3]2+ and 108.0° for the tetrahedral [(ImH)3Zn–S(H)CH3]2+ (Figure S1). Over the 40° range in constrained angles the energy of the [(ImH)2,3Zn(II)–S(H)CH3] systems increases by up to 58 kJ/mol, while a ±10° change about the equilibrium positions requires significantly less energy, <15 kJ/mol. Thus the part of the potential energy well encompassing this physiologically relevant angular motion is shallow. Consistent with this result, variability in coordination angles observed in the many X-ray crystal and NMR structures of pro- and inhibited MMPs reveals that protein conformational forces are sufficient to distort the active-sites from their intrinsically optimum geometries (Table 1, Figure 1).25,44–47
The plots of relative [(ImH)3Zn–S(H)CH3]2+ and [(ImH)3Zn–SCH3]+ energies versus change in S–Zn–N3 angle (Figure 5) reveals an interesting relationship between these systems. First, note that the thiolate plot is shifted to larger angles relative to the thiol plot, owing to the stronger and shorter Zn–S bond forcing ∠SZn(ImH)3 to greater values in the fully optimized thiolate complex (average ∠SZn(ImH)3 =113.2° thiolate vs. 108.0° thiol) (see Figure S1). Also apparent in the rightmost columns of Table 4 and in Figure 5 is the approximately harmonic nature of the molecular distortions along the S–Zn–(ImH)2,3 bending coordinates. The bottom of each well falls near the average S–Zn–(ImH)3 angles of the respective fully optimized structures for both the protonated and deprotonated complexes. For any molecule, there is a thermodynamic driving force to obtain its equilibrium ground state geometry. So a structure, either protonated or deprotonated, that is located anywhere above the potential energy minimum will undergo a spontaneous change to the lowest energy structure, thereby releasing energy in proportion to the height of the structure on the potential well. It appears that the factors indicating whether the PDE of a compound will change with geometry are: 1) the extent of separation between the protonated and deprotonated geometries along the S–Zn–(ImH)2,3 bending coordinate (i.e. how far the bottom of the wells are from each other) and, 2) how high in energy the structure is above that of its conjugate acid or base (magenta triangles = blue squares – red diamonds), which, for a given separation of minima, scales with the steepness of the potential wells. In other words, if the coordination geometries of the protonated and deprotonated structures are very similar, and the structures are energetically the same height from the minimum of their acid/base conjugate, any change in geometry will affect both structures equally and the PDE/acidity will not be a steep function of geometry.
Figure 5.
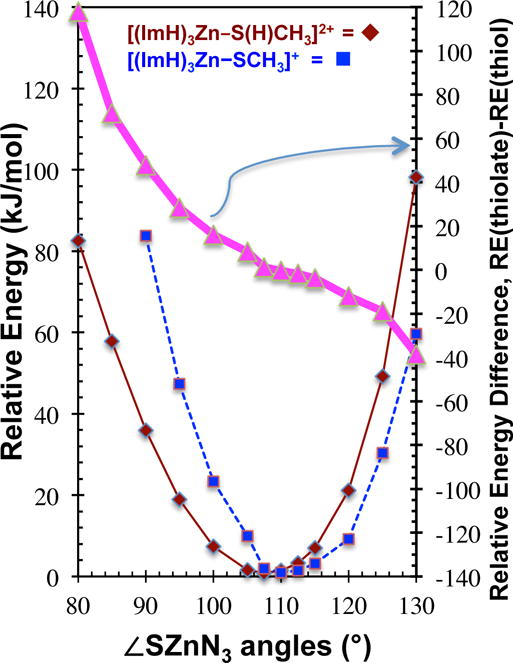
Relative energy versus S–Zn–N3 angle for [(ImH)3Zn–S(H)CH3]2+, (red solid diamonds) and [(ImH)3Zn–SCH3]+, (blue square dashes) systems. Energies are with respect to the fully optimized geometries at the M05-2X/cc-pVDZ level of theory. Also shown is the difference in relative energies (magenta triangles = blue squares – red diamonds), RE([(ImH)3Zn–SCH3]+) – RE([(ImH)3Zn–S(H)CH3]2+). Points are connected by straight lines for ease of viewing. For clarity, 80° and 85° points missing from thiolate plot.
