

Introduction
It was a distinct honor to present a lecture celebrating Ed Charlton, who died in 2010. Ed was a good friend and colleague, a wonderful story-teller and family man. Ed also made many significant contributions to the pain research and pain management community. Most notably, Ed edited the Third Edition of the IASP Core Curriculum for Professional Education in Pain and co-edited the Proceedings of the VIth World Congress on Pain. My relationship with Ed flourished when I invited him to take over as Section Editor for the Clinical Notes section of PAIN. This was a difficult position, as the requirements for publication were never clearly articulated. In his inimitable way, Ed articulated what he considered the requirements. In an Editorial he wrote with advice to prospective authors of contributions to Clinical Notes: “It may be easier to start by listing the sort of article that the Clinical Notes Section will NOT publish. These include papers about the anecdotal use of a drug on a handful of patients or even a single patient; or papers that include statements like “….further studies are indicated”; or “……controlled studies are proceeding”. Ed’s advice was simple: “Complete the studies before submitting them to PAIN!”. In other words, what PAIN is interested in were studies that readers can rely upon as they assess new approaches to understanding and treating difficult pain conditions. What a wonderful message to send to authors and to the readership of the journal.
There is certainly consensus that treating chronic pain conditions, notably neuropathic pain, is difficult. Although there are numerous pharmacological approaches to the management of neuropathic pain, the results are disappointing. The general consensus is that the best available therapies are effective in less than 50% of patients and in these patients there is less than 50% decrease in pain (18, 34, 36). The pain reduction is better than placebo, but clearly not sufficient. In this review, we will highlight a different perspective on the etiology of neuropathic pain, notably that produced by peripheral nerve damage, and offer a novel approach to management.
The pathophysiology of neuropathic pain
Many years of preclinical study demonstrated that nerve injury provokes complex molecular and biochemical changes in primary afferents, dorsal horn circuits (neurons and notably microglia) and also at higher levels of the neuraxis (3, 4, 29). Many of these biochemical consequences of injury are at the basis of the central sensitization process that underlies the mechanical allodynia and thermal hyperalgesia that are the hallmarks of the neuropathic pain phenotype (55). To what extent these changes also contribute to the ongoing, often burning pain that patients usually describe is not clear, as only recently have animal studies addressed the ongoing pain question (26).
Despite identification of a remarkably large number of molecules that contribute to peripheral and central sensitization (21), few if any new targets have translated into a clinical therapy. In fact, the great majority, if not all approaches to management are directed at the transmission of the pain message itself. A brief review highlights this point. For example, among the most significant findings in patients is that local anesthetic injection of the peripheral afferent (at or proximal to the injury site) often, if not always blocks the pain (for example, See (22). This is true in patients with complex regional pain syndrome or postherpetic neuralgia and even in those with phantom limb pain, using injections into the stump (52). Of course, the pain returns after the anesthetic wears off. It follows from these observations that activity of the injured afferents is a critical contributor to the neuropathic pain condition. But these inputs are now generated on an altered central nervous system, one that has undergone profound biochemical and molecular changes following the injury.
In our opinion the most critical contributor to these central changes are NMDA-mediated, and in many respects these changes are comparable to those that occur in an adaptive learning condition (as in the hippocampus). However, when pain persists, the learning is maladaptive and contributes to the persistence of the pain condition. Unfortunately, the adverse side effect profile of selective NMDA receptor antagonists is unacceptable. Ketamine, a relatively nonspecific NMDA receptor antagonist, has proven useful in some instances, but its adverse effects are definitely problematic (39).
The first-line therapy calls for tricyclic anti-depressants and gabapentinoids (17). The latter targets the alpha2delta subunit of Ca2+ channels, likely in primary afferents. As for the local anesthetics, gabapentinoids are an approach directed at reducing transmission of the “pain” message. To some extent the same is true for opioids (at least at the level of the primary afferent and spinal cord), although opioids are generally considered to have limited utility in neuropathic pain patients (17). A capsaicin injection that transiently ablates nociceptor terminals also exerts its effect by blocking transmission of the pain message (25). On the horizon are more selective blockers that target subtypes of voltage-gated sodium channels, notably the NaV1.7 subtype (15). Other approaches do not act directly on the afferent (e.g. serotonin-norepinephrine reuptake inhibitors), but their aim, in addition to targeting voltage-gated Na channels, is to enhance controls, descending and otherwise, that reduce activity in the pain transmission pathway. The same is true for neuromodulation approaches, e.g. spinal cord stimulation and even transcranial magnetic stimulation.
