

Abstract
Background
Rilpivirine (RPV) is the latest non-nucleoside reverse transcriptase inhibitor (NNRTI) to be FDA-approved to combat HIV-1 infections. NNRTIs inhibit the chemical step in viral DNA synthesis by binding to an allosteric site located about 10 Å from the polymerase active site of reverse transcriptase (RT). Although NNRTIs potently inhibit the replication of WT HIV-1, the binding site is not conserved, and mutations arise in the binding pocket. Doravirine (DOR) is a new a NNRTI in phase III clinical trials.
Methods
Using a single round HIV-1 infection assay, we tested RPV and DOR against a broad panel of NNRTI resistant mutants to determine their respective activities. We also used molecular modeling to determine if the susceptibility profile of each compound was related to how they bind RT.
Results
Several mutants displayed decreased susceptibility to DOR. However, with the exception of E138K, our data suggest that the mutations that reduce the potency of DOR and RPV are non-overlapping. Thus, these two NNRTIs have the potential to be used together in combination therapy. We also show that the location at which DOR and RPV bind with the NNRTI binding pocket of RT correlates with the differences in their respective susceptibility to the panel of NNRTI resistance mutations.
Conclusions
This shows that (1) DOR is susceptible to a number of well-known NNRTI resistance mutations and (2) an understanding of the mutational susceptibilities and binding interactions of NNRTIs with RT could be used to develop pairs of compounds with non-overlapping mutational susceptibilities.
Keywords: HIV-1, Antiviral activity, susceptibility, potency, resistance, nonnucleoside reverse transcriptase inhibitor
Introduction
There are two classes of drugs that block reverse transcription, nucleoside analogs (NRTIs) and NNRTIs. NRTIs are analogs of the deoxynucleosides that are used to synthesize DNA. The NRTIs used to treat HIV-1 infection lack the 3′–OH [1, 2]. If an NRTI is incorporated into the growing viral DNA strand, reverse transcriptase (RT) will not be able to add the next nucleotide, blocking DNA elongation. The five approved NNRTIs all bind in a hydrophobic pocket ~10 Å from the polymerase active site called the NNRTI binding pocket. Once a NNRTI binds within the pocket, structural changes perturb the portion of RT that underlies the nucleic acid substrate, affecting the alignment of the primer terminus and the polymerase active site, inhibiting the chemical step of DNA synthesis [3–5]. NNRTIs potently inhibit the replication of WT HIV-1; however, because the polymerase activity of RT does not directly involve residues within the NNRTI binding pocket, changes in the NNRTI binding pocket can arise that undermine the effectiveness of these compounds. There are five NNRTIs that have been approved for the treatment of HIV-1 infection: nevirapine (NVP, Viramune), delavirdine (DLV, Rescriptor), efavirenz (EFV, Sustiva), etravirine (ETR, Intelence), and RPV (Edurant).
Although there are significant differences in the structures of the first generation NNRTIs (NVP, DLV, and EFV), they all bind in the same hydrophobic pocket of RT [5–8]. Resistance mutations that are selected by one NNRTI often reduce the susceptibility of RT to other first generation NNRTIs without having a large effect on viral replication capacity [1, 2]. A partial list of NNRTI resistance mutations include: L100I, K103N, V106A, E138K, Y181C, Y188L, and H221Y; these mutations can occur singly, or in combinations [2]. Recently, a shift in NNRTI design from more bulky and structurally restricted bi- and tri-cyclic ring compounds to more structurally adaptable compounds has led to the development of the second-generation NNRTIs ETR and RPV (Supp. Fig. 1), which are relatively effective against WT HIV-1 and mutants that carry many of the well-known NNRTI resistance mutations [8–10]. However, in clinical trials individuals on an ETR containing regimen who experienced virological failure were infected with viruses that had a combination of RT mutations including V90I, A98G, L100I, K101E/P, V106I, V179D/F, Y181C/I/V, and G190A/S [11]. In clinical trials that involved RPV and two NRTIs, the RT resistance mutations E138K and M184I/V were selected [12–15]. In addition, viruses that grew out in an in vitro selection with RPV or a compound closely related to RPV had the RT mutations K101E or E40K, D67E, V111A, E138K, Y181C, and M230I, respectively. Thus, it would be useful to develop new NNRTIs that could block the replication of a broader spectrum of resistant variants.
