

Abstract
Background
Ivermectin (IVM) is an antiparasitic agent that has been shown to reduce alcohol intake in mice, suggesting IVM as a potential treatment for alcohol use disorder (AUD). However, the safety profile of IVM administered in combination with an intoxicating dose of alcohol has not been characterized in humans.
Methods
This pilot project sought to provide the first clinical evidence that IVM could be repositioned as an AUD pharmacotherapy by examining 1) the safety of combining IVM (30 mg oral QD) with an intoxicating dose of intravenous alcohol (0.08 g/dl) and 2) the effects of IVM on alcohol cue-induced craving and subjective response to alcohol. Eleven individuals with AUD participated in a randomized, placebo-controlled, crossover study in which they received the study medication, participated in a cue exposure paradigm followed by intravenous alcohol administration, and remained in an inpatient unit overnight for observation.
Results
Ivermectin treatment, vs. placebo, did not increase the number or severity of adverse effects during alcohol administration or throughout the visit. However, IVM did not reduce cue-induced craving nor did it significantly affect subjective response to alcohol.
Conclusions
These results suggest that IVM (30 mg oral, QD) is safe in combination with an intoxicating dose of alcohol, but do not provide evidence that this dose of IVM is effective in reducing alcohol craving or its reinforcing effects. Given the preclinical data suggesting IVM is effective in reducing alcohol consumption in mice, additional studies testing larger samples and alternate dosing regimens are warranted to further characterize the potential efficacy of IVM as an AUD treatment.
Introduction
Of the 18 million people in the United States who suffer from alcohol use disorder (AUD), only an estimated 13% receive specialized treatment for their addiction (Litten et al., 2012; Miller et al., 2011). One factor that undoubtedly contributes to this low treatment rate is the limited number of medications approved by the Food and Drug Administration (FDA) to treat AUD. Further compounding this issue, these few available medications have only modest treatment efficacy (Rosner et al., 2010; Rösner et al., 2010). Recent reviews have outlined the National Institute on Alcohol Abuse and Alcoholism’s (NIAAA’s) vision to improve available pharmacological treatment options to treat AUD (Litten et al., 2012). One such strategy emphasized by the NIAAA is to identify and validate novel molecular targets that can be used to develop additional pharmacotherapies for AUD (Litten et al., 2012).
One novel target, the P2X receptor (P2XR), is a family of cation-permeable ligand gated ion channels activated by synaptically released extracellular adenosine 5′-triphosphate (ATP). This receptor family has garnered significant attention as a possible target for AUD pharmacotherapies [for review see (Franklin et al., 2014)]. Preclinical studies have shown that alcohol acts as a negative allosteric modulator for P2XRs, as low alcohol concentrations (~5 mM) can produce rapid inhibition of P2XR function (Asatryan et al., 2008; Franklin et al., 2014; Kidd et al., 1995; Li et al., 1993). Furthermore, p2rx4 gene expression in the brain of rats is negatively associated with alcohol consumption and preference (Kimpel et al., 2007; Tabakoff et al., 2009). These findings suggest that alcohol alters the function of P2X4Rs, and the P2X4R is involved in alcohol consumption in rodents.
Ivermectin (IVM), a semi-synthetic macrocyclic lactone, is an FDA-approved broad-spectrum antiparasitic avermectin (Geary, 2005). While IVM’s antiparasitic effects are attributed to action on a non-mammalian, glutamate-gated inhibitory chloride channel (Cully et al., 1994; Dent et al., 1997), IVM is also a selective positive allosteric modulator of P2X4Rs (Jelinkova et al., 2006; Silberberg et al., 2007) and acts on P2X4R sites that are thought to be modulated by alcohol (Asatryan et al., 2008; Popova et al., 2010). Recent evidence suggests that IVM blocks the inhibitory effect of alcohol in vitro (Asatryan et al., 2010) and is able to reduce alcohol intake and preference in mice due in part to its action on P2X4Rs (Wyatt et al., 2014; Yardley et al., 2012). The doses of IVM needed to produce these anti-alcohol effects in mice appear to be well-tolerated, safe, and show no evidence of abuse liability (Bortolato et al., 2013). Thus, IVM appears to be a promising, novel therapeutic for AUD.
Despite the promising results in rodents, the efficacy of IVM for the treatment of AUD has not been examined in humans. Additionally, few studies to date have investigated the effects of ethanol on IVM safety and pharmacokinetics. Retrospective self-reports of IVM and alcohol co-use have not found an increased association with serious adverse events (Takougang et al., 2008). Yet, a sub-intoxicating dose of ethanol (Shu et al., 2000) and IVM preparations in an alcoholic solution (Edwards et al., 1988) both increased IVM’s bioavailability, suggesting that alcohol could potentially influence IVM efficacy and adverse effects. Further complicating matters, preclinical studies have indicated that the dose of IVM required for the treatment of AUD may be at least twice as high as the 200 mcg/kg FDA-approved IVM dose (Yardley et al., 2012). Therefore, even though IVM has been shown to be safe and tolerable at 10 times the FDA-recommended dosing (Guzzo et al., 2002), the safety and pharmacokinetic effects of combining a higher-than-approved IVM dose with controlled alcohol administration still needs to be demonstrated before IVM can be developed as an AUD treatment.
Human laboratory models have been useful in medication development for AUD by measuring markers of safety and tolerability and elucidating the biobehavioral mechanisms by which pharmacotherapies may be efficacious (Plebani et al., 2012; Ray et al., 2010a; Ray et al., 2010b). Therefore, the primary goal of this randomized, double blind, placebo-controlled, crossover, human laboratory pilot study was to determine the safety and tolerability of administering IVM (30 mg oral, QD) in combination with an intoxicating dose of alcohol (0.08 g/dl). A secondary study goal was to examine the initial efficacy of IVM in reducing alcohol cue-induced craving and affecting subjective response to alcohol in a sample of non-treatment seeking individuals with AUD. Ivermectin pharmacokinetics were also assessed to ensure that study measures were administered at times corresponding to peak medication bioavailability. This pilot safety study represents the first step in the clinical development of IVM as a treatment for AUD.
Methods
The study protocol and all procedures were approved by the Institutional Review Board of the University of California, Los Angeles and conducted in accordance with the Declaration of Helsinki.
Participants and Screening Procedures
A community sample of non-treatment seeking drinkers was recruited via online and print advertisements in the Los Angeles area. Interested individuals called the laboratory to complete a preliminary telephone-screening interview used to assess general eligibility requirements. Inclusion criteria included the following: 1) aged between 21 and 65 years; 2) met DSM-V criteria for current alcohol use disorder; 3) consumed 48 or more drinks per month; and 4) fluent in English. Exclusion criteria consisted of the following: 1) were currently in a treatment program for alcohol problems, had been in treatment for alcohol use in the 30 days before study enrollment, or were seeking treatment for alcohol use; 2) reported clinically significant alcohol withdrawal symptoms on the Clinical Institute Withdrawal Assessment of Alcohol Scale, Revised; 3) self-reported use of any non-prescription drugs (excluding marijuana) or met DSM-V criteria for a non-alcohol substance use disorder; 4) self-reported diagnosis of or met DSM-V criteria for any psychiatric disorders; 5) being pregnant, as verified by a urine pregnancy test; 6) having a body mass index (BMI) less than 18.5 or greater than 30; and 7) reporting any medical conditions or medications that would be contraindicated with taking IVM.
