

Abstract
ApoE on high-density lipoproteins is primarily responsible for lipid transport and cholesterol homeostasis in the central nervous system (CNS). Normally produced mostly by astrocytes, apoE is also produced under neuropathologic conditions by neurons. ApoE on high-density lipoproteins is critical in redistributing cholesterol and phospholipids for membrane repair and remodeling. The 3 main structural isoforms differ in their effectiveness. Unlike apoE2 and apoE3, apoE4 has markedly altered CNS metabolism, is associated with Alzheimer disease and other neurodegenerative disorders, and is expressed at lower levels in brain and cerebrospinal fluid. ApoE4-expressing cultured astrocytes and neurons have reduced cholesterol and phospholipid secretion, decreased lipid-binding capacity, and increased intracellular degradation. Two structural features are responsible for apoE4 dysfunction: domain interaction, in which arginine-61 interacts ionically with glutamic acid-255, and a less stable conformation than apoE3 and apoE2. Blocking domain interaction by gene targeting (replacing arginine-61 with threonine) or by small-molecule structure correctors increases CNS apoE4 levels and lipid-binding capacity and decreases intracellular degradation. Small molecules (drugs) that disrupt domain interaction, so-called structure correctors, could prevent the apoE4-associated neuropathology by blocking the formation of neurotoxic fragments. Understanding how to modulate CNS cholesterol transport and metabolism is providing important insights into CNS health and disease.
Keywords: Alzheimer disease; apolipoproteins E; astrocytes; cholesterol; lipoproteins, HDL; neurodegenerative diseases; receptors, LDL
Lipoprotein and cholesterol metabolism in the brain involves metabolic pathways that are regulated independently of those in the peripheral circulation and tissues. The blood–brain barrier (BBB) restricts plasma lipids, including cholesterol and plasma lipoproteins, from entering or leaving the central nervous system (CNS). In the CNS, cholesterol is synthesized by astrocytes, oligodendrocytes, microglia, and to a lesser extent neurons. CNS cholesterol homeostasis is maintained by 24S-hydroxycholesterol produced by neurons. CNS lipoproteins redistribute cholesterol and lipids to neurons and other brain cells to maintain neuronal plasticity, which requires repair and remodeling of membranes, organelle biogenesis, and synaptogenesis. ApoE is critical in this process through the formation of apoE-enriched CNS lipoproteins that are high-density lipoprotein (HDL) like but are unlike the major apoAI-enriched HDL in plasma.
Cholesterol Synthesis and Metabolism in the CNS
The brain is the most cholesterol-rich organ; with only 2% of body mass, it contains ≈20% of the body’s cholesterol. About 70% is in myelin, and ≈30% is metabolically active and found in membranes of glial cells and neurons, where it undergoes recycling primarily for neuronal repair and remodeling.1,2
Cholesterol in the brain is synthesized from acetate in situ.1–4 There is essentially no cholesterol that enters the brain from the peripheral circulation. During early development, oligodendrocytes synthesize large quantities of cholesterol for myelination. In adults, when myelination is complete, glial cells and, to a lesser extent, neurons account for the steady-state production of cholesterol. As already stated, cholesterol recycling and redistribution involve an apoE-mediated lipoprotein pathway unique to the CNS. Again, the brain uses a unique mechanism whereby cholesterol is turned over, and excess sterols are ultimately delivered to the liver for secretion into the bile. Cholesterol itself does not exit the brain but instead is converted to a metabolite 24S-hydroxycholesterol. 24S-Hydroxycholesterol is much more soluble than cholesterol and is thought to diffuse across the BBB to enter the plasma, where it is picked up by plasma lipoproteins, transported to the liver, metabolized to bile acids, and excreted. 24S-Hydroxycholesterol is generated by 24-hydroxylase, a P450 enzyme3 in the smooth endoplasmic reticulum (ER) in cortical pyramidal cells, cerebellar Purkinje cells, and hippocampal and thalamic neurons. Little is known about why this process occurs in the smooth ER of neurons and how 24S-hydroxycholesterol exits cells and crosses the BBB.
Cerebrospinal Fluid Constituents Reflect the Cellular Milieu for CNS Metabolism
Many constituents of cerebrospinal fluid (CSF) are transferred across the BBB. CSF concentrations of most plasma-derived proteins are <1% of the plasma concentrations (Tables 1 and 2).5 For example, albumin is transferred across specialized epithelial cells in the choroid plexus by one or more transporters, which bind and deliver the protein to the CSF by transcytosis from the blood.6 ApoAI is also transferred from the blood to the CSF at ≈0.3% of plasma levels (Tables 1 and 2).7–9 ApoE in the brain and CSF is produced by CNS cells. Its concentration is 10% to 20% of that in plasma (Tables 1 and 2).7,10,11 Cholesterol in the CSF and CNS is synthesized de novo in the brain. Thus, cholesterol metabolism and lipoprotein transport pathways are unique and specialized, reflecting the critical importance of lipid homeostasis for CNS structure and function.
Table 1.
Human Plasma and CSF Levels
| ApoE, mg/dL | ApoAI, mg/dL | Albumin,g/L | IgG, g/L | |
|---|---|---|---|---|
| Plasma | ≈3–6 | 90–140 | 40.8±6.05 | 9.3±1.85 |
| CSF | 1.07 | 0.337 | 0.30 | 0.0206 |
| 0.9±0.310 | 0.37±0.188 | |||
| ≈0.711 | 0.37±0.089 | |||
| % of plasma level | ≈10–20 | ≈0.3 | ≈0.75 | ≈0.3 |
CSF indicates cerebrospinal fluid.
Table 2.
Human ApoE Levels (mg/dL) by ApoE Phenotype
Lipoprotein Synthesis and Metabolism in the CNS in Humans
Lipoproteins and their metabolism differ in the peripheral circulation and the CNS (Figure 1). ApoB-containing hepatic and intestinal particles not found in the CNS, including very low-density lipoproteins (VLDL), intermediate-density lipoproteins, low-density lipoproteins (LDL), and chylomicrons, are critical for lipid transport and lipid metabolism in plasma. They participate in absorption of dietary lipid (triglycerides), lipid transport to peripheral cells, generation of fatty acids for storage, and lipid metabolism.12
Figure 1.

The upper portion illustrates the major pathways of plasma lipoprotein metabolism in the peripheral circulation, involving chylomicrons synthesized by the intestine and very low-density lipoproteins (VLDL) synthesized by the liver. The origin of high-density lipo-proteins (HDL) and apoAI and the role of HDL in the redistribution of lipids from cells with excess cholesterol and to the liver for excretion (reverse cholesterol transport) are illustrated. The lower portion contrasts lipoprotein pathways in the brain, including the critical role of apoE in the formation of unique apoE HDL-like lipoproteins that redistribute lipids in the central nervous system. Astrocytes are responsible for the largest production of apoE; normal neurons account for ≈20% of apoE production (enhanced production when neurons are injured or stressed). ABCA1 indicates ATP-binding cassette transporter 1; ABCG1, ATP-binding cassette transporter G1; B, apoB; CE, cholesteryl ester; CETP, cholesteryl ester transfer protein; Chylo, chylomicron; E, apoE; LCAT, lecithin–cholesterol acyltransferase; LDLR, LDL receptor family members; LPL, lipoprotein lipase; PL, phospholipid; and UC, unesterified cholesterol.
Plasma HDL contain primarily apoAI; apoAII, apoE, apoAIV, and apoCs are present at lower levels. Generated from the surface of triglyceride-rich lipoproteins during lipolysis and synthesized by hepatocytes and intestinal enterocytes, apoAI forms a pool of HDL discs—lipid-poor pre–β-HDL that initiate formation of mature HDL particles (HDL3 and HDL2), which grow larger and more enriched in cholesterol and cholesteryl esters. Unesterified cholesterol and phospholipids are transferred by the ATP-binding cassette transporter 1 (ABCA1) in peripheral tissues to HDL; unesterified cholesterol is esterified by lecithin–cholesterol acyltransferase (LCAT). Newly secreted apoAI interacts with ABCA1 primarily on hepatocytes and enterocytes (hepatic ABCA1 activity accounts for ≈80% of HDL formation in vivo).13 Plasma HDL participate in reverse cholesterol transport—delivery of excess cholesterol by the cholesteryl ester transfer protein (CETP) to the liver for excretion. Plasma HDL are a reservoir for apoE, which is transferred to chylomicrons and VLDL to mediate their clearance by the LDL receptor. Although apoE-containing HDL (HDL1) constitute ≤ 10% of total HDL, they have not been extensively studied.14 In lower animals, HDL1 can be a major lipoprotein class, especially in cholesterolfed animals. HDL1 are cholesteryl ester enriched and larger than HDL3 or HDL2.15 In cultured macrophages that synthesize and secrete apoE and are exposed to LCAT, apoE facilitates cholesterol incorporation into HDL particles to form cholesteryl ester–rich cores.16,17 Particle size (>20 nm) corresponds to the layers of cholesteryl ester in the core.18
ApoE is the major apolipoprotein regulating CNS lipid metabolism and, along with apoAI, forms HDL-like particles that help to redistribute cholesterol and phospholipids to cells for repair and remodeling (triglyceride is scarce or absent in the CNS; Figure 1). ApoAI is transferred from plasma to the CNS and is not synthesized in the brain, but apoE is synthesized in the CNS.19 CNS lipoproteins differ from plasma HDL. In the CNS, apoE delivers cholesterol and other lipids to cells through interactions with ≥1 members of the LDL receptor family. Nevertheless, enzymes, transporters, and receptors similar to those in the periphery are used in CNS lipoprotein formation and metabolism, including LCAT, CETP, ABCA1, ABCG1, ABCG4, and the LDL receptor and family members.