DOR (Supp. Fig. 1) is being developed by Merck and is currently in Phase III clinical trials [16, 17]. DOR can effectively inhibit the replication of viruses that carry several prevalent NNRTI-resistance mutations; however, DOR selected mutations in RT that confer reduced susceptibility in experiments done in cultured cells. Mutations that reduced the susceptibility of RT to DOR did not, in general, confer a decrease in susceptibility to RPV and vice-versa, suggesting this difference would make DOR a useful new drug [17]. However, in this initial study, only a limited number of mutants were tested. We tested the ability of DOR and RPV to inhibit the replication of WT HIV-1 and a large number of well-characterized NNRTI-resistant mutants. DOR loses potency against a number of NNRTI-resistant mutants; however, DOR is generally effective against mutants that reduce the potency of RPV, although DOR did lose some potency against E138K. Based on these results, it seems likely that, if DOR was used as the only NNRTI in a regimen, there could be problems with resistance. We suggest an alternative strategy in which DOR would be used in combination with RPV; this would in some sense be similar to strategies in which 2 NRTIs that select non-overlapping mutations (for example, FTC and tenofovir) are used together in cART. Using DOR and RPV together should be effective against HIV-1s that encode most of the well-characterized NNRTI-resistance mutations. It also appears that the difference in the susceptibility of the mutants to DOR and RPV is a result of differences in the way the two compounds bind to RT; this information might be useful in the design of other complementary pairs of NNRTIs.
Material and Methods
NNRTI synthesis
DOR synthesis is described in [16]; RPV synthesis is described in [18].
Cell-Based Assays
Virion production and single-round infectivity assays were used to determine antiviral activity (IC50 values) of the compounds as described [19] and discussed in the supplemental information. Selection of NNRTI mutations in HIV-1 is described in supplemental information.
Vector Constructs
NNRTI resistant mutations in HIV-1 RT are described in supplemental information.
Computer Modeling
All modeling of RPV and DOR in the NNRTI binding pocket is described in supplemental information.
Results
Antiviral activity of RPV and DOR against known NNRTI-resistant HIV-1 mutants
The ability of RPV and DOR to inhibit the replication of WT HIV-1 and drug-resistant mutants was measured using a previously described single-round infection assay [19]. We initially investigated the L100I, K103N, Y181C, Y188L, H221Y, and K103N/Y181C RT mutants. These mutants were chosen because they have been selected in patients and are well distributed around the NNRTI binding pocket [1, 2]. In previous work, RPV potently inhibited infection of both the WT and the mutant viruses with IC50 values of 0.24 ± 0.1 nM and <2 nM, respectively [20]. DOR potently inhibited the replication of the WT HIV-1 vector in the single-cycle assay (0.67 ± 0.14 nM) and L100I mutant (1.14 ± 0.2 nM) as shown in (Fig. 1A). However, the resistant mutants K103N, Y181C, and H221Y showed modest decreases in susceptibility against DOR (ranging from 2–5 nM), and the K103N/Y181C double mutant exhibited a larger drop in susceptibility (11.3 ± 5.9 nM). DOR also showed a considerable loss of potency versus the resistant mutant Y188L, out of the range of our assay (>100 nM).
Figure 1. Antiviral activity of DOR and RPV against HIV-1 vectors that carry well-known NNRTI resistance mutations.