Eligible individuals were invited to the laboratory for an in-person screening visit after a 24-hour abstinence period, where they received a full explanation of study procedures and provided written, informed consent. After consenting, participants were required to blow into a Breathalyzer to demonstrate a breath alcohol concentration (BrAC) of 0.000g/dl, and urine toxicology and pregnancy tests were performed. Participants who tested positive for alcohol, drug use, or pregnancy were excluded from participation. Participants then completed a number of baseline questionnaires and interviews, outlined in the “Measures” section below. Participants received $40 for participating in the screening visit.
Participants deemed eligible following the in-person screening visit were invited to complete a physical exam with the study physician, consisting of medical history, a clinical laboratory panel, and BMI calculation, in order to ensure medical eligibility for the study. Individuals who passed the physical exam were invited to participate in the experimental procedures, detailed below. Participants received $20 for participating in the medical screening visit. A total of 74 participants (20% female) completed the in-person screening visit, 27 of whom were eligible. Seventeen participants were screened by the physician for medical eligibility, 3 of whom were ineligible and 3 of whom decided not to continue with the experimental procedures. Eleven individuals (n = 2 female) were randomized to a medication sequence, and all eleven participants completed both experimental sessions.
Experimental Procedures
Eligible participants completed two experimental sessions in a randomized, counterbalanced, and crossover fashion at the UCLA Clinical and Translational Research Center (CTRC). Participants were asked to abstain from drinking alcohol for 24 hours prior to scheduled sessions; they were also asked to fast the morning of the session. After participants arrived at the laboratory at 8:00 AM, abstinence from alcohol and recreational drugs (excluding marijuana) was immediately verified via Breathalyzer and urine toxicology screen, respectively. Women also provided a negative urine pregnancy test at this time. At 9:00 AM, participants received a single 30 mg dose of IVM (or matched placebo), administered by CTRC nursing staff. Participants ate calorie-controlled, standardized meals at 8:00 AM (breakfast), 11:30 AM (lunch), and 2:30 PM (snack) totaling 70% of daily estimated kilocalories. At regular intervals throughout the day, participants reported subjective adverse effects and alcohol craving and provided blood samples for IVM pharmacokinetics (PK) analysis. In the afternoon, participants completed cue reactivity and alcohol infusion paradigms. Participants who were smokers were allowed smoke breaks as needed until the start of the cue reactivity procedure in order to avoid potential nicotine withdrawal effects on mood.
At 3:00 PM (i.e., 6 hours after medication administration), participants completed a cue reactivity paradigm, following well-established guidelines (Monti et al., 1987; Monti et al., 2001). During this task, participants were asked to listen to a 5 minute guided cue exposure script, during which they were exposed to both a neutral beverage (a glass of water) and their preferred alcoholic beverage in a fixed order to avoid carryover effects. Prior to beginning the paradigm and after each cue exposure, participants completed a questionnaire to assess alcohol craving.
Following the cue reactivity paradigm, at 3:45 PM, participants completed an alcohol infusion session (O’Connor et al., 1998; Ray et al., 2012; Ray et al., 2013). Participants were seated in a reclining chair, and a 6% ethanol solution was administered intravenously in their non-dominant arm. Alcohol was administered intravenously in order to effectively control blood alcohol levels, at the following rates, taking into account participant’s sex and weight: for males, 0.166 ml/min × weight (kg); for females, 0.126 ml/min × weight (kg). Target BrACs were 0.020 g/dl, 0.040g/dl, 0.060g/dl, and 0.080g/dl. At each target BrAC, the infusion rate was reduced to half in order to maintain a stable BrAC level, and participants then completed a battery of subjective measures. Heart rate and blood pressure were also recorded at each target BrAC. After completion of the alcohol infusion and removal of the IV line, participants also completed subjective measures during the descending limb of intoxication at BrAC levels of 0.060g/dl and 0.040g/dl.
Participants were provided a meal following the alcohol infusion procedures and were monitored by CTRC nursing staff overnight. After overnight observation, participants were discharged the following morning (Day 2), returned again the next day (Day 3) for a final blood draw and to complete follow-up questionnaires, and were contacted by phone 7 days after the experimental session to assess potential adverse effects after Day 3. At their second Day 3 visit, participants received an individual session of motivational interviewing, delivered by a licensed clinical psychologist or a clinical psychology PhD student under the supervision of a licensed clinician. Experimental sessions were scheduled at least 7 days apart in order to avoid any carryover effects (17.8 ± 10.9 days, mean ± SD). Participants received $340 for completing all experimental procedures.
Measures
Screening Measures
Participants completed a battery of assessments during the screening process that assessed basic demographics (e.g., age, education), drinking and drug use behavior (e.g., Alcohol Use Disorder Identification Test; AUDIT (Reinert and Allen, 2002)), and depressive symptoms (e.g., Beck Depression Inventory-II; BDI;(Beck, 1996)). The following interviews were also performed: 1) 30-Day Timeline Follow-Back (TLFB;(Sobell and Sobell, 1992)), used to obtain estimates of daily alcohol, cigarette, and marijuana use; 2) Structured Clinical Interview for DSM-V (SCID; (First et al., 1995)), used to assess for AUD and other exclusion criteria, such as non-alcohol substance use or other current psychiatric disorders; and 3) Clinical Institute Withdrawal Assessment of Alcohol Scale, Revised (CIWA-AR; (Sullivan et al., 1989)), a ten-item scale to assess for alcohol withdrawal. All clinical interviews were conducted by masters’ level clinicians under the PI’s supervision.
Arrival and Daily Measures
On the day of each experimental session, upon arrival to the laboratory and before medication administration, baseline depressive symptoms (BDI), anxiety symptoms (Beck Anxiety Inventory; (Beck and Steer, 1990)), alcohol craving (Penn Alcohol Craving Scale; (Flannery et al., 1999)), and overall mood (Profile of Mood States; POMS; (Curran et al., 1995).) were assessed. After medication administration (9:00 AM), alcohol craving and adverse effects were regularly assessed until the start of the cue reactivity paradigm (3:00 PM, 6 hours post-medication). Craving was repeatedly assessed at 0, 2, 4, and 6 hours after medication administration using the Alcohol Urge Questionnaire (AUQ; (Bohn et al., 1995)), which consisted of eight items associated with urge to drink alcohol, rated on a seven-point scale (1=Strongly Disagree, 7=Strongly Agree). Potential medication-related adverse effects were repeatedly assessed using the Systematic Assessment for Treatment Emergent Events (SAFTEE;(Jacobson et al., 1986)) at 0, 1, 2, 4, and 6 hours after medication administration. The SAFTEE is a 24-item checklist in which the participant can identify whether a symptom is present (yes/no), its severity (mild, moderate, severe), and whether it was caused by the medication (yes/no). Participants also completed the AUQ and SAFTEE 24 and 48 hours (Days 2 and 3, respectively) after medication administration and were administered the SAFTEE over the phone 7 days after the experimental session day to monitor whether the participants had experienced any adverse effects after their Day 3 visit.