GABAergic dysfunction and neuropathic pain
In many respects the pathophysiology of neuropathic pain is comparable to that which occurs in patients with seizures., where this is loss of cortical GABAergic inhibitor controls. Indeed the idea that neuropathic pain is an epileptic type condition in the spinal cord is not a new one (31). Not surprisingly, therefore, anticonvulsants are among the more effective pharmacotherapies. Indeed, when a new anticonvulsant is introduced, it is often tested for efficacy against neuropathic pain. There is certainly considerable consensus that peripheral nerve injury results in a significant reduction in GABAergic inhibitory controls at the level of the spinal cord dorsal horn, leading to central sensitization of pain transmission circuits and a resultant hypersensitivity and ongoing pain (27, 45, 57). Not only are the levels of the GABA synthesizing enzyme, glutamic acid decarboxylase, reduced (16), but release of GABA (30) and inhibitory postsynaptic currents in dorsal horn neurons are diminished after peripheral nerve injury (35). However, precisely what underlies these changes in GABAergic inhibitory tone is not clear. For example, some groups reported a frank loss of GABAergic interneurons (12, 35, 43, 48), but this finding has been questioned (40). On the other hand, there is good evidence for alterations in GABAergic receptors (20), which taken together with reduced spinal release of GABA (30) would lead to an overall loss of inhibitory control. Among the most provocative findings is that microglial activation results in a BDNF-mediated shift in the chloride gradient in dorsal horn “pain” transmission neurons, resulting in loss of GABA-mediated inhibition (13). Of course, consistent with the hypothesis that loss of GABAergic inhibition is critical are studies reporting the normalization of pain thresholds by pharmacological restoration of GABAergic transmission (28, 37). Also of interest are approaches to long-term regulation using, for example, trigeminal injection of an adenoviral vector expressing GAD65 (54) or peripheral injection of an HSV vector expressing GAD67 (23).
Treating the “disease” of neuropathic pain
In our opinion, an important alternative to interfering with transmission of the pain message, and until more effective management is achieved by rectifying the pathophysiological molecular and biochemical consequents of nerve damage, is to address the damaged nervous system itself. In this respect, the neuropathic pain condition constitutes a “disease” of the nervous system. The main, and in some conditions, only manifestation of the nerve damage, is chronic pain. If one could treat the “disease”, i.e. by repairing the damage, it may be possible to generate a more complete and prolonged reduction of the neuropathic pain that follows damage.
In several recent studies, we described a very different approach directed at overcoming the loss of GABAergic inhibition that occurs in the spinal cord (4). We first asked whether it is possible to transplant progenitors of GABAergic interneurons into the spinal cord, so as to restore the inhibitory control loss after nerve damage. Our studies followed upon previous reports that demonstrated that all GABergic interneurons of the cerebral cortex derive from the medial ganglionic eminence (MGE) of the forebrain (1, 56). Of particular relevance to our studies was a subsequent report demonstrating that transplanting embryonic MGE cells into the cortex of mice with a K+ channel mutation could reduce seizure incidence in these mice (2). Given the commonalities in the GABAergic contribution to seizures and to neuropathic pain noted above, we hypothesized that transplanting MGE cells into the spinal cord could ameliorate the mechanical hypersensitivity observed in a mouse model of neuropathic pain (7).
There was no assurance that MGE cells would survive in the spinal cord, an environment foreign to their normal developmental location, or that surviving cells would remain and grow at the location of the injection. The latter was a significant concern given the inherent extensive migration ability that occurs after cortical injection. Figure 1a demonstrates that MGE transplants can indeed survive and grow in the adult spinal cord. The cells do migrate from the injection site, but remain within the segment of lumbar cord in which we made injections. What is striking is the incredible axonal and dendritic arborization of the transplanted cells (Figure 1b). Within two weeks of transplant, approximately 90% of the cells assume a neuronal phenotype (established by their immunostaining for NeuN). None of the transplanted cells were immunoreactive for microglial or astrocyte markers. The fact that the cells also immunostained positively for GABA suggests that they retain their cortical neurochemistry. In fact, the cells also co-stain for other markers characteristic of cortical GABAergic interneurons, including parvalbumin and somatostatin. The latter finding is of particular interest, as somatostatin is not found in spinal cord GABAergic interneurons, but rather in a population of superficial dorsal horn excitatory interneurons (41). Thus, despite the foreign environment, the transplanted cells appear to retain their cortical properties.
Figure 1.