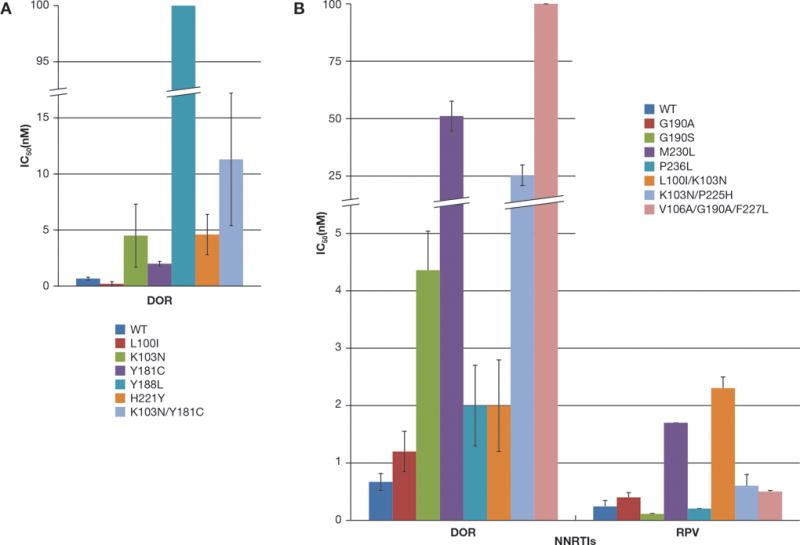
(A) The IC50 values of DOR against WT HIV-1 and several NNRTI-resistant mutants, represented in different colors, were measured using a single round infection assay. (B) The IC50 values of RPV and DOR against WT HIV-1 and several other well-characterized NNRTI resistant mutants, represented in different colors, were measured using a single round infection assay. The IC50 value of DOR against the M230L, K103N/P225H, and V106A/G190A/F227L resistant mutants were >100 nM. Error bars represent the standard deviations of independent experiments, (n=4). The RPV susceptibility data were also used as a control in experiments testing the efficacy of a series of RPV analogs (21).
Antiviral activities of RPV and DOR against other common NNRTI-resistant mutants
We determined the antiviral activities of RPV and DOR against some other known NNRTI-resistant mutants to determine the relative strengths and weaknesses of the two compounds. We chose NNRTI resistant mutants that are known to contribute to virological failure in HIV-infected individuals or mutants that were selected in cell culture: G190A, G190S, M230L, P236L, L100I/K103N, K103N/P225H, and V106A/G190A/F227L [7]. RPV showed potent antiviral activity against all of the tested mutants (<2 nM across the entire panel) [21] whereas DOR was less effective (Fig. 1B). HIV-1 variants G190A, P236L, and L100I/K103N were susceptible to DOR (<2 nM), however the G190S mutant showed a modest reduction in susceptibility (4.6 ± 1.2 nM) while the M230L mutant and the K103N/P225H double mutant had substantial decreases in their respective susceptibilities (51.1 ± 6.5 nM and 25.3 ± 4.5 nM, respectively). In addition, the triple mutant V106A/G190A/F227L was relatively insensitive to DOR (>100 nM). These results suggest that RPV is more broadly effective against many NNRTI-resistant mutants.
Antiviral activities of RPV and DOR against mutants selected by DOR in cell culture
V106A was the major variant selected by DOR treatment of HIV-1 in cultured cells [17]. Some of the viruses selected by DOR in culture contained additional mutations: V106A/F227L, V106A/L234I, and V106A/F227L/L234I [17]. To determine whether there is cross resistance between RPV and DOR and to further compare the overall effectiveness of the compounds, RPV and DOR were screened against the following mutants: V106A, L234I, V106A/F227L, V106/L234I, and V106A/F227L/L234I (Fig. 2). All 5 of these mutants were sensitive to RPV (<0.8 nM) [21]. As expected, L234I (6.8 ± 2.5 nM) showed a modest reduction in susceptibility to DOR, while V106A showed a greater loss of susceptibility (15.6 ± 4 nM). The double and triple mutants V106A/F227L, V106/L234I, and V106A/F227L/L234I all showed a substantial loss of susceptibility to DOR (>100 nM).
Figure 2. Antiviral activities of RPV and DOR against HIV-1 that contains mutations selected by DOR in cell culture.