Cue Reactivity and Alcohol Infusion
Participants completed the AUQ during the cue reactivity paradigm (baseline, post-water cue, and post-alcohol cue). The following measures were administered during the alcohol infusion sessions (0.000 g/dl, 0.020 g/dl, 0.040 g/dl, 0.060 g/dl, and 0.080 g/dl): 1) The AUQ; 2) The Biphasic Alcohol Effects Scale (BAES;(Martin et al., 1993)), a fourteen item scale designed to capture the stimulant and sedative effects of alcohol, each rated on an eleven-point scale (0=Not at all, 10=Extremely); 3) The Drug Effects Questionnaire (DEQ;(Johanson and Uhlenhuth, 1980)), a four item questionnaire that captures subjective effects using the questions “Do you feel any drug effects?”, “Do you like the effects you are feeling right now?”, “Would you like more of the drug right now?”, and “Are you high?” each rated on an eleven-point scale; 4) an 8 item version of the POMS, consisting of eight adjectives rated on a five-point scale (0=Not at all, 4=Extremely) designed to capture four dimensions of mood: tension (consisting of uneasy and anxious), vigor (lively, energetic), negative affect (downhearted, discouraged), and positive affect (cheerful, joyful); and 5) the Subjective High Assessment Scale (SHAS;(Schuckit, 1984)), consisting of 13-items on a 10-point Likert scale ranging from “not at all” to “extremely” used to assess subjective feelings of alcohol intoxication. The SAFTEE was administered at BrAC levels of 0.00 g/dl, 0.040 g/dl, and 0.080 g/dl.
PK Sampling Procedures and Measures
Blood samples (4 ml) were collected from participants’ non-dominant arm at 0, 0.5, 1, 2, 4, 6, 8, 10, 12, 16, 24, and 48 hours post-IVM administration. Plasma samples were analyzed as previously described, where the lowest level of quantification was 5 ng/mL with a range of 1 ng/mL–1000 ng/mL and an accuracy of 90–110% (Yardley et al., 2012). Pharmacokinetic (PK) parameters were calculated using a non-compartmental analysis: 1) Maximum plasma concentration (Cmax; ng/mL) for each subject, 2) area under the curve (AUC) from 0 to 48 hours after IVM administration, 3) time to Cmax (Tmax, time), and 4) half-life (T½ hours).
IVM Dose Selection and Timing of Measures
A single IVM dose of 30 mg was chosen to ensure an optimal balance of participant safety and drug efficacy. Acute administration of a single dose of IVM, ranging from 2.5 – 10 mg/kg, reduced alcohol consumption and preference in mice, with maximum efficacy observed between 5 – 10 mg/kg (Yardley et al., 2012). Also, the lowest IVM dose that was detectable IVM in the brains of mice and reduced alcohol consumption was 2.5 mg/kg (Yardley et al., 2012). In humans, IVM is FDA-approved for a single dose of 200 mcg/kg (e.g., ~13.6 mg for a 150 lb. person) but has been shown to be safe up to 120 mg (Guzzo et al., 2002). However, no human laboratory studies have co-administered an intoxicating dose of alcohol and IVM. Therefore, for this initial pilot study, we selected the lowest dose of IVM that was both effective in reducing alcohol consumption in murine studies (i.e., 3.1 mg/kg; Yardley et al., 2012) and also shown to be safe and tolerable in humans (Guzzo et al., 2002). Using allometric scaling (Anderson and Holford, 2009), the human equivalent dose to 3.1 mg/kg in mice was determined to be 30 mg. The UCLA Research Pharmacy provided IVM and encapsulated the medication into one 30 mg capsule.
The peak effects of IVM in reducing alcohol intake in mice corresponded to the Cmax and Tmax, which occurred ~8 hours after IVM administration (Yardley et al., 2012). As IVM pharmacokinetics are comparable between mice and humans (Guzzo et al., 2002; Yardley et al., 2012), the timing of the alcohol-cue reactivity paradigm was chosen to begin at the time when IVM is likely to be producing central effects (~6 hrs after IVM administration) and the entirety of the alcohol infusion procedures was chosen to correspond with projected peak central effects of 30 mg IVM (i.e., ~7 – 9 hours after IVM administration).
Statistical Analysis
Repeated measures analyses of variance (ANOVA) were used to analyze the effects of IVM on cue-induced craving and subjective response to alcohol infusion. Repeated measures ANOVA were also used to compare arrival mood and craving measures, BrAC levels during infusion, and all repeated daily mood, adverse effect, and craving measures. All repeated measures ANOVAs included dose (IVM or placebo) as a within subject factor. Time was additionally included as a within subject factor, with varying levels, for the following analyses: 1) Post-medication/pre-cue exposure measures: SAFTEE, five levels; AUQ, four levels; 2) Cue exposure: AUQ three levels; 3) Alcohol infusion: SAFTEE, three levels; AUQ, SHAS, DEQ, and BAES, six levels; 4) Post-alcohol infusion, descending limb: AUQ, SHAS, DEQ, and BAES, two levels. The SAFTEE was analyzed as 1) number of adverse effects reported per timepoint (potential range: 0 – 24) and 2) the severity of each reported adverse effect per timepoint (potential range: 1 – 3). The severity of an adverse effect was only analyzed if an adverse effect was reported at that timepoint, resulting in an unequal N for the severity measure across all timepoints for each subject. Because of this, adverse event severity was analyzed separately using individual t-tests for each timepoint. Covariates were considered (e.g., sex, age) but ultimately rejected because in the crossover design participants serve as their own controls. An alpha threshold of 0.05 was set for all statistical analyses. Significant effects were explored with simple effects post hoc testing. All analyses were performed with SPSS 22 for Windows. Power and effect size calculations were performed with SPSS and G*Power version 3.1.9.2. Based on the sample of 11 participants, the current study had 80% power to detect an effect size between 0.2 and 0.4 (Cohen’s f, medium effect size) for subjective measures assessed during the cue reactivity and alcohol infusion paradigms (within participant correlations between 0.63 and 0.92). Cohen’s f is presented in the results section as an index of effect size (f values of 0.10, 0.25, and 0.40 represent small, medium, and large effect sizes, respectively) for outcomes related to the alcohol cue reactivity and alcohol infusion paradigms.
Results
Sample Characteristics and Arrival Measures
Sample characteristics for the 11 participants are reported in Table 1. There were no significant differences between sessions in mood or craving upon arrival to the laboratory (Table 2).
Table 1.
Sample Characteristics
| Variable | Mean (SD) or % | Range |
|---|---|---|
| Age | 38.82 (11.39) | 24 – 58 |
| Sex - % Male | 82% | – |
| Ethnicity - % Caucasian | 46% | – |
| Education (years) | 14.18 (2.27) | 10 – 16 |
| BMI (kg/m2) | 23.90 (2.58) | 20.78 – 29.01 |
| BDI - II | 5.45 (4.61) | 0 – 14 |
| AUDIT | 20.18 (6.16) | 9 – 29 |
| CIWA-AR | 1.55 (2.07) | 0 – 6 |
| DSM-V Current AUD Severity | 2.00 (0.90) | 1 – 3 |
| Number of Drinking Daysa | 18.91 (6.25) | 8 – 27 |
| Drinks per Drinking Daya | 8.90 (3.67) | 3.47 – 14.78 |
| Heavy Drinking Daysa | 13.36 (3.67) | 4 – 24 |
| Cigarette Smokers - % | 55% | – |
| Cigarettes per Daya | 8.62 (6.16) | 2 – 20 |
| Marijuana Smokers - % | 45% | – |
| Number of Days Smoking Marijuanaa | 17.40 (10.60) | 2 – 30 |
Data obtained at behavioral screen.