CNS ApoE: Sites of Synthesis, Secretion, and Regulation
ApoE structure and function have been studied extensively in the periphery and in the brain, and several roles have been defined.20–22 The brain is the second most common site of apoE production (liver accounts for 75%)23; various CNS cells are involved, particularly astrocytes, including specialized astrocytes (Bergmann glia, tanycytes, pituicytes, and retinal Müller cells).24 Primary cultures of mouse astrocytes pulsed with [35S]methionine secrete apoE-containing HDL-like particles.25
Brain apoE expression was mapped in mice by inserting enhanced green fluorescent protein (EGFP) controlled by the endogenous promoter into 1 apoE allele (EGFPapoE mice); the other apoE allele maintains normal cellular physiology.26 Hepatocytes and peripheral macrophages expressed high levels of EGFP, indicating apoE production. About three fourths of astrocytes had high EGFP levels. Astrocyte activation with kainic acid enhanced EGFP expression. In microglia, kainic acid elicited EGFP expression in ≈6% of cells. Microglia in specific brain regions may be more or less active. For example, apoE is produced by microglia in the brains of patients with Alzheimer disease (AD)27 and in the olfactory bulb of lesioned mice.28 Cultured microglia also produce apoE.29
ApoE is produced by injured or stressed neurons.26 In EGFPapoE mice treated with kainic acid, injured neurons produced high levels of apoE as confirmed by EGFP expression and in situ hybridization of apoE mRNA in the hippocampus. Under normal physiological conditions, conditional deletion of Apoe, specifically from neurons in apoE targeted replacement (knockin) mice, decreased cortical and hippocampal apoE levels by 20%.30 Thus, neurons likely synthesize and secrete as much as 20% of CNS apoE.
Furthermore, normally neurons are uniquely poised to rapidly produce apoE. Neurons contain a splicing variant of apoE mRNA not found in astrocytes or hepatocytes.31 In wild-type and apoE targeted replacement mice, cortical and hippocampal neurons retain intron-3 in apoE mRNA and produce low levels of mature apoE mRNA for apoE synthesis under normal conditions. After kainic acid treatment, which causes neurodegeneration, apoE mRNA with intron-3 was markedly decreased in injured hippo-campal neurons, and mature apoE mRNA (lacking the intron) and protein were increased. The neuron-specific regulation of apoE expression demonstrates the critical role of apoE production, presumably for redistribution of cholesterol for cellular repair and maintenance and possibly as a transcription factor.32
Neuronal apoE production is regulated at least in part by an astrocyte-secreted factor or factors through the extracellular signal–regulated kinase pathway. In neuronal cells expressing human apoE3 or apoE4, astrocyte-conditioned medium increases apoE expression 4- to 10-fold.33 Stress- and injury-induced upregulation of neuronal apoE production could be partly mediated by astro-cyte activation (astrocytosis) after acute injury, including traumatic brain injury, oxidative stress, and amyloid-β (Aβ) accumulation. In EGFPapoE mice, smooth muscle cells surrounding large blood vessels in the CNS and cells of the choroid plexus also produce apoE26 as do cultured arterial smooth muscle cells.34
ApoAI in the CNS
ApoAI is not produced in the CNS but is transferred from the peripheral circulation to the CSF to help redistribute cholesterol in the brain. Intravenously injected recombinant, fluorescently labeled human apoAI rapidly appears in CSF.35 ApoAI is primarily transferred in the choroid plexus. Cultured primary human choroid plexus epithelial cells bind, internalize, and transfer apoAI across the cells. Although the choroid plexus contains numerous specific transporters, the apoAI transporter is not known.35
Human CSF Lipoproteins
The major apolipoproteins in the CSF are apoE and apoAI, with lesser amounts of apoAII, apoCs, apoJ, and apoD. CSF lipoproteins float by ultracentrifugation at d=1.063 to 1.21 g/mL with the majority at d=1.09 to 1.15 g/mL. They tend to be small and spherical (≈10–20 nm), like plasma HDL, but are unique HDL-like particles. CSF lipoproteins fractionated by density gradient ultracentrifugation include both apoE and apoAI particles.36 The particles primarily contain phospholipid and cholesterol (≈35% unesterified versus 25% in plasma HDL). Besides phosphatidylcholine (the major phospholipid), the CSF lipoproteins contain more phosphatidylethanolamine. The reason why is unclear. ApoE is sialylated more in CSF than in plasma. ApoE possesses a single carbohydrate attachment site at threonine-194.37 CSF lipoproteins bound avidly to fibroblast LDL receptors (high-affinity binding, >20-fold higher than LDL for the receptors).36
Subfractionation of CSF lipoproteins by apoE and apoAI immunoaffinity columns revealed 2 distinct particles.36 One contained predominantly apoE and was largely spherical (15.4±3 nm) with a few disc-like particles. The other contained apoAI and was spherical and smaller (12.9±2.1 nm). In another study,9 CSF lipoproteins separated by gel filtration had an elution profile equivalent to plasma HDL. Immunoaffinity chromatography with anti-apoE then anti-apoAI resolved 4 lipoproteins: CSF-Lp apoE, CSF-Lp apoAI, CSF-Lp apoE/AI, and Lp (with neither apoE nor apoAI). All particles contained apoD and apoJ. Lp apoE, Lp apoAI, and Lp apoE/AI had similar lipid composition (≈60% protein, 20% to 30% phospholipid, and 14% to 17% cholesterol). Lp apoE particles were larger (18–20 nm) than Lp apoE/AI particles (13–20 nm) and more lipid rich (lipid:protein, 0.76:1). Lp apoAI particles were smaller (13–18 nm).
Isoform-Specific Effects on Human CSF ApoE Levels
Plasma levels of apoE vary with apoE genotype (apoE2>apoE3>apoE4; Tables 1 and 2).38,39 In ≈700 healthy controls, a sensitive ELISA for apoE40 showed a strong gradient (apoE2/2 highest; apoE4/4 lowest). Total apoE was less abundant in apoE4 carriers than in noncarriers (5.6±2.4 versus l7.4±3.1 mg/dL; P<0.001). This gradient was also seen in cognitively impaired subjects.
In >400 cognitive normal subjects, mean plasma apoE was ≈5.7 mg/dL and total CSF apoE was 0.7 mg/dL (range, ≈0.6–0.9 mg/dL; highest in apoE2 and lowest in apoE4 carriers; Tables 1 and 2). In plasma, this gradient reflects decreased LDL receptor binding of apoE2 (and thus higher plasma levels) and the preference of apoE4 for VLDL (accelerates hepatic clearance and results in lower levels). The higher brain levels of apoE2 could also reflect impaired receptor binding. However, the lower apoE4 levels likely reflect increased degradation caused by its unique structural features. ApoE4 preferentially forms a molten globule intermediate that makes it less stable than apoE3,41 and its amino terminus interacts with the carboxyl terminus through an ionic interaction between arginine-61 (Arg-61) and glutamic acid-255 (Figure 2A).42 Domain interaction causes the preferential binding of apoE4 to VLDL in plasma and increases hepatic clearance. In the brain, low apoE4 levels are influenced by the domain interaction, which may be caused by enhanced astrocyte degradation.43 The lower apoE4 levels and thus lower levels of cholesterol transport could affect cholesterol homeostasis and neuronal plasticity.
Figure 2.