The above relationships are illustrated by the relative energy difference plot in Figure 5 (magenta, triangles). The difference plot in Figure 5 was constructed by subtracting the thiol points from the thiolate points. Note the strong resemblance to the PDE curve in Figure 4. The slope of the difference curve provides a quantitative measure of the extent to which the PDE will change as a function of ∠SZn(ImH)3. The following three examples serve to further clarify this relationship. Example 1: Consider a [(ImH)3Zn–S(H)CH3]2+ like-complex with a geometry that is energetically and structurally similar to the geometry minima of the thiol, that is to the left of 108.0° in Figure 5. The resulting PDE is predicted to be higher than that of the fully optimized structure. Example 2: Consider an experimentally determined [(ImH)3Zn–S(H)CH3]2+ complex with a geometry near the bottom of the potential wells, between that of the protonated and deprotonated complexes, between 108.0° and 113.2°. The resulting PDE of the structure will be relatively unchanged, and near its value for the optimized structure. Example 3: Consider a [(ImH)3Zn–S(H)CH3]2+ complex having S–Zn–N3 angles greater than 113.0°, a structure more thiolate like. The resulting PDE is predicted to be lower than that of the optimized geometry. Moreover, the further to the left or right on the plot, the greater will be the change in PDE. Therefore, intentionally restricting the L–Zn–Nn angles to be large or small, as can be done in synthetic model complexes such as the tris(pyrazolyl)borate or cycen/cyclam tetraazamacrocycle structures, could lead to constrained zinc geometries affecting the protonation state, acidity, and chemistry of the models.45,48–51
Three important interatomic distance changes that accompany changes in ∠SZn(ImH)2,3 are listed in Table 4, and include the Zn–S, S–H, and average Zn–NImH bond lengths. Overall, RZn–S decreases significantly, while both RS–H and RZn–N(avg) are less sensitive to ∠SZn(ImH)2,3 for both CNs. Figure 6 illustrates the downward trend in RZn–S and the upward trend in RS–H with increasing ∠SZn(ImH)n. The increase in RS–H is consistent with the lower PDE upon increasing the S–Zn–(ImH)n angles. Importantly, these bond length trends mimic those that occur with the decrease in CN (see Table 2). Although Zn–S BDEs as a function of angle were not calculated here, the results from the CN changes taught us that, in general, the shorter RZn–S is the stronger the Zn–S bond is. Therefore, it is reasonable to suggest that increasing ∠SZn(ImH)2,3 increases the Zn–S bond strength.
Figure 6.

Plots of Zn–S, (triangles) and S–H, (squares) bond lengths as a function of the S–Zn–(ImH)n angles, ∠SZnN2,3 for [(ImH)2Zn–S(H)CH3]2+, open points, and [(ImH)3Zn–S(H)CH3]2+, solid points.
The partial atomic charges on the Zn atom, the imidazole ligands, and the S and H atoms of HSCH3, are also listed in Table 4. The numbers suggest a transfer of negative charge density (electrons) from H and S and into the Zn(II)–(ImH)n moiety with increasing ∠SZn(ImH)n. This charge redistribution is clearly revealed in Figure 7 where H and S partial charges are plotted versus ∠SZn(ImH)n. Although the –S(H) unit becomes more positively charged as ∠SZn(ImH)2,3 increases, the partial charge differences between S and H indicate that the ionic character in the S–H bond decreases, the covalent character increases, with increasing ∠SZn(ImH)2,3 (Table 4 and Figure 7). The end result is that as ∠SZn(ImH)2,3 increases there is an buildup of positive charge on H and, accompanying the accumulation of positive charge on H, there is an increase in RS–H and a concomitant decrease in the S–H proton dissociation energy.
Figure 7.
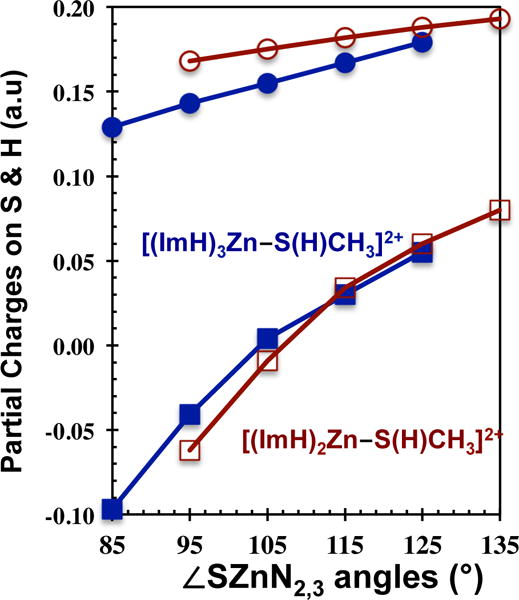
Plots of Partial Charges (atomic units) on S (squares) and H (circles) as a function of the S–Zn–(ImH)2,3 angles, ∠SZnN2,3, for [(ImH)2Zn–S(H)CH3]2+, open red data points, and [(ImH)3Zn–S(H)CH3]2+, solid blue data points.