AUDIT – Alcohol Use Disorder Identification Test; BMI – Body Mass Index; BDI – Beck Depression Inventory; CIWA – Clinical Institute Withdrawal Assessment of Alcohol Scale, Revised
over past 30 days, as determined by TLFB interview
Table 2.
Arrival Measures and Infusion BrAC levels
| Ivermectin | Placebo | |
|---|---|---|
| Arrival Measures | ||
| BAI | 1.64 (2.73) | 0.45 (0.82) |
| BDI | 3.00 (4.22) | 3.00 (4.15) |
| PACS | 13.82 (6.54) | 12.45 (6.64) |
| POMS | ||
| Tension | 0.73 (0.43) | 0.79 (0.79) |
| Vigor | 1.64 (0.69) | 1.36 (0.91) |
| Negative Affect | 0.55 (0.42) | 0.57 (0.32) |
| Positive Affect | 2.54 (1.28) | 2.04 (0.92) |
| Target BrAC (g/dl) | ||
| Infusion | ||
| 0.02 | 0.022 (0.002) | 0.022 (0.003) |
| 0.04 | 0.041 (0.002) | 0.041 (0.013) |
| 0.06 | 0.063 (0.002) | 0.062 (0.002) |
| 0.08 | 0.083 (0.002) | 0.081 (0.001) |
| Post-infusion | ||
| 0.06 | 0.057 (0.003) | 0.059 (0.003) |
| 0.04 | 0.040 (0.002) | 0.040 (0.003) |
| Time to reach target BrAC from previous timepoint (minutes) | ||
| Infusion | ||
| 0.02 | 13 (2) | 12 (2) |
| 0.04 | 18 (4) | 18 (4) |
| 0.06 | 26 (4) | 25 (5) |
| 0.08 | 29 (9) | 28 (5) |
| Post-infusion | ||
| 0.06 | 21 (9) | 22 (6) |
| 0.04 | 55 (21) | 58 (27) |
Data are mean (SD)
BAI – Beck Anxiety Inventory; BDI – Beck Depression Inventory; PACS – Penn Alcohol Craving Scale; POMS – Profile of Mood States, 40 item version POMS was assessed at 9:00 AM, all other Arrival Measures were collected at 8:00 AM.
PK Analysis
Average IVM concentrations across the study timepoints are reported in Figure 1. Mean Cmax was 406.03 ± 398.36 ng/mL (mean ± SD) leading to a mean IVM exposure over 0 to 48 hours (AUC0–48) 5078 ± 4258 (ng*hr/mL). Peak concentration (Tmax) was found 9.09 ± 3.62 hrs after IVM administration, where mean t½ was 15.75 ± 6.86 hrs.
Figure 1. Ivermectin Pharmacokinetics.
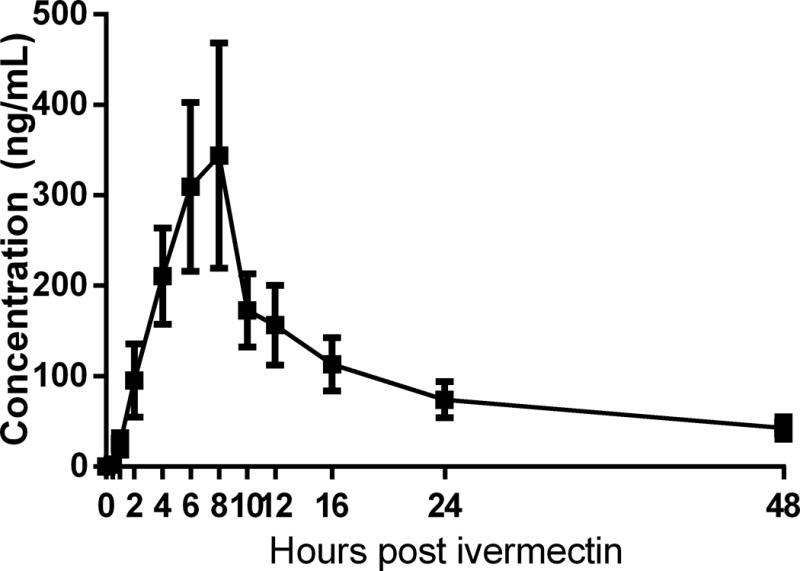
Mean (± SEM) IVM concentrations at baseline (0), 0.5, 1, 2, 4, 6, 8, 10, 12, 16, 24, and 48 hours post-IVM administration. Additional PK measures (means ± SD) are as follows: Cmax = 406.03 ± 398.36 ng/mL; AUC = 5078 ± 4258; Tmax = 9.09 ± 3.62 hrs; T½ = 15.75 ± 6.86 hrs.
Cue-Reactivity and Alcohol Infusion
The alcohol cue significantly increased AUQ craving compared to baseline and the water cue (Time main effect: F(2, 9) = 16.7, p < 0.001, Cohen’s f = 1.30; Post hoc: Alcohol cue > Baseline and Water cue, p < 0.01). IVM, vs. placebo, however, did not affect this response (p = 0.99, Cohen’s f = 0.00; Figure 2).
Figure 2. Alcohol Cue-Reactivity.
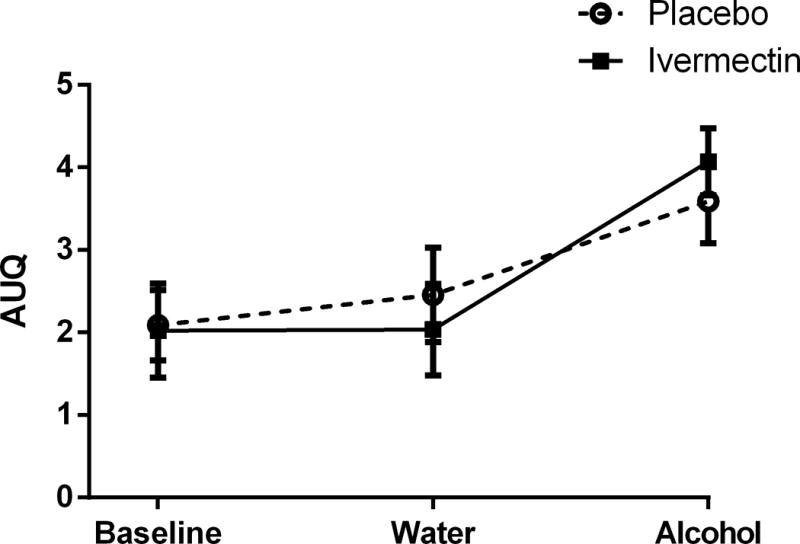
The alcohol cue significantly increased AUQ craving compared to baseline and the water cue. However, IVM, vs. placebo, did not affect AUQ craving during the cue-reactivity paradigm. Data are presented as mean (± SEM).