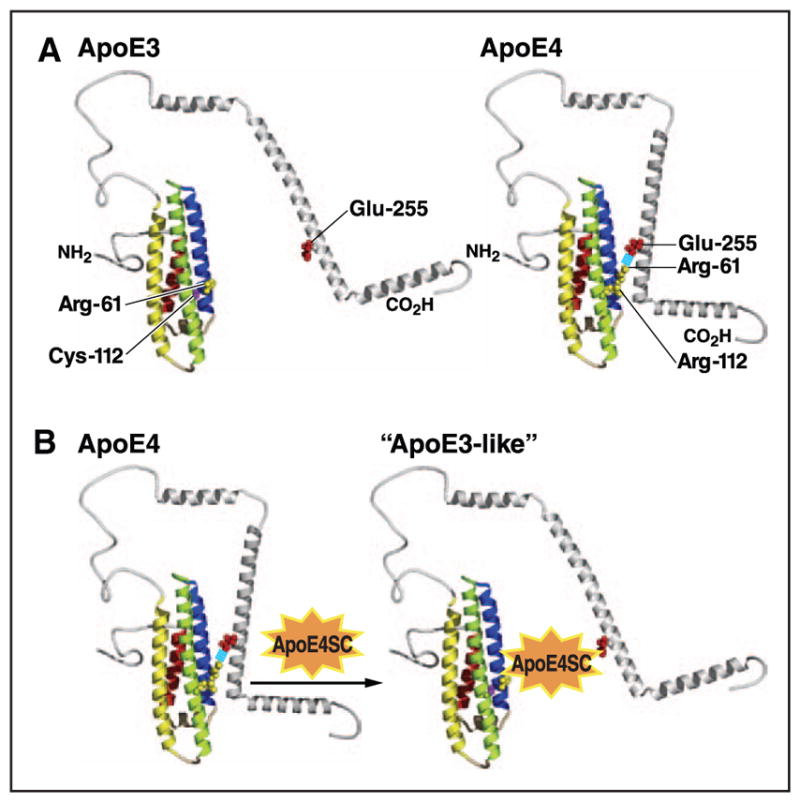
Models of apoE3 and apoE4 structure. A, ApoE4 displays a unique property, referred to as domain interaction, that involves an ionic interaction between arginine (Arg)-61 and glutamic acid (Glu)-255. ApoE3 undergoes domain interaction to a lesser degree. Domain interaction is associated with many altered functional properties that distinguish apoE4 from apoE3. B, Domain interaction can be blocked by small molecules that prevent the ionic interaction and convert apoE4 to an apoE3-like molecule, both structurally and functionally. ApoE4SC indicates apoE4 structure corrector; Cys, cysteine; and Glu, glutamic acid. Modified from Mahley and Huang59 with permission of the publisher. Copyright ©2012, the American Chemical Society.
Mouse Models of Human ApoE Metabolism in the Brain
CSF ApoE Levels in Mouse Models
In an extensive, well-controlled study, plasma and CSF levels were examined in apoE2/2, apoE3/3, and apoE4/4 targeted replacement mice.44 To eliminate spurious results from masked apoE epitopes, multiple methods were used to detect and quantitate apoE, including detergent extraction of plasma, CSF, and brain tissue. Despite similar mRNA levels, plasma levels of apoE2 were the highest and apoE4 were the lowest. CSF apoE levels in the mice had the same gradient as seen in human CSF (apoE2>apoE3>apoE4; Tables 3 and 4). An additional study of targeted replacement mice confirmed these findings, showing a 3- to 4-fold difference between apoE2 and apoE4 levels in CSF,45 which was demonstrated by microdialysis probes in the hippocampus of targeted replacement mice.46 The gradient of apoE levels also occurs in frontal cortex and hippocampus44 (Tables 3 and 4).
Table 3.
ApoE Levels in the CSF and Interstitial Fluid in Target Replacement Mice by ApoE Genotype
| CSF, mg/dL | Interstitial Fluid,mg/dL | ||
|---|---|---|---|
| Genotype | Riddell et al44 | Fryer et al45 | Ulrich et al46 |
| Human apoE2/2 | 0.35±0.06 | 0.55±0.08 | 47.7±15.6 |
| Human apoE3/3 | 0.15±0.03 | 0.20±0.03 | 18.7±5.3 |
| Human apoE4/4 | 0.11±0.03 | 0.13±0.03 | 12.6±1.8 |
CSF indicates cerebrospinal fluid.
Table 4.
ApoE Levels in the Brain in Target Replacement Mice by ApoE Genotype
| Genotype | Frontal Cortex, ng/mg44 | Hippocampus, ng/mg44 |
|---|---|---|
| Human apoE2/2 | ≈120* | ≈180† |
| Human apoE3/3 | ≈90 | ≈160 |
| Human apoE4/4 | ≈75 | ≈150 |
33% higher than apoE3 and 40% higher than apoE4.
12.5% higher than apoE3 and 20% higher than apoE4.
Some studies reported similar apoE levels in apoE4 and apoE3 targeted replacement mice47 and higher apoE4 levels in apoE3/4 humans.48,49 These discrepancies could reflect masked epitopes from variable lipidation and poor extraction from brain samples. Detergent extraction is essential, and data from immunoassays and Western blotting must be analyzed. Moreover, apoE aggregates readily.
Mechanisms for the lower apoE4 levels were studied in cultured primary astrocytes from targeted replacement mice.44 ApoE4/4 mouse astrocytes secreted 28% less apoE and ≈50% less cholesterol than apoE3/3 astrocytes. Cellular retention of apoE4 was not different; however, in pulse-chase studies, [35S]apoE4 disappeared more rapidly than apoE3 (half-life, 49 versus 96 minutes; enhanced degradation of apoE4 will be discussed in greater detail later).
ApoE Structure Modulates ApoE Levels and Metabolism
Mouse apoE has arginine at the site equivalent to residue 112 in human apoE4 but has threonine at the site equivalent to residue 61 and thus lacks domain interaction.42 The same is true for all animals except humans.50 Arg-61 mouse apoE generated by gene targeting displays domain interaction similar to human apoE4 and has lower plasma levels than wild-type mouse apoE,51 and Arg-61 mice have pathological characteristics and behavioral impairments similar to those of mice expressing human apoE4.52
In the brain, Arg-61 mouse apoE behaves like human apoE4. Despite similar mRNA levels, Arg-61 mouse apoE levels are 43% lower overall than wild-type mouse apoE, which behaves more like apoE3,43 and 33% to 47% lower in cortex, hippocampus, and cerebellum, regardless of age or sex. In targeted replacement mice, apoE4 levels are 28% lower than apoE3 levels43 and 26% to 37% lower in cortex, hippocampus, and cerebellum. In primary cultures of astro-cytes,43 secretion of Arg-61 mouse apoE is 46% lower than wild-type mouse apoE, and apoE3/4 cells secrete 25% less apoE4 than apoE3. Thus, domain interaction that occurs in the context of Arg-112 and Arg-61 is the critical structural feature of apoE contributing to the lower apoE levels in the CNS as displayed by the expression of human apoE4 or induced by replacing the normally occurring threonine with arginine at the site equivalent to residue 61 in mouse apoE. Wild-type mouse apoE does not faithfully mimic either human apoE4 or apoE3.
ApoE4 Structure Targets It for Degradation and Decreased Lipid Transport
Domain interaction leads to intracellular degradation and altered lipid-binding capacity. Arg-61 mouse apoE (surrogate for apoE4) elicits an ER stress response in primary cultured astrocytes from the Arg-61 mice, but wild-type mouse apoE (surrogate for apoE3) does not.53 Arg-61 mouse apoE was targeted for degradation, and markers of the unfolded protein response in all 3 unfolded protein response pathways were upregulated. ApoE4 does not activate the ER stress pathway in neurons.
As discussed, neurons are poised for apoE synthesis31 but synthesize less apoE until they are injured or stressed26 by aging, neurotoxins, ischemia, oxidative stress, and likely other environmental and genetic factors that necessitate redistribution of cholesterol and other lipids for repair and remodeling. However, because of domain interaction, apoE4 is recognized by a neuron-specific protease that degrades it to a greater extent than apoE3,54–57 generating neurotoxic carboxyl-terminal fragments of ≈12 to 29 kDa. These fragments escape the secretory pathway, stimulate tau-phosphorylation, and cause mitochondrial dysfunction. The fragmentation is blocked by site-directed mutagenesis (Arg-61 to threonine) or small-molecule structure correctors that prevent domain interaction58,59 (Figure 2B).
ApoE4 synthesized by astrocytes is associated with lipoprotein particles with various degrees of lipidation.25,60–62 Isoform-specific differences in phospholipid and cholesterol secretion from primary cultured astrocytes from targeted replacement mice expressing human apoE3 or apoE4 have been characterized in detail. Similar levels of apoE3 and apoE4 were released, and the particles were of similar size, but apoE3 was associated with a 2- to 3-fold greater release of phospholipid and cholesterol. Thus, apoE3 delivers more cholesterol and phospholipids to cells. Likewise, because of the domain interaction, neuronal cells transfected with apoE4 release ≈25% less phospholipid than cells expressing apoE3.63 When apoE4 domain interaction was disrupted by insertion of threonine at residue 61, phospholipid release from apoE4–threonine-61– and apoE3-expressing cells was the same and greater than from apoE4-expressing cells.
ApoE isoforms differ in their lipid binding and lipoprotein preferences.22,42,64 ApoE3 binds to small HDL, whereas apoE4 binds to large VLDL—at least partially because of domain interaction, which changes the exposure or orientation of the lipid-binding region. When domain interaction is blocked, the lipoprotein-binding preference of apoE4 resembles that of apoE3.
In sum, apoE4 is more susceptible to intracellular degradation in astrocytes and neurons, is present at lower levels in the CNS and CSF, and delivers cholesterol and phospholipid to neurons less efficiently (Figure 3). These alterations are associated with neuropathology, including AD, traumatic brain injury, stroke, and other apoE4-associated neurological disorders.
Figure 3.