4) Conclusions and Further Implications
Through high-quality density functional theory and MP2 calculations performed on a series of [(ImH)0-5Zn(II)–S(H)CH3] complexes, we have demonstrated that both the coordination number and the coordination geometry have major impacts on the Zn–S(H)CH3 bond strength and the S–H proton dissociation energy. Predictably, when a ligand, in this case –S(H)CH3, is coordinated to a transition metal ion, the coordinated ligand becomes “activated” toward greater acidity, (i.e. a lower pKa), and a reduced proton dissociation energy. Our calculations reveal that as the Zn(II) CN increases, the influence on the PDE becomes considerably and quantifiably less, albeit still substantial, even for CN=6, wherein the S–H PDE in [(ImH)5Zn(II)–S(H)CH3] is almost one-half the intrinsic value as that for the uncoordinated S(H)CH3. Accompanying the increase in PDE, the Zn–S bond becomes longer and weaker as CN increases. Perhaps less obvious is that at a constant CN, the geometry about the Zn(II) center also changes the thiol’s PDE to a significant extent. In both [(ImH)2Zn(II)–S(H)CH3] and [(ImH)3Zn(II)–S(H)CH3], as the S–Zn–(ImH)2,3 angles increase the S–H PDE significantly decreases, making them more acidic as ∠SZn(ImH)n increases. Accompanying the reduction in proton dissociation energy, either through CN or geometry, the S–H bond becomes less ionic, the bond lengthens, and the atomic charge on H becomes more positive.
When considering Zn–S(H)R ligation in proteins, the coupling of ligand acidity to conformational changes may play a role the aforementioned activation of proMMPs wherein proton transfer to the coordinated cysteine thiolate appears to be crucial. Additionally, in the quest to develop new drugs/inhibitors for the zinc enzymes, sulfur coordination by zinc-binding-groups contributes strongly to the energetic stability of the inhibitor complex and, as shown in the present study, their protonation state is vital.52 To date, the potential role of active site conformational reorganization in regulating its protonation state via modulation of its pKa and, therefore, its catalytic mechanism has not been discussed for the MMPs. Based on the analysis presented herein, the experimental [(His)3Zn–L] active sites shown in Figure 1 should have different PDEs when L=thiol. Since the mean ∠NZnN is the greatest in the 1SLM structure (the ∠SZnN3 would be the smallest), we predict that it would have the highest PDE (left side of Figure 4 and Figure 5 plots). Likewise, the 1BIW structure would have the lowest PDE (right side of Figure 4 and Figure 5 plots), since the mean ∠NZnN are the smallest and the imidazoles closest together. When a methylthiol is bound to the Zn(II) in the three structures of Figure 1, after removing the inhibitors and performing a constrained optimization at the experimental geometries, we find that the results are completely consistent with the above predictions. The 1BIW structure has a S–H PDE of 722 kJ/mol while 1SLM has a PDE = 765 kJ/mol (M05-2X/cc-pVDZ values for De). Although the experimental structures of MMPs indicate both 4- and 5-coordinate Zn(II) centers having different geometries, our results point toward changes in both the Zn(II) CN and coordination geometry conspiring to modulate the acidity and, therefore, the active-site hydrolytic chemistries.
A recent study has suggested the dynamic nature of the active site zinc geometry may be linked to the activation of homocystein in methionine synthases, an enzyme with a catalytic zinc center comprising His and Cys ligation,53 as well as other enzymes.54,55 If experimental pKa differences within the different zinc enzymes derive from variability in the CN or as a result of the flexibility in the coordination spheres of their catalytic zinc centers, it should be possible to exploit active site pKa differences to gain specificity in their inhibition through judicious choice of ZBGs with the appropriate acidity, as has been suggested.56 Additionally, it may be possible to design inhibition strategies that involve ternary complexes, including co-inhibitors, that modulate the coordination geometry at the catalytic zinc center.