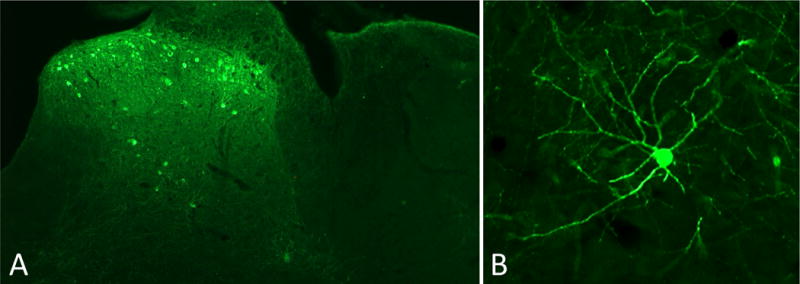
A: Intraspinal transplantation of embryonic GABAergic progenitor cells derived from the medial ganglionic eminence of the mouse cortex. The cells were transplanted unilaterally in the lumbar enlargement and the mouse survived 4 weeks. The cells, which express GFP (green), are concentrated in the superficial dorsal horn and in the region of lamina V. Note that there is no spread of the cells to the contralateral side of the cord.
B: High magnification of a single GFP-expressing transplanted neuron illustrates its extensive arborization (axonal and dendritic), which is characteristic of cortical GABAergic inhibitory interneurons.
Perhaps the most important finding of our earlier studies is the extent to which the transplanted cells integrate into host circuitry. Our first demonstration of this property relied on our previous development of transgenic mice in which we can initiate Cre-dependent expression of a transneuronal tracer, wheat germ agglutinin (WGA), in host neurons (8, 9). Using these mice, we demonstrated transneuronal transfer of the WGA into transplanted MGE cells (10). In some of these studies we used a mouse line in which expression of the Cre-dependent WGA is induced only following nerve injury (6). Taking advantage of the fact that neuropeptide Y is only expressed in DRG neurons after peripheral nerve injury, predominantly in myelinated afferents (24, 53), we were able to direct expression of the WGA to myelinated afferent. In these mice, we found transfer of the tracer into dorsal horn MGE cells, demonstrating that the transplanted cells indeed integrate into host circuit, in this case, receiving a primary afferent input from, myelinated primary afferents. This connection is of particular interest as myelinated, cutaneous sensory neurons convey innocuous peripheral inputs that can drive a low threshold-mediated inhibitory control in the spinal cord, a major postulate of the Gate Control Theory (4, 32).
Using a different approach we next asked whether the transplants also establish connections with host spinal cord neurons. In these studies we took advantage of the retrograde transneuronal transport features of pseudorabies virus (14). We injected the virus into the parabrachial nucleus (PB) of the dorsolateral pons, a major target of dorsal horn projection neurons, including many in lamina I, and mapped the retrograde spread of the virus into the spinal cord. Our detection of MGE cells retrogradely infected by the virus established that some transplanted cells must be presynaptic to the PB-projecting spinal neurons. However, because the pseudorabies virus crosses multiple synapses, we could not conclude that there is a direct connection between transplanted cells and projection neurons, but this seems likely.
More recently, we sought a more direct demonstration that there are indeed synaptic connections between the transplanted neurons and the host. We initiated an extensive set of ultrastructural studies that has already provided unequivocal evidence, not only that host neurons are presynaptic to transplanted cells, but also that MGE cells form synapses with host neurons. Of particular interest is our finding that transplanted neurons also interact with one another. Taken together, these observations establish that the transplanted neurons are profoundly integrated into the host circuitry. A key question, of course, is whether the neurons are recapitulating altered host circuits (whether anatomical or functionally altered), or whether the cells will form connections with any host element. It is our impression that the latter is the case, and as described below, we believe that this integration is both functional and serves to reestablish a level of inhibition that is “normal”. Finally, and most importantly, we demonstrated that the connections established by the transplants are functional. For example, peripheral injections of formalin, a noxious stimulus, can induce expression of Fos (i.e. activate) in transplanted MGE cells. In other words, the transplants survive, integrate and are functionally connected with dorsal horn circuits.
In the next series of studies we asked whether the transplants could mitigate the major behavioral features of different preclinical neuropathic pain models. In the first set of studies (10), we used the sciatic nerve injury model of neuropathic pain, in which two of the three branches of the sciatic nerve are transected, leaving the tibial nerve intact (44). In this model, mice develop a profound hypersensitivity (assayed using von Frey hairs), at one day after surgery. After manifesting hypersensitivity we made multiple rostrocaudally-spaced injections of MGE cells into the lumbar spinal cord, targeting the dorsal horn. Although we began with approximately 50,000 cells, we estimate that less than 10% survive. Whether some cells are lost in the injection protocol, or undergo an early apoptotic death is not known.