ApoE4 is a major therapeutic target to prevent or slow Alzheimer disease and several other neurodegenerative disorders. ApoE4 is synthesized primarily by astrocytes but to a lesser extent by neurons (injury to neurons stimulates apoE production). I, ApoE4 synthesis by neurons triggers neuron-specific proteolysis, generating a series of neurotoxic fragments that escape the secretory pathway and stimulate tau-phosphorylation and mitochondrial dysfunction. The unique structural feature of apoE4—domain interaction—is responsible for stimulating the proteolysis, and blocking apoE4 domain interaction with apoE4 structure correctors prevents the formation and the neurotoxic effects of the fragments and protects against neurodegeneration and impaired cognition. II, The accelerated degradation of apoE4 by astrocytes (and to a lesser extent the increased proteolysis of apoE4 in neurons) results in decreased apoE4 levels in the cerebrospinal fluid (CSF) and central nervous system (CNS). [It has been postulated that this contributes to decreased amyloid-β (Aβ) clearance.] In addition to lower levels of apoE4, apoE4 has a decreased lipid-binding capacity. ApoE4 domain interaction is also associated with decreased apoE4 levels and decreased affinity for lipids. ApoE4 structure correctors can restore the apoE4 levels and enhance lipid binding. ER indicates endoplasmic reticulum.
Role of ABCA1 in HDL Formation
ApoAI, the major apolipoprotein in plasma HDL metabolism, participates in a cholesterol redistribution process called reverse cholesterol transport (Figure 1). Lipidation of apoAI in peripheral tissues, including hepatocytes and enterocytes, involves ABCA1-mediated transport of cholesterol and phospholipid from cell membranes (Figure 1).13 ABCA1 is found in a wide range of cells in the body, including astrocytes and neurons in the brain.
In the CNS, ABCA1 participates in cholesterol efflux to apoE to form HDL-like apoE-containing particles enriched in phospholipid and unesterified cholesterol.65 In apoE-producing macrophages, apoE interacts with ABCA1 to form unesterified cholesterol–rich HDL; apoAI, if present, enhances the efficiency of this process.66 ABCA1 is important in mouse CNS.67,68 ABCA1 deletion reduces apoE levels in plasma, brain, and CSF by >80% and is associated with the formation of small apoE-containing lipoproteins. In addition, cultured astrocytes lacking ABCA1 form only small, cholesterol-poor apoE-containing lipoproteins. Thus, ABCA1 in mice is critical for apoE lipidation and HDL formation.
The importance of ABCA1 in human CNS lipid metabolism is not completely clear. In Tangier disease, mutations in both alleles and the absence of ABCA1 activity reduce plasma HDL cholesterol and apoAI by 40% to 50% and decrease apoAI HDL lipidation69,70; plasma apoE levels are unchanged or increased.71 However, Tangier disease patients do not have CNS problems (eg, no increased risk of dementia). A study of patients with 10 single-nucleotide polymorphisms in ABCA1, including those associated with altered plasma HDL levels, failed to show an association with CNS apoE levels.10 If ABCA1 activity was rate limiting for apoE–HDL lipidation, one would expect decreased apoE–HDL levels as observed in the mice.
Alternatively, other transporters could assume the role of ABCA1 in the CNS. For example, ABCA7, a relative of ABCA1, serves as a phospholipid transporter that seems to mediate cholesterol removal.72 Importantly, ABCA7 is highly expressed in the brain, and in a genome-wide association study, it was shown to be a risk factor for AD.73,74 As will be discussed, ABCG1 and ABCG4 could also compensate for a deficiency of ABCA1 activity and mediate apoE–HDL lipidation.
Role of ABCG1 in Peripheral Tissues and the CNS
In cultured cells, ABCA1 and ABCG1 lipidate nascent HDL, possibly with ABCA1 initiating cholesterol and phospholipid transfer to apoAI or apoE to form discs, which are further lipidated by ABCG1. However, neither deletion nor overex-pression of ABCG1 in mice affects plasma lipid levels significantly.75 Interestingly, ABCG1 and its relative ABCG4 may function more importantly in the CNS than the periphery. Both are widely expressed in neurons and astrocytes (ABCG4 primarily expressed in the brain).76 These transporters display overlapping roles in sterol efflux from cells to HDL. Deletion of either ABCG1 or ABCG4 did not affect CNS sterol levels, whereas deletion of both transporters resulted in a 2- to 3-fold increase of sterols, including desmosterol, lanosterol, lathosterol, and 27-OH cholesterol.76 Thus, the variety of lipid transporters displaying both unique and overlapping roles in efflux and transport within the CNS further illustrates the critical role of lipid homeostasis occurring in situ and restricted by the BBB.
Role of LDL Receptor Family Members in ApoE Delivery of Cholesterol to CNS Cells
The LDL receptor and the LDL receptor–related protein 1 are primarily responsible for lipoprotein binding and cholesterol delivery by apoE to neurons and glia.77,78 The VLDL receptor and apoE receptor 2 participate in reelin modulation of neuronal development of cerebral cortex and in reelin-mediated intracellular signaling but not cholesterol transport.79,80 LDL receptors are most highly expressed in glia and to a lesser extent in neurons. LDL receptor–related protein 1 receptors are more highly expressed in neurons.81 Heparan sulfate proteoglycans on neuronal and glial cell membranes may facilitate binding to the LDL receptor–related protein 1 or to sequester apoE molecules to enrich the lipoproteins for receptor-mediated uptake as in the liver.82 In mice, brain and CSF apoE levels are increased by LDL receptor deletion45 and decreased by LDL receptor overexpression.83 Likewise, conditional deletion of LDL receptor–related protein 1 in forebrain neurons increases apoE levels.84
Role of LCAT in Cholesteryl Ester Formation in the CNS
LCAT transfers a fatty acid from phospholipid to unesterified cholesterol to form cholesteryl esters. ApoAI activates LCAT to a greater extent than apoE.85 In an artificial recombinant system lacking apoAI, apoE4 was less efficient in activating LCAT to form cholesteryl esters and in converting discoidal particles to spherical apoE-containing lipoproteins.86 The unique structure and conformation assumed by apoE4 on these apoE discs and small particles may alter the access of LCAT to the phospholipid acyl chains. Less formation of cholesteryl esters in apoE4-containing lipoproteins could impair maturation of CNS HDL, limiting the availability of cholesterol and other lipids for cells. Core formation is essential for HDL to optimally acquire cholesterol from cells and to increase its cholesterol-carrying capacity.
Role of CETP in the CNS
In plasma, CETP mediates cholesteryl ester transfer from HDL to lower density lipoproteins during reverse cholesterol transport. In human CSF, cholesteryl ester transfer activity is present at a concentration of ≈12% that in plasma, indicating that it is made in the brain. CETP activity is present in culture medium from neuroblastoma cells, astrocytes, and choroid plexus.87 Its role in the CNS is undefined. There are no lower density lipoproteins (eg, LDL) to serve as choles-teryl ester acceptors and no triglyceride-rich lipoproteins (eg, VLDL) for exchange of triglyceride for cholesteryl esters. In a highly artificial system of apoE-recombinant lipid discs, CETP caused apoE-containing lipoproteins to fuse, forming large particles.86 Thus, CETP may facilitate cholesteryl ester transfer among the CNS lipoproteins, resulting in fusion of apoE- and apoAI-containing particles, or its function could be unrelated to CNS lipoprotein metabolism.
Other Apolipoproteins in the Brain and CSF
ApoJ
ApoJ (clusterin) is a 75- to 80-kDa glycoprotein in the plasma HDL fraction and in several tissues, including brain. Normally produced by astrocytes,62 apoJ is markedly upregulated after stress or injury and produced by both astrocytes and neurons. In traumatic brain injury, this upregulation helps protect against oxidative stress and neuroinflammation.88,89 However, apoJ seems to play multiple roles in complement activation, oxidative stress, apoptosis, and cancer.65,90 It also serves as a chaperone in protein unfolding pathways and disposal of misfolded proteins.91 Much more needs to be learned about apoJ’s role in cholesterol transport and lipid delivery to CNS cells.
ApoD
ApoD, a 29-kDa protein that is transported on plasma HDL, has little homology with other apolipoproteins involved in lipid transport and is a member of the lipocalin family. It is produced in the brain and is associated with CSF HDL. ApoD is produced by astrocytes and oligodendrocytes. Its importance as a cholesterol transporter is unknown.65,88 ApoD has been shown to be cardioprotective against experimentally induced myocardial infarction in mice and can protect cultured cardiomyocytes from hypoxia by inhibiting oxidation.92 ApoD might play such a role in the brain where neurodegeneration is often accompanied by oxidative stress.
ApoE-Associated Neuropathology
AD, the leading cause of dementia, is increasing in prevalence secondary to aging populations worldwide.93,94 No effective therapy to prevent or reverse the cognitive impairment exists. During the past 25 years, the amyloid hypothesis has been the main AD research focus77,78,95–97; however, several clinical trials using a variety of drugs to decrease amyloid plaques and the formation of Aβ peptides have failed. Undoubtedly, multiple factors involving several pathways must be considered in AD pathogenesis. This includes targeting apoE4 as a therapeutic approach.21,58,59,93,94
ApoE4 is the major genetic risk factor for AD98,99 (65% to 80% of all patients with AD have at least 1 apoE4 allele). ApoE4 increases the risk many fold and decreases the age of onset by 8 years for each apoE4 allele. It also plays a key role in poor clinical outcome after traumatic brain injury and stroke and in severity or progression in frontotemporal dementia, Lewy body disease, and other neurological disorders.21,58,59,93,94 ApoE4 is associated with deposition of amyloid; however, it remains to be proved that amyloid or Aβ is causative.77,78 Clearly, amyloid pathology does not explain the apoE4-associated neuropathology seen in the neurological disorders lacking amyloid pathology.