There were no significant differences between target infusion and post-infusion BrAC levels or time to reach BrAC targets between sessions (Table 2). Table 3 lists the number of individual adverse effects experienced during alcohol infusion as measured by the SAFTEE. During alcohol infusion, the average number of reported adverse effects (p = 0.71, Cohen’s f = 0.12) and severity of each reported adverse effect (p’s > 0.44, Cohen’s d’s > 0.01 and < 0.47) did not differ between IVM and placebo sessions (Figure 3). Alcohol infusion increased DEQ “like,” “feel,” “high,” and “more,” and SHAS intoxication and decreased POMS tension (Time main effects, p’s < 0.05, Cohen’s f’s > 0.66 and < 1.70; Figure 4), but did not affect AUQ craving, BAES stimulation or sedation, or any other POMS subscale (p’s > 0.09, Cohen’s f’s > 0.25 and < 0.47). However, IVM, vs. placebo, did not affect subjective response on any measure during alcohol infusion or during the post-infusion, declining BrAC timepoints (p’s > 0.19, Cohen’s f’s > 0.05 and < 0.20). None of the sample characteristics in Table 1 were significant covariates of the responses to IVM.
Table 3.
Number of individual SAFTEE adverse effects reported during alcohol infusion
| Adverse Effect | Placebo | Ivermectin | ||||
|---|---|---|---|---|---|---|
| 0.00 | 0.04 | 0.08 | 0.00 | 0.04 | 0.08 | |
| 1. Abdominal pain or cramps | 0 | 1 | 1 | 0 | 0 | 0 |
| 2. Yellow eyes | 0 | 0 | 0 | 0 | 0 | 0 |
| 3. Nausea or vomiting | 0 | 0 | 0 | 0 | 0 | 0 |
| 4. Irritability or anger | 1 | 0 | 0 | 0 | 0 | 0 |
| 5. Increased desire for sex | 1 | 0 | 1 | 1 | 2 | 1 |
| 6. Nervousness | 1 | 0 | 0 | 1 | 0 | 0 |
| 7. Ringing in the ears | 0 | 0 | 0 | 0 | 0 | 0 |
| 8. Decrease in appetite | 0 | 0 | 0 | 0 | 0 | 0 |
| 9. Depression | 0 | 0 | 0 | 0 | 0 | 0 |
| 10. Fatigue | 1 | 2 | 2 | 1 | 1 | 2 |
| 11. Difficulty in staying awake | 3 | 3 | 4 | 3 | 2 | 2 |
| 12. Increase in appetite | 0 | 1 | 3 | 1 | 2 | 4 |
| 13. Blurred vision | 1 | 1 | 1 | 0 | 1 | 1 |
| 14. Drowsiness | 2 | 4 | 2 | 2 | 4 | 5 |
| 15. Headache | 0 | 0 | 0 | 0 | 1 | 0 |
| 16. Night sweats | 0 | 0 | 0 | 0 | 0 | 0 |
| 17. Mental confusion | 0 | 0 | 0 | 0 | 0 | 0 |
| 18. Anxiety | 0 | 0 | 0 | 0 | 1 | 0 |
| 19. Joint or muscle pain | 0 | 0 | 0 | 0 | 0 | 0 |
| 20. Dizziness | 0 | 0 | 0 | 0 | 1 | 2 |
| 21. Sexual problems | 0 | 0 | 0 | 0 | 0 | 0 |
| 22. Difficulty sleeping | 0 | 0 | 0 | 0 | 0 | 0 |
| 23. Fever or chills | 0 | 0 | 0 | 0 | 0 | 0 |
| 24. Decreased desire for sex | 0 | 0 | 0 | 0 | 0 | 0 |
Figure 3. Adverse Effects During Alcohol Infusion.
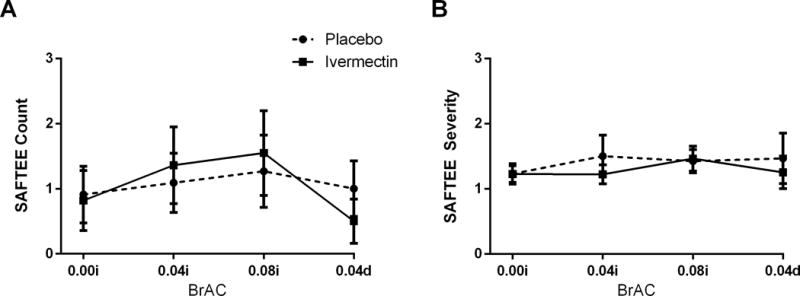
The average number and severity of reported adverse effects on the SAFTEE was not affected by alcohol and did not differ between IVM and placebo sessions. On the x-axis, 0.00i, 0.04i, and 0.08i refer to BrAC during alcohol infusion, while 0.04d refers to BrAC post-alcohol infusion. Data are presented as mean (± SEM).
Figure 4. Subjective Response to Alcohol.
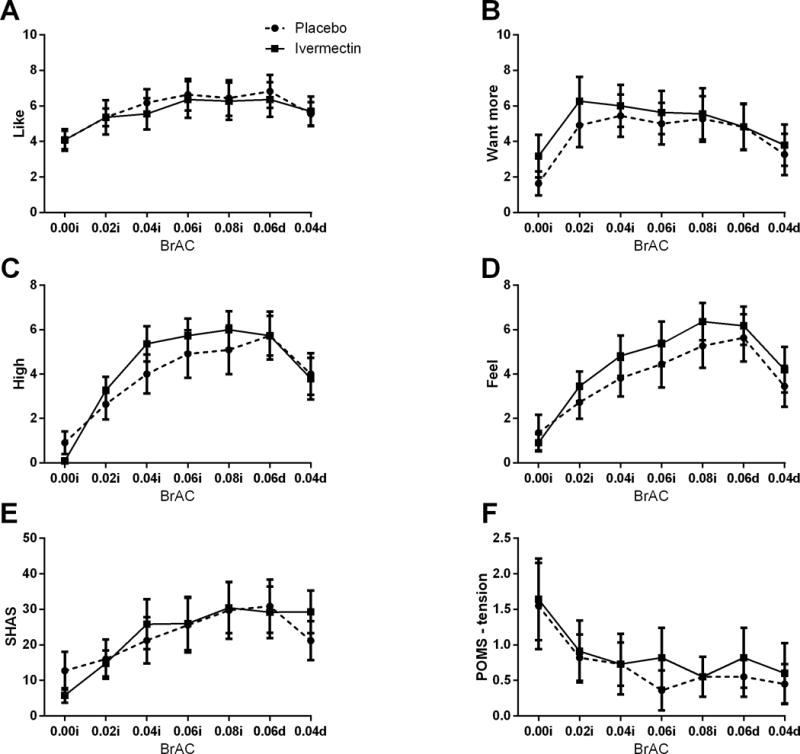
Alcohol infusion increased DEQ “like,” “feel,” “high,” and “more,” and SHAS intoxication and decreased POMS tension. On the x-axis, 0.00i, 0.02i, 0.04i, and 0.08i refer to BrAC during alcohol infusion, whereas 0.06d and 0.04d refer to BrAC post-alcohol infusion. Data are presented as mean (± SEM).
Daily Measures
For the first six hours post-medication administration, IVM, vs. placebo, did not affect AUQ craving, nor the average number or severity of reported adverse effects. Similarly, IVM, vs. placebo, did not affect AUQ craving or either SAFTEE outcome 24 and 48 hours post medication administration. Finally, the average number and severity of reported adverse effects collected seven days after the experimental session did not differ between IVM and placebo sessions.