Animals were tested for mechanical responsiveness weekly by an investigator blind to whether the mice received MGE cells or medium. When the code was broken four weeks after the transplant, we found that mechanical thresholds in the MGE transplanted animals had gradually returned to baseline. In recent studies, we find that the cells survive and that the recovery persists for at least 6 months. Interestingly, we did not find that threshold recovery correlated with the number of surviving cells. Rather we assume that the recovery is more a function of the extensive axonal arbors that arise from the transplanted cells. In parallel studies, we assayed for levels of GAD, which as noted above, decrease significantly in the dorsal horn after peripheral nerve injury. We found that the transplant returned GAD levels to those of the contralateral, intact side of the cord. The fact that we did not detect levels above baseline provides further evidence that the transplants normalize biochemical and functional processes. This finding suggests that the transplanted neurons, through a process that is not at all clear, are sensitive to the magnitude of the GABAergic dysfunction and respond accordingly. In other words, there is a kind of GABAstat that establishes a target level of inhibition that the cells achieve. This observation differs greatly from the effects produced if one administers a GABA agonist at the level of the cord. Thus, intrathecal injection of baclofen or muscimol can increase thresholds well above baseline, i.e., an analgesic action is readily attained (51). Taken together these findings suggest that the transplants do not function as a sophisticated cellular GABAergic pump but rather modify the “disease” process, namely the nerve injury-induced loss of GABAergic inhibition.
More recently, we asked whether the transplants can also overcome the hypersensitivity that occurs in a very different, chemotherapy-induced model of neuropathic pain. Paclitaxel produces a systemic, peripheral neuropathy manifest behaviorally as mechanical hypersensitivity (19) and in some instances thermal hyperalgesia (46). Although the behavioral readout in this model is comparable to that following traumatic peripheral nerve injury, its basis differs considerably, at least at the doses we used in our studies. First, in contrast to traumatic injury (5, 50), there is no induction of the transcription factor, ATF3, in affected sensory neurons of the dorsal root ganglion. Second, the dramatic dorsal horn microglial activation that is produced after peripheral nerve transection (33, 49) does not occur in the chemotherapy model. And finally, and of particular importance to our study, is that GAD levels are not decreased in this model. Whether the hypersensitivity reflects a different pathology in GABAergic circuits (e.g. alteration in receptor function) is not clear. Importantly, however, even though GAD levels are normal in the paclitaxel animals, we found that the transplants completely reversed the paclitaxel-induced mechanical hypersensitivity and heat hyperalgesia, in a topographic manner (11). Thus, transplants unilaterally injected into the lumbar enlargement normalized mechanical and heat threshold of the ipsilateral hindlimb, but there was no change in the contralateral hindpaw. Furthermore, GAD levels were not increased above normal in these animals, which is consistent with our contention that the transplants do not merely “add” GABA to the spinal cord, but rather, maintain levels at close to normal, regardless of the starting point.
Our paclitaxel studies also more directly addressed the particular contribution of GABA to the therapeutic effects of the MGE cell transplants. Thus, in a separate set of experiments we transplanted cells from mice that are deficient for the vesicular GABA transporter (VGAT; (11). These mice synthesize GABA, but cannot store and then release it because VGAT is required for transport of the GABA into synaptic vesicles. In these mice the transplants survived and integrated, however, we found no effect on paclitaxel-induced mechanical allodynia or thermal hyperalgesia. This finding clearly demonstrates that the effects of the transplants require a GABA contribution. Indeed our preliminary electrophysiological recordings from transplanted neurons, in a spinal cord slice preparation, established that the transplanted cells are functional and exhibit properties characteristic of GABAergic interneurons and that their inhibitory effects involve an action at the GABA-A receptor.
Treating the “disease” of neuropathic itch
More recently, we asked whether the transplants could be beneficial in a very different condition that is triggered by GABAergic dysfunction. Ross and colleagues (42) reported that mice with a deletion of the gene that codes for the Bhlhb5 transcription factor develop a relentless syndrome of scratching, resulting in self-inflicted skin lesions. This neuropathic itch condition parallels the neuropathic pain that follows peripheral nerve damage. Bhlhb5 is required for the development and survival of a subpopulation of GABAergic interneurons in the dorsal horn of the lumbar spinal cord. We hypothesized, therefore, that replenishing the pool of inhibitory interneurons via intraspinal transplantation of MGE cells could restore some level of GABAergic function in the Bhlhb5 mice and thus reduce the neuropathic itch that these mice develop. In these studies, we performed the transplants in spinal cord segments corresponding to the dermatomes where the skin lesions manifest. We monitored spontaneous scratching as well as severity of the lesions over time. Within 2 weeks of transplantation, we observed a remarkable reduction in the size and severity of the skin lesions, and this result correlated with a decrease in scratching (7). Interestingly, although the transplants did not normalize the very large decrease of GAD levels in the spinal cord of Bhlhb5 mice, they were nonetheless able to restore inhibitory controls, we presume as a result of the extensive axonal arborization of the transplanted neurons.