Supplementary Material
Acknowledgments
The authors are grateful to the American Heart Association (0525734Z, D.P.L.; 0550089Z, K.R.R), and to the Oklahoma State Regent for Higher Ed (PRC# 0802032, DPL), and to the NCRR (P20RR15566, KRR) for financial support of this work.
ABBREVIATIONS
- DFT
density functional theory
- MP2
second-order perturbation theory
- MMP
matrix metalloproteinase
- ImH
imidazole
- CN
coordination number
Footnotes
Supporting Information.
Molecular coordinates in xyz format for the optimized geometries in Figure 2 at the M05-2X/cc-pVTZ level. Also, xyz coordinates for optimized [(ImH)3Zn–S(H)CH3]2+ and [(ImH)3Zn–SCH3]+, and figures with molecular parameters, at the M05-2X/cc-pVDZ level. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Coleman JE. Zinc Proteins: Enzymes, Storage Proteins, Transcription Factors, and Replication Proteins. Annu Rev Biochem. 1992;61:897–946. doi: 10.1146/annurev.bi.61.070192.004341. [DOI] [PubMed] [Google Scholar]
- 2.Andreini C, Banci L, Bertini I, Rosato A. Counting the Zinc-Proteins Encoded in the Human Genome. J Proteome Res. 2006;5:196–201. doi: 10.1021/pr050361j. [DOI] [PubMed] [Google Scholar]
- 3.Sousa SF, Lopes AB, Fernandes PA, Ramos MJ. The Zinc Proteome: A Tale of Stability and Functionality. Dalton Trans. 2009:7946–7956. doi: 10.1039/b904404c. [DOI] [PubMed] [Google Scholar]
- 4.Auld DS. The Ins and Outs of Biological Zinc Sites. Biometals. 2009;22:141–148. doi: 10.1007/s10534-008-9184-1. [DOI] [PubMed] [Google Scholar]
- 5.Zastrow ML, Pecoraro VL. Designing Hydrolytic Zinc Metalloenzymes. Biochemistry. 2014;53:957–978. doi: 10.1021/bi4016617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laitaoja M, Valjakka J, Janis J. Zinc Coordination Spheres in Protein Structures. Inorg Chem. 2013;52:10983–10991. doi: 10.1021/ic401072d. [DOI] [PubMed] [Google Scholar]
- 7.Yao S, Flight RM, Rouchka EC, Moseley HNB. A Less-Biased Analysis of Metalloproteins Reveals Novel Zinc Coordination Geometries. Proteins. 2015;83:1470–1487. doi: 10.1002/prot.24834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sternlicht MD, Werb Z. How Matrix Metalloproteinases Regulate Cell Behavior. Annu Rev cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Visse R, Nagase H. Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases: Structure, Function, and Biochemistry. Circ Res. 2003;92:827–839. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- 10.Bonnans C, Chou J, Werb Z. Remodelling the Extracellular Matrix in Development and Disease. Nat Rev Mol Cell Biol. 2014;15:786–801. doi: 10.1038/nrm3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindsey ML. MMP Induction and Inhibition in Myocardial Infarction. Heart Fail Rev. 2004;9:7–19. doi: 10.1023/B:HREV.0000011390.44039.b7. [DOI] [PubMed] [Google Scholar]
- 12.Wang M, Kim SH, Monticone RE, Lakatta EG. Matrix Metalloproteinases Promote Arterial Remodeling in Aging, Hypertension, and Atherosclerosis. Hypertension. 2015;65:698–703. doi: 10.1161/HYPERTENSIONAHA.114.03618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen M, Lee J, Basu R, Sakamuri SSVP, Wang X, Fan D, Kassiri Z. Divergent Roles of Matrix Metalloproteinase 2 in Pathogenesis of Thoracic Aortic Aneurysm. Arterioscler Thromb Vasc Biol. 2015;35:888–898. doi: 10.1161/ATVBAHA.114.305115. [DOI] [PubMed] [Google Scholar]