ApoE is envisioned to affect 2 major pathways affecting neurodegeneration: (1) a direct effect of apoE4 expression in neurons on the generation of neurotoxic fragments and (2) an indirect effect with lower levels of apoE4 and decreased lipid transport in the CNS associated with apoE4 (Figure 3). The unique structure of apoE4 (ie, domain interaction) drives these outcomes.58,93
Our working hypothesis is that apoE4 sets the stage for various second hits (eg, aging, oxidative stress, excitotoxicity, ischemia, traumatic brain injury, and increased Aβ) that injure or stress neurons. Injured neurons synthesize apoE (Figure 3).58,59,93 However, apoE4 undergoes enhanced neuron-specific proteolysis, which generates neurotoxic fragments (≈12–29 kDa) that enter the cytosol, causing tau-hyperphosphorylation54,56,57,100 and mitochondrial dysfunction101,102 and ultimately neurodegeneration. Fragments containing the receptor-binding region (residues 136–150) and the lipid-binding region (residues 240–270) are the minimal structure of apoE responsible for neurotoxicity. The receptor-binding region serves as a protein translocation domain, and the lipid-binding region mediates fragment interaction with mitochondria.103 The initial proteolytic cleavage results in the formation of a 29-kDa fragment lacking the carboxyl-terminal 27 to 30 amino acids; subsequent cleavage removes various regions of the amino terminus.54 Blocking apoE4 domain interaction by site-directed mutagenesis (Arg-61 to threonine) or by small molecules (apoE structure correctors) prevents the ionic interaction of Arg-61 and glutamic acid-255, the generation of neurotoxic fragments, and the detrimental effects of apoE4 in cultured neurons and in apoE4 transgenic and targeted replacement mouse models.58,59,101,102 Preclinical studies to develop apoE structure correctors as a therapeutic approach to prevent apoE4-associated neuropathology are in progress. Furthermore, inhibiting the neuronal protease would be an additional approach to preventing the generation of the apoE4 neurotoxic fragments.
An alternative therapeutic approach targets the lower levels and impaired lipidation of apoE4 in the CNS (Figure 3). ApoE4 is degraded more effectively than apoE3/2 in both astrocytes (ER stress activation)53 and neurons (neuron-specific proteolysis).54–57 The result is lower levels of apoE4. ApoE4 also displays a lower lipid-binding capacity and lower affinity for lipids.63 Thus, apoE4 is associated with decreased lipid transport in the CNS and possible inefficient neuronal repair and remodeling. Importantly, both of these processes—apoE levels and lipidation—are controlled by apoE4 domain interaction. ApoE4 conformation and its stability are responsible for the differences in lipid and lipoprotein binding.104–106 Blocking apoE4 domain interaction prevents neuronal and astrocytic degradation and allows enhanced lipidation to occur.
Modulation of this pathway includes stimulating apoE expression with bexarotene, a retinoid x receptor agonist. An initial study with bexarotene decreased amyloid plaques and improved cognitive function in an AD mouse model,107 but this has not been confirmed in other studies.108–112 Such agonists alter the expression not only of apoE but also of ABCA1 and numerous other proteins. It remains to be determined whether increasing apoE4 is beneficial, or more likely, detrimental. Other groups are considering approaches to enhance apoE4 lipidation by modulating ABC transporter activity. At present, there is no way to selectively enhance the activity of one or more of the lipid transporters in the CNS. The rationale behind enhancing apoE4 lipidation is the possibility that apoE4 HDL may have an enhanced ability to clear Aβ in the CNS; however, this remains to be proven.
Clearly, elucidation of apoE structure and function has led to understanding isoform-specific effects in normal and pathological processes. Unraveling CNS lipid and lipoprotein metabolism has contributed to the development of critically important therapeutic approaches in treating/preventing apoE4-associated neuropathology.
Highlights.
Central nervous system lipoproteins and their metabolism are unique, reflecting the isolation of the brain from the peripheral circulation by the blood–brain barrier.
ApoE is synthesized in the brain, forms high-density lipoprotein–like particles, and is responsible for the transport and redistribution of cholesterol to neurons and other cells for repair and remodeling.
ApoE has isoform-specific effects on cholesterol homeostasis under normal and neuropathological conditions. ApoE4 is less abundant, has less affinity for high-density lipoprotein–like particles, and transports less cholesterol than apoE3 or apoE2.
Unique structural features of apoE4, including domain interaction (ionic interaction between arginine-61 and glutamic acid-255) and conformational instability, are responsible for these effects.
Domain interaction accelerates degradation in astrocytes under normal conditions, and in injured neurons it is associated with neuropathology from neurotoxic fragments of apoE4. Small-molecule structure correctors that block apoE4 domain interaction convert apoE4 to apoE3/2-like molecules and block the detrimental effects of apoE4, including the generation of neurotoxic fragments.
Acknowledgments
We thank Sylvia Richmond and Charlotte Pfeiffer for article preparation, Stephen Ordway and Gary Howard for editorial assistance, and John C.W. Carroll for graphics.
Sources of Funding
This work was supported, in part, by the award (number 101786) from The Wellcome Trust Seeding Drug Discovery.
Nonstandard Abbreviations and Acronyms
- Aβ
amyloid-β
- ABCA1
ATP-binding cassette transporter 1
- ABCG1
ATP-binding cassette transporter G1
- AD
Alzheimer disease
- Arg-61
arginine-61
- BBB
blood–brain barrier
- CETP
cholesteryl ester transfer protein
- CNS
central nervous system
- CSF
cerebrospinal fluid
- EGFP
enhanced green fluorescent protein
- ER
endoplasmic reticulum
- HDL
high-density lipoproteins
- LCAT
lecithin–cholesterol acyltransferase
- LDL
low-density lipoproteins
- VLDL
very low-density lipoproteins
Footnotes
Disclosures
Dr Mahley is a shareholder and serves on the scientific advisory board and as a consultant for E-Scape Bio, Inc.
References
- 1.Dietschy JM, Turley SD. Cholesterol metabolism in the brain. Curr Opin Lipidol. 2001;12:105–112. doi: 10.1097/00041433-200104000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Dietschy JM, Turley SD. Thematic review series: brain lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res. 2004;45:1375–1397. doi: 10.1194/jlr.R400004-JLR200. [DOI] [PubMed] [Google Scholar]
- 3.Russell DW, Halford RW, Ramirez DM, Shah R, Kotti T. Cholesterol 24-hydroxylase: an enzyme of cholesterol turnover in the brain. Annu Rev Biochem. 2009;78:1017–1040. doi: 10.1146/annurev.biochem.78.072407.103859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vance JE, Hayashi H. Formation and function of apolipoprotein E-containing lipoproteins in the nervous system. Biochim Biophys Acta. 2010;1801:806–818. doi: 10.1016/j.bbalip.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 5.Ganrot K, Laurell CB. Measurement of IgG and albumin content of cere-brospinal fluid, and its interpretation. Clin Chem. 1974;20:571–573. [PubMed] [Google Scholar]
- 6.Liddelow SA, Dzie˛gielewska KM, Møllgård K, Whish SC, Noor NM, Wheaton BJ, Gehwolf R, Wagner A, Traweger A, Bauer H, Bauer HC, Saunders NR. Cellular specificity of the blood-CSF barrier for albumin transfer across the choroid plexus epithelium. PLoS One. 2014;9:e106592. doi: 10.1371/journal.pone.0106592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roher AE, Maarouf CL, Sue LI, Hu Y, Wilson J, Beach TG. Proteomics-derived cerebrospinal fluid markers of autopsy-confirmed Alzheimer’s disease. Biomarkers. 2009;14:493–501. doi: 10.3109/13547500903108423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song H, Saito K, Seishima M, Noma A, Urakami K, Nakashima K. Cerebrospinal fluid apo E and apo A-I concentrations in early- and late-onset Alzheimer’s disease. Neurosci Lett. 1997;231:175–178. doi: 10.1016/s0304-3940(97)00558-2. [DOI] [PubMed] [Google Scholar]
- 9.Koch S, Donarski N, Goetze K, Kreckel M, Stuerenburg HJ, Buhmann C, Beisiegel U. Characterization of four lipoprotein classes in human cere-brospinal fluid. J Lipid Res. 2001;42:1143–1151. [PubMed] [Google Scholar]