Summary and future directions
Taken together, our transplantation studies demonstrate that an inhibitory, cell-based therapy is a powerful strategy for the management of neuropathic pain and itch. Even more significant perhaps is that the transplantation approach differs considerably from traditional pharmacological manipulations. In many respects, the ability to repair a nerve injury-induced dysfunctional nervous system is disease-modifying. Furthermore, because the MGE-derived GABA release is restricted to the site of transplant, in a synaptic manner, many of the common side effects associated with systemic or intrathecal administration of GABAergic compounds can be avoided. If this therapy is to be adaptable to the management of neuropathic pain and itch in patients, the next step is to evaluate the utility of human cell transplantation. In this regard, recent studies have made considerable advances in generating pluripotent cells that exhibit properties similar to those of mouse MGE cells (38, 47), including robust synaptic integration into host circuitry and the development of a GABAergic phenotype. We have now initiated studies using human embryonic stem cells transplanted into immunocompromised mice and are very encouraged by the ability of these cells to integrate into the host spinal cord. Given the many neuropathic pain and itch conditions that remain intractable when managed with traditional therapies, the possibility of their management by a cell transplantation approach is exciting.
Acknowledgments
This work was supported by grants from the NIH: NS78326, NS14627, DA29204 and a grant from the Wellcome Trust. AIB is on the Scientific Advisory Board of Neurona.
References
- 1.Alvarez-Dolado M, Calcagnotto ME, Karkar KM, Southwell DG, Jones-Davis DM, Estrada RC, Rubenstein JL, Alvarez-Buylla A, Baraban SC. Cortical inhibition modified by embryonic neural precursors grafted into the postnatal brain. J Neurosci. 2006;26:7380–7389. doi: 10.1523/JNEUROSCI.1540-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baraban SC, Southwell DG, Estrada RC, Jones DL, Sebe JY, Alfaro-Cervello C, Garcia-Verdugo JM, Rubenstein JL, Alvarez-Buylla A. Reduction of seizures by transplantation of cortical GABAergic interneuron precursors into Kv1.1 mutant mice. Proc Natl Acad Sci (USA) 2009;106:15472–15477. doi: 10.1073/pnas.0900141106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Braz J, Solorzano C, Wang X, Basbaum AI. Transmitting pain and itch messages: a contemporary view of the spinal cord circuits that generate gate control. Neuron. 2014;82:522–536. doi: 10.1016/j.neuron.2014.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braz JM, Basbaum AI. Differential ATF3 expression in dorsal root ganglion neurons reveals the profile of primary afferents engaged by diverse noxious chemical stimuli. Pain. 2010;150:290–301. doi: 10.1016/j.pain.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Braz JM, Basbaum AI. Triggering genetically-expressed transneuronal tracers by peripheral axotomy reveals convergent and segregated sensory neuron-spinal cord connectivity. Neuroscience. 2009;163:1220–1232. doi: 10.1016/j.neuroscience.2009.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braz JM, Juarez-Salinas D, Ross SE, Basbaum AI. Transplant restoration of spinal cord inhibitory controls ameliorates neuropathic itch. J Clin Invest. 2014;124:3612–3616. doi: 10.1172/JCI75214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braz JM, Nassar MA, Wood JN, Basbaum AI. Parallel “pain” pathways arise from subpopulations of primary afferent nociceptor. Neuron. 2005;47:787–793. doi: 10.1016/j.neuron.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 9.Braz JM, Rico B, Basbaum AI. Transneuronal tracing of diverse CNS circuits by Cre-mediated induction of wheat germ agglutinin in transgenic mice. Proc Natl Acad Sci (USA) 2002;99:15148–15153. doi: 10.1073/pnas.222546999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Braz JM, Sharif-Naeini R, Vogt D, Kriegstein A, Alvarez-Buylla A, Rubenstein JL, Basbaum AI. Forebrain GABAergic neuron precursors integrate into adult spinal cord and reduce injury-induced neuropathic pain. Neuron. 