- 14.Spinale FG. Matrix Metalloproteinases: Regulation and Dysregulation in the Failing Heart. Circ Res. 2002;90:520–530. doi: 10.1161/01.res.0000013290.12884.a3. [DOI] [PubMed] [Google Scholar]
- 15.Kawasaki Y, Xu Z-Z, Wang X, Park JY, Zhuang Z-Y, Tan P-H, Gao Y-J, Roy K, Corfas G, Lo EH, et al. Distinct Roles of Matrix Metalloproteases in the Early- and Late-Phase Development of Neuropathic Pain. Nat Med. 2008;14:331–336. doi: 10.1038/nm1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Overall CM, López-Otín C. Strategies for MMP Inhibition in Cancer: Innovations for the Post-Trial Era. Nat Rev Cancer. 2002;2:657–672. doi: 10.1038/nrc884. [DOI] [PubMed] [Google Scholar]
- 17.Lee M, Fridman R, Mobashery S. Extracellular Proteases as Targets for Treatment of Cancer Metastases. Chem Soc Rev. 2004;33:401–409. doi: 10.1039/b209224g. [DOI] [PubMed] [Google Scholar]
- 18.Cathcart JM, Cao J. MMP Inhibitors: Past, Present and Future. Front Biosci. 2015;20:1164–1178. doi: 10.2741/4365. [DOI] [PubMed] [Google Scholar]
- 19.Coussens LM, Fingleton B, Matrisian LM. Matrix Metalloproteinase Inhibitors and Cancer: Trials and Tribulations. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 20.Tallant C, Garcia-Castellanos R, Baumann U, Gomis-Ruth FX. On the Relevance of the Met-Turn Methionine in Metzincins. J Biol Chem. 2010;285:13951–13957. doi: 10.1074/jbc.M109.083378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Butler GS, Tam EM, Overall CM. The Canonical Methionine 392 of Matrix Metalloproteinase 2 (Gelatinase A) Is Not Required for Catalytic Efficiency or Structural Integrity: Probing The Role of the Methionine-Turn in the Metzincin Metalloprotease Superfamily. J Biol Chem. 2004;279:15615–15620. doi: 10.1074/jbc.M312727200. [DOI] [PubMed] [Google Scholar]
- 22.Pelmenschikov V, Siegbahn PEM. Catalytic Mechanism of Matrix Metalloproteinases: Two-Layered ONIOM Study. Inorg Chem. 2002;41:5659–5666. doi: 10.1021/ic0255656. [DOI] [PubMed] [Google Scholar]
- 23.Whittaker M, Floyd CD, Brown P, Gearing AJH. Design and Therapeutic Application of Matrix Metalloproteinase Inhibitors. Chem Rev. 1999;99:2735–2776. doi: 10.1021/cr9804543. [DOI] [PubMed] [Google Scholar]
- 24.Kotra LP, Cross JB, Shimura Y, Fridman R, Schlegel HB, Mobashery S. Insight into the Complex and Dynamic Process of Activation of Matrix Metalloproteinases. J Am Chem Soc. 2001;123:3108–3113. doi: 10.1021/ja001896a. [DOI] [PubMed] [Google Scholar]
- 25.Rosenblum G, Meroueh S, Toth M, Fisher JF, Fridman R, Mobashery S, Sagi I. Molecular Structures and Dynamics of the Stepwise Activation Mechanism of a Matrix Metalloproteinase Zymogen: Challenging the Cysteine Switch Dogma. J Am Chem Soc. 2007;129:13566–13574. doi: 10.1021/ja073941l. [DOI] [PubMed] [Google Scholar]
- 26.Natchus MG, Cheng M, Wahl CT, Pikul S, Almstead NG, Bradley RS, Taiwo YO, Mieling GE, Dunaway CM, Snider CE, et al. Design and Synthesis of Conformationally-Constrained MMP Inhibitors. Bioorganic & Med Chem Lett. 1998;8:2077–2080. doi: 10.1016/s0960-894x(98)00370-9. [DOI] [PubMed] [Google Scholar]
- 27.Spurlino JC, Smallwood AM, Carlton DD, Banks TM, Vavra KJ, Johnson JS, Cook ER, Falvo J, Wahl RC, Pulvino TA, et al. 1.56 Å Structure of Mature Truncated Human Fibroblast Collagenase. Proteins: Struct Funct Bioinforma. 1994;19:98–109. doi: 10.1002/prot.340190203. [DOI] [PubMed] [Google Scholar]