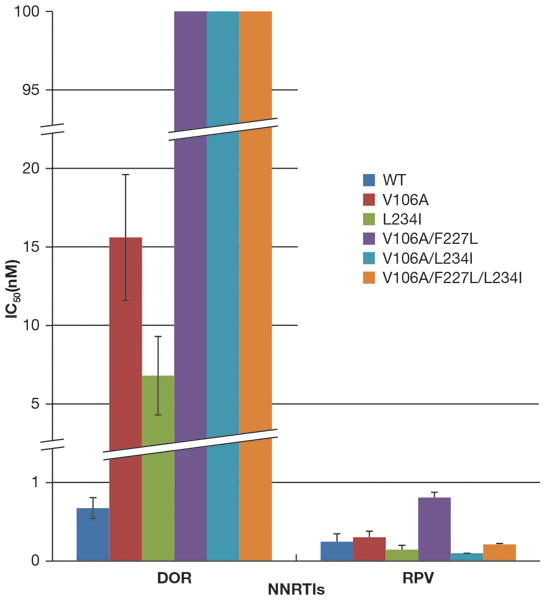
The IC50 values of RPV and DOR against vectors that carry WT RT and mutants selected by DOR in cell culture were measured using a single-round infection assay. Error bars represent the standard deviations of independent experiments (n=4). The IC50 values of the graph have a maximum value of 100 nM and to better illustrate the lower IC50 values, the Y-axis was broken twice between 1 and 5 nM and 20 and 95 nM, there are similar breaks in the appropriate bars. The IC50 value of DOR against the V106A/F227L, V106A/L234I, and V106A/F227L/L234I resistant mutants were beyond the point of detection in our single round infection assay (>100 nM). The RPV susceptibility data were also used as a control in experiments testing the efficacy of a series of RPV analogs (21).
Antiviral activities of RPV and DOR against mutants selected by RPV
The E138K substitution in RT is the most common NNRTI-resistance mutation seen in patients who fail RPV-containing regimens [13]. Because many of the commonly used cART regimens include either 3TC or FTC, some isolates from virological failures also contained the M184V/I mutation. K101E is a mutation that is commonly selected in patients who fail regimens that include ETR [11], a compound that is structurally related to RPV, and that we selected with RPV in vitro. In addition, we selected HIV-1 RT mutations in cell culture with a closely related RPV analog (unpublished observations): E40K, D67E, V111A, E138K, Y181C, and M230I. As described above, both compounds were tested against M230L (which should be similar to M230I). The effects of Y181C on the susceptibility of the vector to DOR and RPV were discussed earlier. We screened a panel of mutants that included: E40K, D67E, K101E, V111A, E138K, M184I, M184V, K101E/M184I, K101E/M184V, E138K/M184I, and E138K/M184V (Fig. 3) to determine whether these mutants are susceptible to DOR. DOR potently inhibited the replication of E40K, K101E, V111A, M184I, M184V, K101E/M184I, K101E/M184V, E138K/M184I, and E138K/M184V (all <3 nM). The E138K mutant showed a modest decrease in susceptibility to DOR (13.9 ± 2.4 nM), while D67E variant showed a larger decrease in susceptibility (46 ± 14 nM). Interestingly, the variant K101E was the only RPV-associated single mutant which conferred a small, but measurable decrease in susceptibility to RPV (2.6 ± 1.6 nM), while the rest of the mutants selected by RPV remained sensitive in our assay (< 1 nM) [21]. The ability of RPV to inhibit these E138K/M184I/V RT mutants in tissue culture has been reported previously [15]. RPV has been reported to select for another resistance pathway that involves K101P and Y181I, and K101P/V179I [13, 17]. We tested DOR and RPV against these resistant mutants (Fig. 4); K101P HIV-1 showed a slight increase in susceptibility to DOR (1.0 ± 0.27 nM) and a modest decrease in susceptibility to RPV (6.2 ± 1.6 nM). The Y181I mutant was also sensitive to DOR (0.63 ± 0.28 nM) but this mutant showed a reduced susceptibility to RPV (8.8 ± 0.12 nM). The double mutant K101P/V179I showed an increase in susceptibility to DOR (1.5 ± 0.4 nM); however, this mutant had a significant loss in susceptibility to RPV (93.5 ± 12.1 nM) [21].
Figure 3. Antiviral activities of RPV and DOR against HIV-1 that contains mutations selected by RPV during cART and in cell culture.