Discussion
The current pilot laboratory study was the first to examine the safety and initial efficacy of co-administration of a single 30 mg dose of IVM and intravenous alcohol infusion. This dose of IVM was safe and well tolerated both on its own and during the alcohol infusion. The number and severity of reported adverse effects were low and did not differ from the placebo session. Conversely, IVM showed no efficacy at this dose in reducing alcohol cue-induced craving or basal alcohol craving throughout the day, nor did it affect subjective response to alcohol infusion. These findings are an important first step in developing IVM as a treatment for AUD and may inform future studies testing IVM or other members of the avermectin family as potential treatments for AUD.
The primary goal of this phase 1 pilot study, to establish the safety of IVM (30 mg) independently and in combination with alcohol, was successful. Confirming the safety of a novel medication is a required, and often difficult, first step in the clinical development of any drug (Litten et al., 2012). A fundamental challenge in translational medication development is transitioning a novel medication from preclinical research to successfully testing the medication in a phase 1 trial (Litten et al., 2012). The gap between preclinical and phase 1 testing, often referred to as the “Valley of Death,” has obstructed progression in the development of numerous promising novel medications (Litten et al., 2012). Therefore, establishing the safety and tolerability combining IVM at a dosage above the FDA-approved quanity with an intoxicating dose of alcohol is a necessary landmark in developing IVM for treating AUD.
Ivermectin did not reduce basal or cue-induced craving and failed to affect subjective response to alcohol, all of which are considered potential markers of medication efficacy (Ray et al., 2010b). There are several potential factors that may have contributed to these null results, but it seems particularly likely that a higher IVM dose may be needed to produce anti-alcohol effects in humans. As the maximum IVM concentration (Cmax: 406.03 ± 398.36 ng/mL) occurred at a time (Tmax: 9.09 ± 3.62 hrs) when subjective response to alcohol was being measured, we would have expected to detect a medication effect if this IVM dose was effective. Taken together, these results suggest that the 30 mg IVM dose administered in the present study, while higher than the FDA-approved dose, may not have been high enough to affect response to alcohol and alcohol craving. In rodents, single, acute IVM doses, ranging from 2.5 – 10 mg/kg, reduced alcohol consumption and preference, with maximum efficacy observed between 5 – 10 mg/kg (Yardley et al., 2012). The absence of studies in the literature reporting the safety of co-administering an intoxicating dose of alcohol and IVM led us to select the lowest IVM dose that produced anti-alcohol effects in mice and was also safe in humans (Guzzo et al., 2002). However, given the safety profile of IVM that was demonstrated in the present study and by others (Guzzo et al., 2002), future studies should consider examining a higher dose of IVM that is comparable to the optimal range of 5 – 10 mg/kg observed in mice. An equivalent dose within this range may increase the likelihood of detecting anti-alcohol use effects without producing safety and tolerability concerns.
An additional factor that might have contributed to null efficacy findings may relate to the choice of measures administered in the current study. Forced alcohol administration and alcohol cue-reactivity are reliable methods used to measure subjective response to alcohol and alcohol craving, respectively (Ray et al., 2016). However, several promising pharmacotherapies for AUD have not reduced the positive subjective effects of alcohol in the laboratory (reviewed elsewhere, e.g. Ray et al., 2016). Thus, IVM could feasibly have utility in treating AUD without affecting subjective response to alcohol. For example, it may instead act by reducing motivation to consume alcohol or alleviating protracted withdrawal symptoms. The current design would not capture such mechanisms of action. Given that IVM reduces alcohol self-administration in rodents (Yardley et al., 2012) and is also a positive allosteric modulator at GABA-A receptors (Bortolato et al., 2013), both aforementioned behavioral markers may be sensitive to the effects of IVM. To capture the full translational potential of IVM, future studies should consider employing both a higher dose of IVM and including additional paradigms, such as an alcohol self-administration session, in their design.
A comparison of the IVM pharmacokinetic parameters that were obtained in this study with other human and rodent IVM studies may also aid in the interpretation of the present findings. Our IVM pharmacokinetic results in humans were generally similar to those reported in a mouse study that found IVM was effective in reducing alcohol consumption (Yardley et al., 2012); however, the total IVM exposure (as measured by AUC) in the current study was comparatively lower, which may in part explain the discrepant medication effects between the two studies (Yardley et al., 2012). In comparison to human study that administered a single 30 mg IVM dose (Guzzo et al., 2002), the Cmax (~56% greater) and Tmax (~97% greater) reported in the present study were considerably greater than previously reported values. One possibility for the noticeable discrepency between these values is that prior studies have found alcohol increases plasma concentration of IVM (Edwards et al., 1988; Shu et al., 2000). Although the increase in IVM bioavailability did not correspond to increases in adverse effects or medication efficacy, future alcohol studies administering IVM doses greater than 30 mg should consider this altered pharmacokinetic profile.
The results of the present study should be examined in light of its strengths and limitations. The strengths of the study include its highly translational nature, its within subject and crossover design, and its use of well-established human laboratory paradigms with putative clinical significance as outcome measures (e.g., alcohol cue-exposure and alcohol infusion). Additionally, all experimental procedures being conducted in a highly controlled inpatient environment allowed the regular assessment of adverse effects for 24 continuous hours. As with all pilot studies, the primary study limitation is a small sample size that potentially limited our ability to detect significance. While the crossover design may have mitigated some loss in statistical power, the high variability in pharmacotherapy response that is typically observed in such studies may have eliminated these gains. The alcohol infusion did not significantly change ratings of BAES stimulation or sedation, AUQ craving, and POMS negative mood, positive mood, or vigor despite producing medium effect sizes on these outcomes (Cohen’s f’s > 0.25 and < 0.47), which may be indicative of a lack of statistical power. Importantly, this lack of alcohol effect could have contributed to null IVM effects on many of these measures (i.e., if there is no alcohol effect, there is no effect for IVM to augment or diminish). Yet, alcohol did produce sizeable increases on the SHAS and DEQ want, feel, like, and high items, as well as a decrease on POMS tension (Cohen’s f’s > 0.66 and < 1.70, large effect sizes), providing several measures that could have captured potential IVM effects. Furthermore, IVM produced only small, nonsignificant effects on subjective response to alcohol or to alcohol-related cues (Cohen’s f’s > 0.00 and < 0.20) and, therefore, we are confident that the null IVM findings reported in the manuscript were primarily due to a lack of medication effect rather than a lack of alcohol effect or low statistical power.
In summary, the current pilot study found that IVM (30 mg QD) was safe in combination with an intoxicating dose of alcohol but did not display efficacy in reducing alcohol craving or affecting subjective response to alcohol. We advise against interpreting the null initial efficacy results of this pilot study as an indication that IVM is not a promising AUD pharmacotherapy or that the P2XR family is not a promising target for treating this disorder. Indeed, other members from IVM’s class of drugs, such as abamectin, have also been shown to decrease alcohol intake and preference in mice through actions at the P2X4R (Asatryan et al., 2014). The study and its results as a whole are promising for several reasons: it’s an excellent example of a translational study, it provides support for the safety of IVM, and it identified methodological changes that future studies should employ when testing this medication for AUD (e.g., higher dosage, additional measures). These strengths speak to the importance of using human laboratory studies to effectively translate preclinical findings and fostering working collaborations between preclinical and clinical scientists to facilitate the development of novel treatments for AUD. Given the paucity and limited efficacy of available pharmacotherapies, as well as IVM’s strong preclinical findings and its safety and tolerability, IVM and other avermectins warrant investigation as potential AUD pharmacotherapies.