2012;74:663–675. doi: 10.1016/j.neuron.2012.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Braz JM, Wang X, Guan Z, Rubenstein JL, Basbaum AI. Transplant-mediated enhancement of spinal cord GABAergic inhibition reverses paclitaxel-induced mechanical and heat hypersensitivity. Pain. 2015;156:1084–1091. doi: 10.1097/j.pain.0000000000000152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Castro-Lopes JM, Tavares I, Coimbra A. GABA decreases in the spinal cord dorsal horn after peripheral neurectomy. Brain Res. 1993;620:287–291. doi: 10.1016/0006-8993(93)90167-l. [DOI] [PubMed] [Google Scholar]
- 13.De Koninck Y. Altered chloride homeostasis in neurological disorders: a new target. Curr Opin Pharmacol. 2007;7:93–99. doi: 10.1016/j.coph.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 14.DeFalco J, Tomishima M, Liu H, Zhao C, Cai X, Marth JD, Enquist L, Friedman JM. Virus-assisted mapping of neural inputs to a feeding center in the hypothalamus. Science. 2001;291:2608–2613. doi: 10.1126/science.1056602. [DOI] [PubMed] [Google Scholar]
- 15.Dib-Hajj SD, Yang Y, Black JA, Waxman SG. The Na(V)1.7 sodium channel: from molecule to man. Nat Rev Neurosci. 2013;14:49–62. doi: 10.1038/nrn3404. [DOI] [PubMed] [Google Scholar]
- 16.Eaton MJ, Plunkett JA, Karmally S, Martinez MA, Montanez K. Changes in GAD- and GABA- immunoreactivity in the spinal dorsal horn after peripheral nerve injury and promotion of recovery by lumbar transplant of immortalized serotonergic precursors. J Chem Neuroanat. 1998;16:57–72. doi: 10.1016/s0891-0618(98)00062-3. [DOI] [PubMed] [Google Scholar]
- 17.Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, Gilron I, Haanpaa M, Hansson P, Jensen TS, Kamerman PR, Lund K, Moore A, Raja SN, Rice AS, Rowbotham M, Sena E, Siddall P, Smith BH, Wallace M. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol. 2015;14:162–173. doi: 10.1016/S1474-4422(14)70251-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Finnerup NB, Sindrup SH, Jensen TS. The evidence for pharmacological treatment of neuropathic pain. Pain. 2010;150:573–581. doi: 10.1016/j.pain.2010.06.019. [DOI] [PubMed] [Google Scholar]
- 19.Flatters SJ, Xiao WH, Bennett GJ. Acetyl-L-carnitine prevents and reduces paclitaxel-induced painful peripheral neuropathy. Neurosci Lett. 2006;397:219–223. doi: 10.1016/j.neulet.2005.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fukuoka T, Tokunaga A, Kondo E, Miki K, Tachibana T, Noguchi K. Change in mRNAs for neuropeptides and the GABA(A) receptor in dorsal root ganglion neurons in a rat experimental neuropathic pain model. Pain. 1998;78:13–26. doi: 10.1016/S0304-3959(98)00111-0. [DOI] [PubMed] [Google Scholar]
- 21.Gangadharan V, Kuner R. Pain hypersensitivity mechanisms at a glance. Dis Model Mech. 2013;6:889–895. doi: 10.1242/dmm.011502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gracely RH, Lynch SA, Bennett GJ. Painful neuropathy: altered central processing maintained dynamically by peripheral input. Pain. 1992;51:175–194. doi: 10.1016/0304-3959(92)90259-E. [DOI] [PubMed] [Google Scholar]
- 23.Hao S, Mata M, Wolfe D, Huang S, Glorioso JC, Fink DJ. Gene transfer of glutamic acid decarboxylase reduces neuropathic pain. Ann Neurol. 2005;57:914–918. doi: 10.1002/ana.20483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kashiba H, Noguchi K, Ueda Y, Senba E. Neuropeptide Y and galanin are coexpressed in rat large type A sensory neurons after peripheral transection. Peptides. 1994;15:411–416. doi: 10.1016/0196-9781(94)90197-x. [DOI] [PubMed] [Google Scholar]
- 25.Katz NP, Mou J, Paillard FC, Turnbull B, Trudeau J, Stoker M. Predictors of Response in Patients with Post-herpetic Neuralgia and HIV-associated Neuropathy Treated with the 8% Capsaicin Patch (Qutenza(R)) Clin J Pain. 2014 doi: 10.1097/AJP.0000000000000186. [DOI] [PubMed] [Google Scholar]