- 28.Becker JW, Marcy AI, Rokosz LL, Axel MG, Burbaum JJ, Fitzgerald PMD, Cameron PM, Esser CK, Hagmann WK, Hermes JD, et al. Stromelysin-1: Three-Dimensional Structure of the Inhibited Catalytic Domain and of the C-Truncated Proenzyme. Protein Sci. 1995;4:1966–1976. doi: 10.1002/pro.5560041002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sommerhalter M, Lieberman RL, Rosenzweig AC. X-Ray Crystallography and Biological Metal Centers: Is Seeing Believing? Inorg Chem. 2005;44:770–778. doi: 10.1021/ic0485256. [DOI] [PubMed] [Google Scholar]
- 30.Davis AM, Teague SJ, Kleywegt GJ. Application and Limitations of X-ray Crystallographic Data in Structure-Based Ligand and Drug Design. Angew Chem Int Ed. 2003;42:2718–2736. doi: 10.1002/anie.200200539. [DOI] [PubMed] [Google Scholar]
- 31.Frisch Mj, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, et al. Gaussian 09, Revision A. 02. Gaussian Inc.; Wallingford, CT: 2009. [Google Scholar]
- 32.Dennington R, Keith T, Millam J. GaussView, Version 5. Semichem Inc.; Shawnee Mission, KS: 2009. [Google Scholar]
- 33.Zhao Y, Schultz NE, Truhlar DG. Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions. J Chem Theory Comput. 2006;2:364–382. doi: 10.1021/ct0502763. [DOI] [PubMed] [Google Scholar]
- 34.Woon DE, Dunning TH. Gaussian Basis Sets for Use in Correlated Molecular Calculations. III. The Atoms Aluminum through Argon. J Chem Phys. 1993;98:1358–1371. [Google Scholar]
- 35.Dunning TH. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J Chem Phys. 1989;90:1007–1023. [Google Scholar]
- 36.Balabanov NB, Peterson KA. Systematically Convergent Basis Sets for Transition Metals. I. All-Electron Correlation Consistent Basis Sets for the 3d Elements Sc–Zn. J Chem Phys. 2005;123:064107. doi: 10.1063/1.1998907. [DOI] [PubMed] [Google Scholar]
- 37.Bartmess JE, Scott JA, McIver RT. Scale of Acidities in the Gas Phase from Methanol to Phenol. J Am Chem Soc. 1979;101:6046–6056. [Google Scholar]
- 38.Sorkin A, Truhlar DG, Amin EA. Energies, Geometries, and Charge Distributions of Zn Molecules, Clusters, and Biocenters from Coupled Cluster, Density Functional, and Neglect of Diatomic Differential Overlap Models. J Chem Theory Comput. 2009;5:1254–1265. doi: 10.1021/ct900038m. [DOI] [PubMed] [Google Scholar]
- 39.Amin EA, Truhlar DG. Zn Coordination Chemistry: Development of Benchmark Suites for Geometries, Dipole Moments, and Bond Dissociation Energies and Their Use To Test and Validate Density Functionals and Molecular Orbital Theory. J Chem Theory Comput. 2008;4:75–85. doi: 10.1021/ct700205n. [DOI] [PubMed] [Google Scholar]
- 40.Weaver MN, Merz KM, Ma D, Kim HJ, Gagliardi L. Calculation of Heats of Formation for Zn Complexes: Comparison of Density Functional Theory, Second Order Perturbation Theory, Coupled-Cluster and Complete Active Space Methods. J Chem Theory Comput. 2013;9:5277–5285. doi: 10.1021/ct400856g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Addison AW, Rao TN, Reedijk J, van Rijn J, Verschoor GC. Synthesis, Structure, and Spectroscopic Properties of copper(II) Compounds Containing Nitrogen-Sulphur Donor Ligands; the Crystal and Molecular Structure of aqua[1,7-bis(N-Methylbenzimidazol-2′-Yl)-2,6-dithiaheptane]copper(II) Perchlorate. J Chem Soc, Dalton Trans. 1984:1349–1356. [Google Scholar]
- 42.Lin Y, Lim C. Factors Governing the Protonation State of Zn-Bound Histidine in Proteins: A DFT/CDM Study. J Am Chem Soc. 2004;126:2602–2612. doi: 10.1021/ja038827r. [DOI] [PubMed] [Google Scholar]