Acknowledgments
Funding sources: This work was supported by the following grants: UCLA CTSI UL1TR000124 (L.A.R.), USC CTSI NIH/NCRR/NCATS UL1TR000130, AA022448 (D.L.D.), F32 AA023449 (M.M.Y.), TRDRP 23FT-0102 (D.J.O.R.), as well as the USC School of Pharmacy.
Footnotes
Conflicts of interest: Lara Ray is a paid consultant for GSK and has received medication from Pfizer and Medicinova. Daryl Davies is an inventor on a patent for the use of IVM for the treatment of AUD.
References
- Anderson BJ, Holford NH. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab Pharmacokinetics. 2009;24:25–36. doi: 10.2133/dmpk.24.25. [DOI] [PubMed] [Google Scholar]
- Asatryan L, Popova M, Perkins DI, Trudell JR, Alkana RL, Davies DL. Ivermectin antagonizes ethanol inhibition in P2X4 receptors. J Pharmacol Exp Ther. 2010;334:720–728. doi: 10.1124/jpet.110.167908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asatryan L, Popova M, Woodward JJ, King BF, Alkana RL, Davies DL. Roles of ectodomain and transmembrane regions in ethanol and agonist action in purinergic P2X2 and P2X3 receptors. Neuropharmacology. 2008;55:835–843. doi: 10.1016/j.neuropharm.2008.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asatryan L, Yardley MM, Khoja S, Trudell JR, Hyunh N, Louie SG, Petasis NA, Alkana RL, Davies DL. Avermectins differentially affect ethanol intake and receptor function: Implications for developing new therapeutics for alcohol use disorders. Int J Neuropsyhcopharmacol. 2014;17:907–916. doi: 10.1017/S1461145713001703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck AT, Steer RA. Manual for the Beck anxiety inventory 1990 [Google Scholar]
- Beck AT, Steer RA, Brown G. Manual for the Beck Depression Inventory-II. San Antonio, TX: 1996. [Google Scholar]
- Bohn MJ, Krahn DD, Staehler BA. Development and initial validation of a measure of drinking urges in abstinent alcoholics. Alcohol Clin Exp Res. 1995;19:600–606. doi: 10.1111/j.1530-0277.1995.tb01554.x. [DOI] [PubMed] [Google Scholar]
- Bortolato M, Yardley M, Khoja S, Godar SC, Asatryan L, Finn DA, Alkana RL, Louie SG, Davies DL. Pharmacological insights into the role of P2X4 receptors in behavioral regulation: Lessons from ivermectin. Int J Neuropsychopharmacol. 2013;16:1059–1070. doi: 10.1017/S1461145712000909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cully DF, Vassilatis DK, Liu KK, Paress PS, Van der Ploeg LHT, Schaeffer JM, Arena JP. Cloning of an avermectin-sensitive glutamate-gated chloride channel from Caenorhabditis elegans. Nature. 1994;371:707–711. doi: 10.1038/371707a0. [DOI] [PubMed] [Google Scholar]
- Curran SL, Andrykowski MA, Studts JL. Short Form of the Profile of Mood States (POMS-SF): Psychometric information. Psychological Assessment. 1995;7:80. [Google Scholar]
- Dent JA, Davis MW, Avery L. avr-15 encodes a chloride channel subunit that mediates inhibitory glutamatergic neurotransmission and ivermectin sensitivity in Caenorhabditis elegans. EMBO J. 1997;16:5867–5879. doi: 10.1093/emboj/16.19.5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards G, Dingsdale A, Helsby N, Orme ME, Breckenridge AM. The relative systemic availability of ivermectin after administration as capsule, tablet, and oral solution. Eur J Clin Pharmacol. 1988;35:681–684. doi: 10.1007/BF00637608. [DOI] [PubMed] [Google Scholar]
- First MB, Spitzer RL, Gibbon M, Williams JBW, Davies M, Borus J, Howes MJ, Kane J, Pope HG. The structured clinical interview for DSM-III-R Personality-Disorders. J Pers Disord. 1995;9:92–104. [Google Scholar]
- Flannery BA, Volpicelli JR, Pettinati H. Psychometric properties of the Penn Alcohol Craving Scale. Alcohol Clin Exp Res. 1999;23:1289–1295. [PubMed] [Google Scholar]
- Franklin KM, Asatryan L, Jakowec MW, Trudell JR, Bell JR, Davies DL. P2X4 receptors (P2X4Rs) represent a novel target for the development of drugs to prevent and/or treat alcohol use disorders. Front Neurosci. 2014;8 doi: 10.3389/fnins.2014.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geary TG. Ivermectin 20 years on: Maturation of a wonder drug. Trends Parasitol. 2005;21:530–532. doi: 10.1016/j.pt.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Guzzo CA, Furtek CI, Porras AG, Chen C, Tipping R, Clineschmidt CM, Sciberras DG, Hsieh JY, Lasseter KC. Safety, tolerability, and pharmacokinetics of escalating high doses of ivermectin in healthy adult subjects. J Clin Pharmacol. 2002;42:1122–1133. doi: 10.1177/009127002401382731. [DOI] [PubMed] [Google Scholar]
- Jacobson AF, Goldstein BJ, Dominguez RA, Steinbook RM. Interrater agreement and intraclass reliability measures of SAFTEE in psychopharmacologic clinical trials. Psychopharmacol Bull. 1986;22:382–388. [PubMed] [Google Scholar]
- Jelinkova I, Yan Z, Liang Z, Moonat S, Teisinger J, Stojilkovic SS, Zemkova H. Identification of P2X4 receptor-specific residues contributing to the ivermectin effects on channel deactivation. Biochem Biophys Res Commun. 2006;349:619–625. doi: 10.1016/j.bbrc.2006.08.084. [DOI] [PubMed] [Google Scholar]
- Johanson CE, Uhlenhuth EH. Drug preference and mood in humans: Diazepam. Psychopharmacology. 1980;71:269–273. doi: 10.1007/BF00433061. [DOI] [PubMed] [Google Scholar]
- Kidd EJ, Grahames BA, Simon J, Michel AD, Barnard EA, Humphrey PPA. Localization of P2X purinoceptor transcripts in the rat nervous system. Mol Pharmacol. 1995;48:569–573. [PubMed] [Google Scholar]
- Kimpel MW, Strother WN, McClintick JN, Carr LG, Liang T, Edenberg HJ, McBride WJ. Functional gene expression differences between inbred alcohol-preferring and -non-preferring rats in five brain regions. Alcohol. 2007;41:95–132. doi: 10.1016/j.alcohol.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Peoples RW, Weight FF. Ethanol inhibits a neuronal ATP-gated ion channel. Mol Pharmacol. 1993;44:871–875. [PubMed] [Google Scholar]