- 10.Wahrle SE, Shah AR, Fagan AM, Smemo S, Kauwe JS, Grupe A, Hinrichs A, Mayo K, Jiang H, Thal LJ, Goate AM, Holtzman DM. Apolipoprotein E levels in cerebrospinal fluid and the effects of ABCA1 polymorphisms. Mol Neurodegener. 2007;2:7. doi: 10.1186/1750-1326-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cruchaga C, Kauwe JS, Nowotny P, Bales K, Pickering EH, Mayo K, Bertelsen S, Hinrichs A, Fagan AM, Holtzman DM, Morris JC, Goate AM Alzheimer’s Disease Neuroimaging Initiative. Cerebrospinal fluid APOE levels: an endophenotype for genetic studies for Alzheimer’s disease. Hum Mol Genet. 2012;21:4558–4571. doi: 10.1093/hmg/dds296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mahley RW, Weisgraber KH, Bersot TP. Disorders of lipid metabolism. In: Kronenberg HM, Melmed S, Polonsky KS, Larsen PR, editors. Williams Textbook of Endocrinology. Philadelphia: Saunders; 2008. pp. 1589–1653. [Google Scholar]
- 13.Timmins JM, Lee JY, Boudyguina E, Kluckman KD, Brunham LR, Mulya A, Gebre AK, Coutinho JM, Colvin PL, Smith TL, Hayden MR, Maeda N, Parks JS. Targeted inactivation of hepatic Abca1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of apoA-I. J Clin Invest. 2005;115:1333–1342. doi: 10.1172/JCI23915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weisgraber KH, Mahley RW. Subfractionation of human high density lipoproteins by heparin-Sepharose affinity chromatography. J Lipid Res. 1980;21:316–325. [PubMed] [Google Scholar]
- 15.Mahley RW. Atherogenic lipoproteins and coronary artery disease: concepts derived from recent advances in cellular and molecular biology. Circulation. 1985;72:943–948. doi: 10.1161/01.CIR.72.5.943. [DOI] [PubMed] [Google Scholar]
- 16.Gordon V, Innerarity TL, Mahley RW. Formation of cholesterol- and apoprotein E-enriched high density lipoproteins in vitro. J Biol Chem. 1983;258:6202–6212. [PubMed] [Google Scholar]
- 17.Koo C, Innerarity TL, Mahley RW. Obligatory role of cholesterol and apolipoprotein E in the formation of large cholesterol-enriched and receptor-active high density lipoproteins. J Biol Chem. 1985;260:11934–11943. [PubMed] [Google Scholar]
- 18.Atkinson D, Tall AR, Small DM, Mahley RW. Structural organization of the lipoprotein HDLc from atherosclerotic swine. Structural features relating the particle surface and core. Biochemistry. 1978;17:3930–3933. doi: 10.1021/bi00612a007. [DOI] [PubMed] [Google Scholar]
- 19.Linton MF, Gish R, Hubl ST, Bütler E, Esquivel C, Bry WI, Boyles JK, Wardell MR, Young SG. Phenotypes of apolipoprotein B and apolipo-protein E after liver transplantation. J Clin Invest. 1991;88:270–281. doi: 10.1172/JCI115288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 21.Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci U S A. 2006;103:5644–5651. doi: 10.1073/pnas.0600549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mahley RW, Rall SC., Jr Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet. 2000;1:507–537. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]
- 23.Elshourbagy NA, Liao WS, Mahley RW, Taylor JM. Apolipoprotein E mRNA is abundant in the brain and adrenals, as well as in the liver, and is present in other peripheral tissues of rats and marmosets. Proc Natl Acad Sci U S A. 1985;82:203–207. doi: 10.1073/pnas.82.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boyles JK, Pitas RE, Wilson E, Mahley RW, Taylor JM. Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J Clin Invest. 1985;76:1501–1513. doi: 10.1172/JCI112130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pitas RE, Boyles JK, Lee SH, Foss D, Mahley RW. Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim Biophys Acta. 1987;917:148–161. doi: 10.1016/0005-2760(87)90295-5. [DOI] [PubMed] [Google Scholar]
- 26.Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, Huang Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci. 2006;26:4985–4994. doi: 10.1523/JNEUROSCI.5476-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uchihara T, Duyckaerts C, He Y, Kobayashi K, Seilhean D, Amouyel P, Hauw JJ. ApoE immunoreactivity and microglial cells in Alzheimer’s disease brain. Neurosci Lett. 1995;195:5–8. doi: 10.1016/0304-3940(95)11763-M. [DOI] [PubMed] [Google Scholar]
- 28.Nathan BP, Nisar R, Randall S, Short J, Sherrow M, Wong GK, Struble RG. Apolipoprotein E is upregulated in olfactory bulb glia following peripheral receptor lesion in mice. Exp Neurol. 2001;172:128–136. doi: 10.1006/exnr.2001.7762. [DOI] [PubMed] [Google Scholar]
- 29.Xu Q, Li Y, Cyras C, Sanan DA, Cordell B. Isolation and characterization of apolipoproteins from murine microglia. Identification of a low density lipoprotein-like apolipoprotein J-rich but E-poor spherical particle. J Biol Chem. 2000;275:31770–31777. doi: 10.1074/jbc.M002796200. [DOI] [PubMed] [Google Scholar]
- 30.Knoferle J, Yoon SY, Walker D, Leung L, Gillespie AK, Tong LM, Bien-Ly N, Huang Y. Apolipoprotein E4 produced in GABAergic interneurons causes learning and memory deficits in mice. J Neurosci. 2014;34:14069–14078. doi: 10.1523/JNEUROSCI.2281-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu Q, Walker D, Bernardo A, Brodbeck J, Balestra ME, Huang Y. Intron-3 retention/splicing controls neuronal expression of apolipoprotein E in the CNS. J Neurosci. 2008;28:1452–1459. doi: 10.1523/JNEUROSCI.3253-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Theendakara V, Peters-Libeu CA, Spilman P, Poksay KS, Bredesen DE, Rao RV. Direct transcriptional effects of apolipoprotein E. J Neurosci. 2016;36:685–700. doi: 10.1523/JNEUROSCI.3562-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harris FM, Tesseur I, Brecht WJ, Xu Q, Mullendorff K, Chang S, Wyss-Coray T, Mahley RW, Huang Y. Astroglial regulation of apolipoprotein E expression in neuronal cells. Implications for Alzheimer’s disease. J Biol Chem. 2004;279:3862–3868. doi: 10.1074/jbc.M309475200. [DOI] [PubMed] [Google Scholar]
- 34.Majack RA, Castle CK, Goodman LV, Weisgraber KH, Mahley RW, Shooter EM, Gebicke-Haerter PJ. Expression of apolipoprotein E by cultured vascular smooth muscle cells is controlled by growth state. J Cell Biol. 1988;107:1207–1213. doi: 10.1016/0304-3940(95)11763-M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stukas S, Robert J, Lee M, et al. Intravenously injected human apo-lipoprotein A-I rapidly enters the central nervous system via the choroid plexus. J Am Heart Assoc. 2014;3:e001156. doi: 10.1161/JAHA.114.001156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pitas RE, Boyles JK, Lee SH, Hui D, Weisgraber KH. Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E(LDL) receptors in the brain. J Biol Chem. 1987;262:14352–14360. [PubMed] [Google Scholar]
- 37.Wernette-Hammond ME, Lauer SJ, Corsini A, Walker D, Taylor JM, Rall SC., Jr Glycosylation of human apolipoprotein E. The carbohydrate attachment site is threonine 194. J Biol Chem. 1989;264:9094–9101. [PubMed] [Google Scholar]
- 38.Utermann G. Genetic polymorphism of apolipoprotein E – impact on plasma lipoprotein metabolism. In: Crepaldi G, Tiengo A, Baggio G, editors. Diabetes, Obesity and Hyperlipidemias – III. Amsterdam: Elsevier Science Publishers; 1985. pp. 1–28. [Google Scholar]
- 39.Rasmussen KL, Tybjaerg-Hansen A, Nordestgaard BG, Frikke-Schmidt R. Plasma levels of apolipoprotein E and risk of dementia in the general population. Ann Neurol. 2015;77:301–311. doi: 10.1002/ana.24326. [DOI] [PubMed] [Google Scholar]
- 40.Gupta VB, Wilson AC, Burnham S, et al. AIBL Research Group. Follow-up plasma apolipoprotein E levels in the Australian Imaging, Biomarkers and Lifestyle Flagship Study of Ageing (AIBL) cohort. Alzheimers Res Ther. 2015;7:16. doi: 10.1186/s13195-015-0105-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morrow JA, Hatters DM, Lu B, Hochtl P, Oberg KA, Rupp B, Weisgraber KH. Apolipoprotein E4 forms a molten globule. A potential basis for its association with disease. J Biol Chem. 2002;277:50380–50385. doi: 10.1074/jbc.M204898200. [DOI] [PubMed] [Google Scholar]
- 42.Dong LM, Weisgraber KH. Human apolipoprotein E4 domain interaction. Arginine 61 and glutamic acid 255 interact to direct the preference for very low density lipoproteins. J Biol Chem. 1996;271:19053–19057. doi: 10.1074/jbc.271.32.19053. [DOI] [PubMed] [Google Scholar]