- 43.Linder DP, Rodgers KR. A Theoretical Study of Imidazole-and Thiol-Based Zinc Binding Groups Relevant to Inhibition of Metzincins. J Phys Chem B. 2004;108:13839–13849. [Google Scholar]
- 44.Rockwell A, Melden M, Copeland RA, Hardman K, Decicco CP, DeGrado WF. Complementarity of Combinatorial Chemistry and Structure-Based Ligand Design: Application to the Discovery of Novel Inhibitors of Matrix Metalloproteinases. J Am Chem Soc. 1996;118:10337–10338. [Google Scholar]
- 45.Parkin G. Synthetic Analogues Relevant to the Structure and Function of Zinc Enzymes. Chem Rev. 2004;104:699–768. doi: 10.1021/cr0206263. [DOI] [PubMed] [Google Scholar]
- 46.Moy FJ, Chanda PK, Chen J, Cosmi S, Edris W, Levin JI, Rush TS, Wilhelm J, Powers R. Impact of Mobility on Structure-Based Drug Design for the MMPs. J Am Chem Soc. 2002;124:12658–12659. doi: 10.1021/ja027391x. [DOI] [PubMed] [Google Scholar]
- 47.Maret W, Li Y. Coordination Dynamics of Zinc in Proteins. Chem Rev. 2009;109:4682–4707. doi: 10.1021/cr800556u. [DOI] [PubMed] [Google Scholar]
- 48.Martin DP, Blachly PG, Marts AR, Woodruff TM, de Oliveira CAF, McCammon JA, Tierney DL, Cohen SM. “Unconventional” Coordination Chemistry by Metal Chelating Fragments in a Metalloprotein Active Site. J Am Chem Soc. 2014;136:5400–5406. doi: 10.1021/ja500616m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koziol L, Valdez CA, Baker SE, Lau EY, Floyd WC, Wong SE, Satcher JH, Lightstone FC, Aines RD. Toward a Small Molecule, Biomimetic Carbonic Anhydrase Model: Theoretical and Experimental Investigations of a Panel of Zinc(II) Aza-Macrocyclic Catalysts. Inorg Chem. 2012;51:6803–6812. doi: 10.1021/ic300526b. [DOI] [PubMed] [Google Scholar]
- 50.He H, Puerta DT, Cohen SM, Rodgers KR. Structural and Spectroscopic Study of Reactions between Chelating Zinc-Binding Groups and Mimics of the Matrix Metalloproteinase and Disintegrin Metalloprotease Catalytic Sites: The Coordination Chemistry of Metalloprotease Inhibition. Inorg Chem. 2005;44:7431–7442. doi: 10.1021/ic050723p. [DOI] [PubMed] [Google Scholar]
- 51.Jacobsen FE, Cohen SM. Using Model Complexes To Augment and Advance Metalloproteinase Inhibitor Design. Inorg Chem. 2004;43:3038–3047. doi: 10.1021/ic035388o. [DOI] [PubMed] [Google Scholar]
- 52.Day JA, Cohen SM. Investigating the Selectivity of Metalloenzyme Inhibitors. J Med Chem. 2013;56:7997–8007. doi: 10.1021/jm401053m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koutmos M, Pejchal R, Bomer TM, Matthews RG, Smith JL, Ludwig ML. Metal Active Site Elasticity Linked to Activation of Homocysteine in Methionine Synthases. Proc Natl Acad Sci United States Am. 2008;105:3286–3291. doi: 10.1073/pnas.0709960105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu R, Hu P, Wang S, Cao Z, Zhang Y. Flexibility of Catalytic Zinc Coordination in Thermolysin and HDAC8: A Born–Oppenheimer Ab Initio QM/MM Molecular Dynamics Study. J Chem Theory Comput. 2010;6:337–343. doi: 10.1021/ct9005322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Arnaud CH. Chemical & Engineering News Archive. American Chemical Society; Apr 27, 2009. Enzymes’ Many Movements; pp. 34–36. [Google Scholar]
- 56.Johnson LL, Pavlovsky AG, Johnson AR, Janowicz JA, Man C-F, Ortwine DF, Purchase CF, White AD, Hupe DJ. A Rationalization of the Acidic pH Dependence for Stromelysin-1 (Matrix Metalloproteinase-3) Catalysis and Inhibition. J Biol Chem. 2000;275:11026–11033. doi: 10.1074/jbc.275.15.11026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.