- Litten RZ, Egli M, Heilig M, Cui C, Fertig JB, Ryan ML, Falk DE, Moss H, Huebner R, Noronha A. Medications development to treat alcohol dependence: A vision for the next decade. Addict Biol. 2012;17:513–527. doi: 10.1111/j.1369-1600.2012.00454.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin CS, Earleywine M, Musty RE, Perrine MW, Swift RM. Development and validation of the Biphasic Alcohol Effects Scale. Alcohol Clin Exp Res. 1993;17:140–146. doi: 10.1111/j.1530-0277.1993.tb00739.x. [DOI] [PubMed] [Google Scholar]
- Miller PM, Book SW, Stewart SH. Medical Treatment of Alcohol Dependence: A Systematic Review. Int J Psychiat Med. 2011;42:227–266. doi: 10.2190/PM.42.3.b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti PM, Binkoff JA, Abrams DB, Zwick WR, Nirenberg TD, Liepman MR. Reactivity of alcoholics and nonalcoholics to drinking cues. J Abnorm Psychol. 1987;96:122–126. doi: 10.1037//0021-843x.96.2.122. [DOI] [PubMed] [Google Scholar]
- Monti PM, Rohsenow DJ, Swift RM, Gulliver SB, Colby SM, Mueller TI, Brown RA, Gordon A, Abrams DB, Niaura RS, Asher MK. Naltrexone and cue exposure with coping and communication skills training for alcoholics: Treatment process and 1-year outcomes. Alcohol Clin Exp Res. 2001;25:1634–1647. [PubMed] [Google Scholar]
- O’Connor S, Morzorati S, Christian J, Li TK. Clamping breath alcohol concentration reduces experimental variance: Application to the study of acute tolerance to alcohol and alcohol elimination rate. Alcohol Clin Exp Res. 1998;22:202–210. [PubMed] [Google Scholar]
- Plebani JG, Ray LA, Morean ME, Corbin WR, MacKillop J, Amlung M, King AC. Human laboratory paradigms in alcohol research. Alcohol Clin Exp Res. 2012;36:972–983. doi: 10.1111/j.1530-0277.2011.01704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popova M, Asatryan L, Ostrovskaya O, Wyatt RL, Li K, Alkana RL, Davies DL. A point mutation in the ectodomain-transmembrane 2 interface eliminates the inhibitory effects of ethanol in P2X4 receptors. J Neurochem. 2010;112:307–317. doi: 10.1111/j.1471-4159.2009.06460.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray LA, Bujarski S, Chin PF, Miotto K. Pharmacogenetics of naltrexone in asian americans: A randomized placebo-controlled laboratory study. Neuropsychopharmacol. 2012;37:445–455. doi: 10.1038/npp.2011.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray LA, Bujarski S, MacKillop J, Courtney KE, Monti PM, Miotto K. Subjective response to alcohol among alcohol-dependent individuals: Effects of the mu-opioid receptor (OPRM1) gene and alcoholism severity. Alcohol Clin Exp Res. 2013;37(Suppl 1):E116–124. doi: 10.1111/j.1530-0277.2012.01916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray LA, Hutchison KE, Tartter M. Application of human laboratory models to pharmacotherapy development for alcohol dependence. Curr Pharm Des. 2010a;16:2149–2158. doi: 10.2174/138161210791516422. [DOI] [PubMed] [Google Scholar]
- Ray LA, Mackillop J, Monti PM. Subjective responses to alcohol consumption as endophenotypes: Advancing behavioral genetics in etiological and treatment models of alcoholism. Subst Use Misuse. 2010b;45:1742–1765. doi: 10.3109/10826084.2010.482427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray LA, Bujarski S, Roche DJ. Subjective Response to Alcohol as a Research Domain Criterion. Alcohol Clin Exp Res. 2016;40:6–17. doi: 10.1111/acer.12927. [DOI] [PubMed] [Google Scholar]
- Reinert DF, Allen JP. The Alcohol Use Disorders Identification Test (AUDIT): A Review of Recent Research. Alcohol Clin Exp Res. 2002;26:272–279. [PubMed] [Google Scholar]
- Rosner S, Hackl-Herrwerth A, Leucht S, Vecchi S, Srisurapanont M, Soyka M. Opioid antagonists for alcohol dependence. Cochrane Database Syst Rev. 2010:CD001867. doi: 10.1002/14651858.CD001867.pub3. [DOI] [PubMed] [Google Scholar]
- Rösner S, Hackl-Herrwerth A, Leucht S, Lehert P, Vecchi S, Soyka M. Acamprosate for alcohol dependence. The Cochrane Library; 2010. [DOI] [PubMed] [Google Scholar]
- Schuckit MA. Subjective responses to alcohol in sons of alcoholics and control subjects. Arch Gen Psychiat. 1984;41:879–884. doi: 10.1001/archpsyc.1984.01790200061008. [DOI] [PubMed] [Google Scholar]
- Shu EN, Onwujekwe EO, Okonkwo PO. Do alcoholic beverages enhance availability of ivermectin? Eur J Clin Pharmacol. 2000;56(5):437–438. doi: 10.1007/s002280000120. [DOI] [PubMed] [Google Scholar]
- Silberberg SD, Li M, Swartz KJ. Ivermectin interaction with transmembrane helices reveals widespread rearrangements during opening of P2X receptor channels. Neuron. 2007;54:263–274. doi: 10.1016/j.neuron.2007.03.020. [DOI] [PubMed] [Google Scholar]
- Sobell LC, Sobell MB. Measuring alcohol consumption. Springer; 1992. Timeline follow-back; pp. 41–72. [Google Scholar]
- Sullivan JT, Sykora K, Schneiderman J, Naranjo CA, Sellers EM. Assessment of Alcohol Withdrawal: The revised Clinical Institute Withdrawal Assessment for Alcohol Scale (CIWA-Ar) Brit J Addict. 1989;84:1353–1357. doi: 10.1111/j.1360-0443.1989.tb00737.x. [DOI] [PubMed] [Google Scholar]
- Tabakoff B, Saba L, Printz M, Flodman P, Hodgkinson C, Goldman D, Koob G, Richardson H, Kechris K, Bell RL, Hubner N, Heinig M, Pravenec M, Mangion M, Legault L, Dongier M, Conigrave KM, Whitfield JB, Saunders JB, Grant B, Hoffman PL. Genetical genomic determinants of alcohol consumption in rats and humans. BMC Biology. 2009;7:70. doi: 10.1186/1741-7007-7-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takougang I, Ngogang J, Sihom F, Ntep M, Kamgno J, Eyamba A, Zouré H, Noma M, Amazigo U. Does alcohol consumption increase the risk of severe adverse events to ivermectin treatment. Afr J Pharm Pharmacol. 2008;2:077–082. [Google Scholar]
- Wyatt LR, Finn DA, Yardley MM, Khoja S, Asatryan L, Alkana RL, Davies DL. Contribution of P2X4 receptors to ethanol intake in male C57BL/6 mice. Neurochem Res. 2014;39:1127–1139. doi: 10.1007/s11064-014-1271-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yardley M, Wyatt L, Khoja S, Asatryan L, Ramaker MJ, Finn DA, Alkana RL, Huynh N, Louie SG, Petasis NA, Bortolato M, Davies DL. Ivermectin reduces alcohol intake and preference in mice. Neuropharmacol. 2012;63:190–201. doi: 10.1016/j.neuropharm.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]