- 26.King T, Porreca F. Preclinical assessment of pain: improving models in discovery research. Curr Top Behav Neurosci. 2014;20:101–120. doi: 10.1007/7854_2014_330. [DOI] [PubMed] [Google Scholar]
- 27.Kingery WS, Fields RD, Kocsis JD. Diminished dorsal root GABA sensitivity following chronic peripheral nerve injury. Exp Neurol. 1988;100:478–490. doi: 10.1016/0014-4886(88)90033-7. [DOI] [PubMed] [Google Scholar]
- 28.Knabl J, Witschi R, Hosl K, Reinold H, Zeilhofer UB, Ahmadi S, Brockhaus J, Sergejeva M, Hess A, Brune K, Fritschy JM, Rudolph U, Mohler H, Zeilhofer HU. Reversal of pathological pain through specific spinal GABAA receptor subtypes. Nature. 2008;451:330–334. doi: 10.1038/nature06493. [DOI] [PubMed] [Google Scholar]
- 29.Kuner R. Spinal excitatory mechanisms of pathological pain. Pain. 2015;156(Suppl 1):S11–17. doi: 10.1097/j.pain.0000000000000118. [DOI] [PubMed] [Google Scholar]
- 30.Lever I, Cunningham J, Grist J, Yip PK, Malcangio M. Release of BDNF and GABA in the dorsal horn of neuropathic rats. Eur J Neurosci. 2003;18:1169–1174. doi: 10.1046/j.1460-9568.2003.02848.x. [DOI] [PubMed] [Google Scholar]
- 31.Loeser JD, Ward AA., Jr Some effects of deafferentation on neurons of the cat spinal cord. Arch Neurol. 1967;17:629–636. doi: 10.1001/archneur.1967.00470300071012. [DOI] [PubMed] [Google Scholar]
- 32.Melzack R, Wall PD. Pain mechanisms: a new theory. Science. 1965;150:971–979. doi: 10.1126/science.150.3699.971. [DOI] [PubMed] [Google Scholar]
- 33.Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 2009;10:23–36. doi: 10.1038/nrn2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moore A, Derry S, Eccleston C, Kalso E. Expect analgesic failure; pursue analgesic success. BMJ. 2013;346:f2690. doi: 10.1136/bmj.f2690. [DOI] [PubMed] [Google Scholar]
- 35.Moore KA, Kohno T, Karchewski LA, Scholz J, Baba H, Woolf CJ. Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. J Neurosci. 2002;22:6724–6731. doi: 10.1523/JNEUROSCI.22-15-06724.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moore RA, Derry S, Wiffen PJ. Challenges in design and interpretation of chronic pain trials. Br J Anaesth. 2013;111:38–45. doi: 10.1093/bja/aet126. [DOI] [PubMed] [Google Scholar]
- 37.Munro G, Ahring PK, Mirza NR. Developing analgesics by enhancing spinal inhibition after injury: GABAA receptor subtypes as novel targets. Trends Pharmacol Sci. 2009;30:453–459. doi: 10.1016/j.tips.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 38.Nicholas CR, Chen J, Tang Y, Southwell DG, Chalmers N, Vogt D, Arnold CM, Chen YJ, Stanley EG, Elefanty AG, Sasai Y, Alvarez-Buylla A, Rubenstein JL, Kriegstein AR. Functional maturation of hPSC-derived forebrain interneurons requires an extended timeline and mimics human neural development. Cell Stem Cell. 2013;12:573–586. doi: 10.1016/j.stem.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Niesters M, Martini C, Dahan A. Ketamine for chronic pain: risks and benefits. Br J Clin Pharmacol. 2014;77:357–367. doi: 10.1111/bcp.12094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Polgar E, Hughes DI, Riddell JS, Maxwell DJ, Puskar Z, Todd AJ. Selective loss of spinal GABAergic or glycinergic neurons is not necessary for development of thermal hyperalgesia in the chronic constriction injury model of neuropathic pain. Pain. 2003;104:229–239. doi: 10.1016/s0304-3959(03)00011-3. [DOI] [PubMed] [Google Scholar]
- 41.Proudlock F, Spike RC, Todd AJ. Immunocytochemical study of somatostatin, neurotensin, GABA, and glycine in rat spinal dorsal horn. J Comp Neurol. 1993;327:289–297. doi: 10.1002/cne.903270210. [DOI] [PubMed] [Google Scholar]