- 43.Ramaswamy G, Xu Q, Huang Y, Weisgraber KH. Effect of domain interaction on apolipoprotein E levels in mouse brain. J Neurosci. 2005;25:10658–10663. doi: 10.1523/JNEUROSCI.1922-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Riddell DR, Zhou H, Atchison K, et al. Impact of apolipoprotein E (ApoE) polymorphism on brain ApoE levels. J Neurosci. 2008;28:11445–11453. doi: 10.1523/JNEUROSCI.1972-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fryer JD, Demattos RB, McCormick LM, O’Dell MA, Spinner ML, Bales KR, Paul SM, Sullivan PM, Parsadanian M, Bu G, Holtzman DM. The low density lipoprotein receptor regulates the level of central nervous system human and murine apolipoprotein E but does not modify amyloid plaque pathology in PDAPP mice. J Biol Chem. 2005;280:25754–25759. doi: 10.1074/jbc.M502143200. [DOI] [PubMed] [Google Scholar]
- 46.Ulrich JD, Burchett JM, Restivo JL, Schuler DR, Verghese PB, Mahan TE, Landreth GE, Castellano JM, Jiang H, Cirrito JR, Holtzman DM. In vivo measurement of apolipoprotein E from the brain interstitial fluid using microdialysis. Mol Neurodegener. 2013;8:13. doi: 10.1186/1750-1326-8-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sullivan PM, Mace BE, Maeda N, Schmechel DE. Marked regional differences of brain human apolipoprotein E expression in targeted replacement mice. Neuroscience. 2004;124:725–733. doi: 10.1016/j.neuroscience.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 48.Fukumoto H, Ingelsson M, Gårevik N, Wahlund LO, Nukina N, Yaguchi Y, Shibata M, Hyman BT, Rebeck GW, Irizarry MC. APOE epsilon 3/epsilon 4 heterozygotes have an elevated proportion of apolipoprotein E4 in cerebrospinal fluid relative to plasma, independent of Alzheimer’s disease diagnosis. Exp Neurol. 2003;183:249–253. doi: 10.1016/S0014-4886(03)00088-8. [DOI] [PubMed] [Google Scholar]
- 49.Wahrle SE, Holtzman DM. Differential metabolism of ApoE iso-forms in plasma and CSF. Exp Neurol. 2003;183:4–6. doi: 10.1016/S0014-4886(03)00185-7. [DOI] [PubMed] [Google Scholar]
- 50.Weisgraber KH. Apolipoprotein E distribution among human plasma lipo-proteins: role of the cysteine-arginine interchange at residue 112. J Lipid Res. 1990;31:1503–1511. [PubMed] [Google Scholar]
- 51.Raffai RL, Dong LM, Farese RV, Jr, Weisgraber KH. Introduction of human apolipoprotein E4 “domain interaction” into mouse apolipoprotein E. Proc Natl Acad Sci U S A. 2001;98:11587–11591. doi: 10.1073/pnas.201279298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhong N, Scearce-Levie K, Ramaswamy G, Weisgraber KH. Apolipoprotein E4 domain interaction: synaptic and cognitive deficits in mice. Alzheimers Dement. 2008;4:179–192. doi: 10.1016/j.jalz.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhong N, Ramaswamy G, Weisgraber KH. Apolipoprotein E4 domain interaction induces endoplasmic reticulum stress and impairs astro-cyte function. J Biol Chem. 2009;284:27273–27280. doi: 10.1074/jbc.M109.014464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang Y, Liu XQ, Wyss-Coray T, Brecht WJ, Sanan DA, Mahley RW. Apolipoprotein E fragments present in Alzheimer’s disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc Natl Acad Sci U S A. 2001;98:8838–8843. doi: 10.1073/pnas.151254698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harris FM, Brecht WJ, Xu Q, Tesseur I, Kekonius L, Wyss-Coray T, Fish JD, Masliah E, Hopkins PC, Scearce-Levie K, Weisgraber KH, Mucke L, Mahley RW, Huang Y. Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer’s disease-like neurodegeneration and behavioral deficits in transgenic mice. Proc Natl Acad Sci U S A. 2003;100:10966–10971. doi: 10.1073/pnas.1434398100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brecht WJ, Harris FM, Chang S, Tesseur I, Yu GQ, Xu Q, Dee Fish J, Wyss-Coray T, Buttini M, Mucke L, Mahley RW, Huang Y. Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J Neurosci. 2004;24:2527–2534. doi: 10.1523/JNEUROSCI.4315-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harris FM, Brecht WJ, Xu Q, Mahley RW, Huang Y. Increased tau phos-phorylation in apolipoprotein E4 transgenic mice is associated with activation of extracellular signal-regulated kinase: modulation by zinc. J Biol Chem. 2004;279:44795–44801. doi: 10.1074/jbc.M408127200. [DOI] [PubMed] [Google Scholar]
- 58.Mahley RW, Huang Y. Apolipoprotein e sets the stage: response to injury triggers neuropathology. Neuron. 2012;76:871–885. doi: 10.1016/j.neuron.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mahley RW, Huang Y. Small-molecule structure correctors target abnormal protein structure and function: structure corrector rescue of apolipoprotein E4-associated neuropathology. J Med Chem. 2012;55:8997–9008. doi: 10.1021/jm3008618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Beffert U, Danik M, Krzywkowski P, Ramassamy C, Berrada F, Poirier J. The neurobiology of apolipoproteins and their receptors in the CNS and Alzheimer’s disease. Brain Res Brain Res Rev. 1998;27:119–142. doi: 10.1016/S0165-0173(98)00008-3. [DOI] [PubMed] [Google Scholar]
- 61.LaDu MJ, Gilligan SM, Lukens JR, Cabana VG, Reardon CA, Van Eldik LJ, Holtzman DM. Nascent astrocyte particles differ from lipoproteins in CSF. J Neurochem. 1998;70:2070–2081. doi: 10.1046/j.1471-4159.1998.70052070.x. [DOI] [PubMed] [Google Scholar]
- 62.Fagan AM, Holtzman DM, Munson G, Mathur T, Schneider D, Chang LK, Getz GS, Reardon CA, Lukens J, Shah JA, LaDu MJ. Unique lipo-proteins secreted by primary astrocytes from wild type, apoE (−/−), and human apoE transgenic mice. J Biol Chem. 1999;274:30001–30007. doi: 10.1074/jbc.274.42.30001. [DOI] [PubMed] [Google Scholar]
- 63.Xu Q, Brecht WJ, Weisgraber KH, Mahley RW, Huang Y. Apolipoprotein E4 domain interaction occurs in living neuronal cells as determined by fluorescence resonance energy transfer. J Biol Chem. 2004;279:25511–25516. doi: 10.1074/jbc.M311256200. [DOI] [PubMed] [Google Scholar]
- 64.Ye S, Huang Y, Müllendorff K, Dong L, Giedt G, Meng EC, Cohen FE, Kuntz ID, Weisgraber KH, Mahley RW. Apolipoprotein (apo) E4 enhances amyloid beta peptide production in cultured neuronal cells: apoE structure as a potential therapeutic target. Proc Natl Acad Sci U S A. 2005;102:18700–18705. doi: 10.1073/pnas.0508693102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vitali C, Wellington CL, Calabresi L. HDL and cholesterol handling in the brain. Cardiovasc Res. 2014;103:405–413. doi: 10.1093/cvr/cvu148. [DOI] [PubMed] [Google Scholar]
- 66.Yancey PG, Yu H, Linton MF, Fazio S. A pathway-dependent on apoE, ApoAI, and ABCA1 determines formation of buoyant high-density lipoprotein by macrophage foam cells. Arterioscler Thromb Vasc Biol. 2007;27:1123–1131. doi: 10.1161/ATVBAHA.107.139592. [DOI] [PubMed] [Google Scholar]
- 67.Hirsch-Reinshagen V, Zhou S, Burgess BL, Bernier L, McIsaac SA, Chan JY, Tansley GH, Cohn JS, Hayden MR, Wellington CL. Deficiency of ABCA1 impairs apolipoprotein E metabolism in brain. J Biol Chem. 2004;279:41197–41207. doi: 10.1074/jbc.M407962200. [DOI] [PubMed] [Google Scholar]
- 68.Wahrle SE, Jiang H, Parsadanian M, Legleiter J, Han X, Fryer JD, Kowalewski T, Holtzman DM. ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J Biol Chem. 2004;279:40987–40993. doi: 10.1074/jbc.M407963200. [DOI] [PubMed] [Google Scholar]
- 69.Rust S, Rosier M, Funke H, Real J, Amoura Z, Piette JC, Deleuze JF, Brewer HB, Duverger N, Denèfle P, Assmann G. Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat Genet. 1999;22:352–355. doi: 10.1038/11921. [DOI] [PubMed] [Google Scholar]
- 70.Assmann G, von Eckardstein A, Bryan Brewer H. Familial analphalipoproteinemia: Tangier disease. In: Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson KM, Mitchell G, editors. OMMBID. New York, NY: The McGraw-Hill Companies, Inc; 2014. [Google Scholar]
- 71.Asztalos BF, Brousseau ME, McNamara JR, Horvath KV, Roheim PS, Schaefer EJ. Subpopulations of high density lipoproteins in homozygous and heterozygous Tangier disease. Atherosclerosis. 2001;156:217–225. doi: 10.1016/S0021-9150(00)00643-2. [DOI] [PubMed] [Google Scholar]
- 72.Quazi F, Molday RS. Differential phospholipid substrates and directional transport by ATP-binding cassette proteins ABCA1, ABCA7, and ABCA4 and disease-causing mutants. J Biol Chem. 2013;288:34414–34426. doi: 10.1074/jbc.M113.508812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hollingworth P, Harold D, Sims R, et al. Alzheimer’s Disease Neuroimaging Initiative; CHARGE Consortium; EADI1 Consortium. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vardarajan BN, Ghani M, Kahn A, et al. Rare coding mutations identified by sequencing of Alzheimer disease genome-wide association studies loci. Ann Neurol. 2015;78:487–498. doi: 10.1002/ana.24466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Burgess BL, Parkinson PF, Racke MM, et al. ABCG1 influences the brain cholesterol biosynthetic pathway but does not affect amyloid precursor protein or apolipoprotein E metabolism in vivo. J Lipid Res. 2008;49:1254–1267. doi: 10.1194/jlr.M700481-JLR200. [DOI] [PubMed] [Google Scholar]