- 42.Ross SE, Mardinly AR, McCord AE, Zurawski J, Cohen S, Jung C, Hu L, Mok SI, Shah A, Savner EM, Tolias C, Corfas R, Chen S, Inquimbert P, Xu Y, McInnes RR, Rice FL, Corfas G, Ma Q, Woolf CJ, Greenberg ME. Loss of inhibitory interneurons in the dorsal spinal cord and elevated itch in Bhlhb5 mutant mice. Neuron. 2010;65:886–898. doi: 10.1016/j.neuron.2010.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scholz J, Broom DC, Youn DH, Mills CD, Kohno T, Suter MR, Moore KA, Decosterd I, Coggeshall RE, Woolf CJ. Blocking caspase activity prevents transsynaptic neuronal apoptosis and the loss of inhibition in lamina II of the dorsal horn after peripheral nerve injury. J Neurosci. 2005;25:7317–7323. doi: 10.1523/JNEUROSCI.1526-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shields SD, Eckert WA, 3rd, Basbaum AI. Spared nerve injury model of neuropathic pain in the mouse: a behavioral and anatomic analysis. J Pain. 2003;4:465–470. doi: 10.1067/s1526-5900(03)00781-8. [DOI] [PubMed] [Google Scholar]
- 45.Sivilotti L, Nistri A. GABA receptor mechanisms in the central nervous system. Prog Neurobiol. 1991;36:35–92. doi: 10.1016/0301-0082(91)90036-z. [DOI] [PubMed] [Google Scholar]
- 46.Smith SB, Crager SE, Mogil JS. Paclitaxel-induced neuropathic hypersensitivity in mice: responses in 10 inbred mouse strains. Life Sci. 2004;74:2593–2604. doi: 10.1016/j.lfs.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 47.Southwell DG, Nicholas CR, Basbaum AI, Stryker MP, Kriegstein AR, Rubenstein JL, Alvarez-Buylla A. Interneurons from embryonic development to cell-based therapy. Science. 2014;344:1240622. doi: 10.1126/science.1240622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sugimoto T, Bennett GJ, Kajander KC. Transsynaptic degeneration in the superficial dorsal horn after sciatic nerve injury: effects of a chronic constriction injury, transection, and strychnine. Pain. 1990;42:205–213. doi: 10.1016/0304-3959(90)91164-E. [DOI] [PubMed] [Google Scholar]
- 49.Tsuda M, Beggs S, Salter MW, Inoue K. Microglia and intractable chronic pain. Glia. 2013;61:55–61. doi: 10.1002/glia.22379. [DOI] [PubMed] [Google Scholar]
- 50.Tsujino H, Kondo E, Fukuoka T, Dai Y, Tokunaga A, Miki K, Yonenobu K, Ochi T, Noguchi K. Activating transcription factor 3 (ATF3) induction by axotomy in sensory and motoneurons: A novel neuronal marker of nerve injury. Mol Cell Neurosci. 2000;15:170–182. doi: 10.1006/mcne.1999.0814. [DOI] [PubMed] [Google Scholar]
- 51.Ugarte SD, Homanics GE, Firestone LL, Hammond DL. Sensory thresholds and the antinociceptive effects of GABA receptor agonists in mice lacking the beta3 subunit of the GABA(A) receptor. Neuroscience. 2000;95:795–806. doi: 10.1016/s0306-4522(99)00481-9. [DOI] [PubMed] [Google Scholar]
- 52.Vaso A, Adahan HM, Gjika A, Zahaj S, Zhurda T, Vyshka G, Devor M. Peripheral nervous system origin of phantom limb pain. Pain. 2014;155:1384–1391. doi: 10.1016/j.pain.2014.04.018. [DOI] [PubMed] [Google Scholar]
- 53.Verge VM, Richardson PM, Wiesenfeld-Hallin Z, Hokfelt T. Differential influence of nerve growth factor on neuropeptide expression in vivo: a novel role in peptide suppression in adult sensory neurons. J Neurosci. 1995;15:2081–2096. doi: 10.1523/JNEUROSCI.15-03-02081.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vit JP, Ohara PT, Sundberg C, Rubi B, Maechler P, Liu C, Puntel M, Lowenstein P, Castro M, Jasmin L. Adenovector GAD65 gene delivery into the rat trigeminal ganglion produces orofacial analgesia. Mol Pain. 2009;5:42. doi: 10.1186/1744-8069-5-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain. 2011;152:S2–15. doi: 10.1016/j.pain.2010.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu Q, Cobos I, De La Cruz E, Rubenstein JL, Anderson SA. Origins of cortical interneuron subtypes. J Neurosci. 2004;24:2612–2622. doi: 10.1523/JNEUROSCI.5667-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang AL, Hao JX, Seiger A, Xu XJ, Wiesenfeld-Hallin Z, Grant G, Aldskogius H. Decreased GABA immunoreactivity in spinal cord dorsal horn neurons after transient spinal cord ischemia in the rat. Brain Res. 1994;656:187–190. doi: 10.1016/0006-8993(94)91383-8. [DOI] [PubMed] [Google Scholar]