- 76.Wang N, Yvan-Charvet L, Lütjohann D, Mulder M, Vanmierlo T, Kim TW, Tall AR. ATP-binding cassette transporters G1 and G4 mediate cholesterol and desmosterol efflux to HDL and regulate sterol accumulation in the brain. FASEB J. 2008;22:1073–1082. doi: 10.1096/fj.07-9944com. [DOI] [PubMed] [Google Scholar]
- 77.Bu G. Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009;10:333–344. doi: 10.1038/nrn2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Holtzman DM, Herz J, Bu G. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006312. doi: 10.1101/cshperspect.a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J. Reeler/disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell. 1999;97:689–701. doi: 10.1016/S0092-8674(00)80782-5. [DOI] [PubMed] [Google Scholar]
- 80.Hirota Y, Kubo K, Katayama K, Honda T, Fujino T, Yamamoto TT, Nakajima K. Reelin receptors ApoER2 and VLDLR are expressed in distinct spatiotemporal patterns in developing mouse cerebral cortex. J Comp Neurol. 2015;523:463–478. doi: 10.1002/cne.23691. [DOI] [PubMed] [Google Scholar]
- 81.Rebeck GW, Reiter JS, Strickland DK, Hyman BT. Apolipoprotein E in sporadic Alzheimer’s disease: allelic variation and receptor interactions. Neuron. 1993;11:575–580. doi: 10.1016/0896-6273(93)90070-8. [DOI] [PubMed] [Google Scholar]
- 82.Mahley RW, Ji ZS. Remnant lipoprotein metabolism: key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. J Lipid Res. 1999;40:1–16. [PubMed] [Google Scholar]
- 83.Kim J, Castellano JM, Jiang H, Basak JM, Parsadanian M, Pham V, Mason SM, Paul SM, Holtzman DM. Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular A beta clearance. Neuron. 2009;64:632–644. doi: 10.1016/j.neuron.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu Q, Zerbinatti CV, Zhang J, Hoe HS, Wang B, Cole SL, Herz J, Muglia L, Bu G. Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron. 2007;56:66–78. doi: 10.1016/j.neuron.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen CH, Albers JJ. Activation of lecithin: cholesterol acyltransferase by apolipoproteins E-2, E-3, and A-IV isolated from human plasma. Biochim Biophys Acta. 1985;836:279–285. doi: 10.1016/0005-2760(85)90131-6. [DOI] [PubMed] [Google Scholar]
- 86.Rye KA, Bright R, Psaltis M, Barter PJ. Regulation of reconstituted high density lipoprotein structure and remodeling by apolipoprotein E. J Lipid Res. 2006;47:1025–1036. doi: 10.1194/jlr.M500525-JLR200. [DOI] [PubMed] [Google Scholar]
- 87.Albers JJ, Tollefson JH, Wolfbauer G, Albright RE., Jr Cholesteryl ester transfer protein in human brain. Int J Clin Lab Res. 1992;21:264–266. doi: 10.1007/BF02591657. [DOI] [PubMed] [Google Scholar]
- 88.Elliott DA, Weickert CS, Garner B. Apolipoproteins in the brain: implications for neurological and psychiatric disorders. Clin Lipidol. 2010;51:555–573. doi: 10.2217/CLP.10.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Huang Z, Cheng C, Jiang L, Yu Z, Cao F, Zhong J, Guo Z, Sun X. Intraventricular apolipoprotein ApoJ infusion acts protectively in traumatic brain injury. J Neurochem. 2016;136(5):1017–1025. doi: 10.1111/jnc.13491. [DOI] [PubMed] [Google Scholar]
- 90.Trougakos IP, Gonos ES. Clusterin/apolipoprotein J in human aging and cancer. Int J Biochem Cell Biol. 2002;34:1430–1448. doi: 10.1016/S1357-2725(02)00041-9. [DOI] [PubMed] [Google Scholar]
- 91.Wyatt AR, Yerbury JJ, Wilson MR. Structural characterization of cluster-in-chaperone client protein complexes. J Biol Chem. 2009;284:21920–21927. doi: 10.1074/jbc.M109.033688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tsukamoto K, Mani DR, Shi J, Zhang S, Haagensen DE, Otsuka F, Guan J, Smith JD, Weng W, Liao R, Kolodgie FD, Virmani R, Krieger M. Identification of apolipoprotein D as a cardioprotective gene using a mouse model of lethal atherosclerotic coronary artery disease. Proc Natl Acad Sci U S A. 2013;110:17023–17028. doi: 10.1073/pnas.1315986110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J Lipid Res. 2009;50(suppl):S183–S188. doi: 10.1194/jlr.R800069-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 2009;63:287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu CC, Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–118. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.LaDu MJ, Falduto MT, Manelli AM, Reardon CA, Getz GS, Frail DE. Isoform-specific binding of apolipoprotein E to beta-amyloid. J Biol Chem. 1994;269:23403–23406. [PubMed] [Google Scholar]
- 98.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apo-lipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 99.Roses AD. Apolipoprotein E alleles as risk factors in Alzheimer’s disease. Annu Rev Med. 1996;47:387–400. doi: 10.1146/annurev.med.47.1.387. [DOI] [PubMed] [Google Scholar]
- 100.Andrews-Zwilling Y, Bien-Ly N, Xu Q, Li G, Bernardo A, Yoon SY, Zwilling D, Yan TX, Chen L, Huang Y. Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J Neurosci. 2010;30:13707–13717. doi: 10.1523/JNEUROSCI.4040-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen HK, Ji ZS, Dodson SE, Miranda RD, Rosenblum CI, Reynolds IJ, Freedman SB, Weisgraber KH, Huang Y, Mahley RW. Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. J Biol Chem. 2011;286:5215–5221. doi: 10.1074/jbc.M110.151084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen HK, Liu Z, Meyer-Franke A, et al. Small molecule structure correctors abolish detrimental effects of apolipoprotein E4 in cultured neurons. J Biol Chem. 2012;287:5253–5266. doi: 10.1074/jbc.M111.276162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chang S, ran Ma T, Miranda RD, Balestra ME, Mahley RW, Huang Y. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc Natl Acad Sci U S A. 2005;102:18694–18699. doi: 10.1073/pnas.0508254102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dong LM, Wilson C, Wardell MR, Simmons T, Mahley RW, Weisgraber KH, Agard DA. Human apolipoprotein E. Role of arginine 61 in mediating the lipoprotein preferences of the E3 and E4 isoforms. J Biol Chem. 1994;269:22358–22365. [PubMed] [Google Scholar]
- 105.Morrow JA, Segall ML, Lund-Katz S, Phillips MC, Knapp M, Rupp B, Weisgraber KH. Differences in stability among the human apolipoprotein E isoforms determined by the amino-terminal domain. Biochemistry. 2000;39:11657–11666. doi: 10.1021/bi000099m. [DOI] [PubMed] [Google Scholar]
- 106.Nguyen D, Dhanasekaran P, Nickel M, Nakatani R, Saito H, Phillips MC, Lund-Katz S. Molecular basis for the differences in lipid and lipoprotein binding properties of human apolipoproteins E3 and E4. Biochemistry. 2010;49:10881–10889. doi: 10.1021/bi1017655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ, Brunden KR, Wilson DA, Landreth GE. ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.LaClair KD, Manaye KF, Lee DL, Allard JS, Savonenko AV, Troncoso JC, Wong PC. Treatment with bexarotene, a compound that increases apolipoprotein-E, provides no cognitive benefit in mutant APP/PS1 mice. Mol Neurodegener. 2013;8:18. doi: 10.1186/1750-1326-8-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fitz NF, Cronican AA, Lefterov I, Koldamova R. Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science. 2013;340:924–92c. doi: 10.1126/science.1235809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Price AR, Xu G, Siemienski ZB, Smithson LA, Borchelt DR, Golde TE, Felsenstein KM. Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science. 2013;340:924–92d. doi: 10.1126/science.1234089. [DOI] [PubMed] [Google Scholar]
- 111.Tesseur I, Lo AC, Roberfroid A, Dietvorst S, Van Broeck B, Borgers M, Gijsen H, Moechars D, Mercken M, Kemp J, D’Hooge R, De Strooper B. Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science. 2013;340:924–92e. doi: 10.1126/science.1233937. [DOI] [PubMed] [Google Scholar]
- 112.Veeraraghavalu K, Zhang C, Miller S, Hefendehl JK, Rajapaksha TW, Ulrich J, Jucker M, Holtzman DM, Tanzi RE, Vassar R, Sisodia SS. Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science. 2013;340:924–92f. doi: 10.1126/science.1235505. [DOI] [PubMed] [Google Scholar]