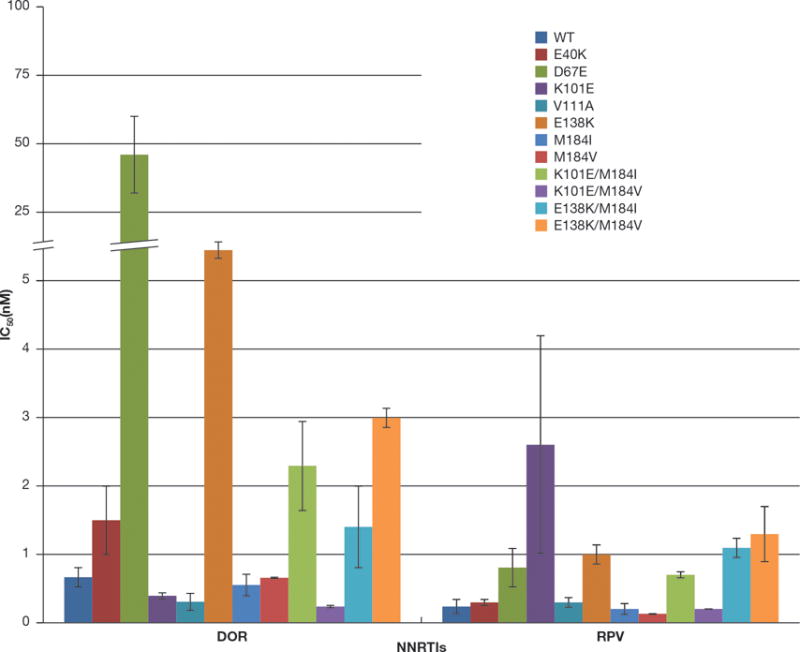
The IC50 values of RPV and DOR against viruses that contain WT RT and mutations selected by RPV in cell culture (E40K, D67E, K101E, and V111A) and in infected individuals during cART (K101E, E138K, M184I, M184V, K101E/M184I, K101E/M184V, E138K/M184I, and E138K/M184V) were measured using a single-round infection assay. Error bars represent the standard deviations of independent experiments (n=4). The IC50 values of the graph have a maximum value of 100 nM and to better illustrate the lower IC50 values, the Y-axis was broken between 5 and 25 nM, there are equivalent breaks in the appropriate bar(s). The RPV susceptibility data were also used as a control in experiments testing the efficacy of a series of RPV analogs (21).
Figure 4. Antiviral Activities of RPV and DOR against HIV-1 that contains additional mutations selected by RPV in vivo.
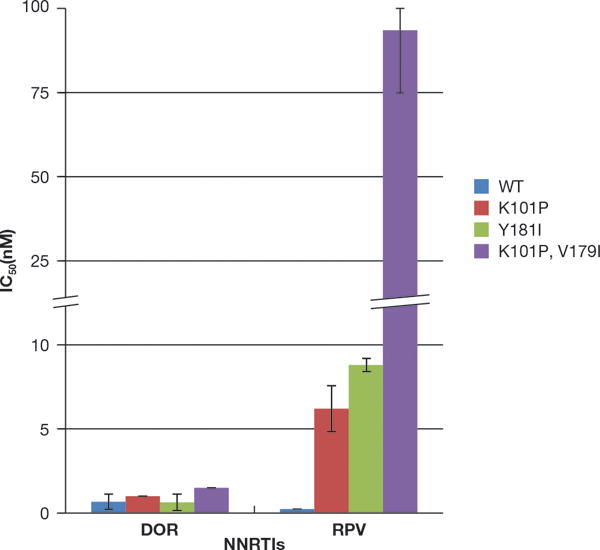
The IC50 values of RPV and DOR against viruses that contain WT RT and mutations selected by RPV in HIV-infected individuals were measured using a single round infection assay. Error bars represent the standard deviations of independent experiments (n=4). The IC50 values of the graph have a maximum value of 100 nM and to better illustrate the lower IC50 values, the Y-axis was broken between 10 and 25 nM and in the appropriate bar. The RPV susceptibility data were also used as a control in experiments testing the efficacy of a series of RPV analogs (21).
Binding of RPV and DOR in the NNRTI binding pocket
Structural studies can be used to understand the binding of inhibitors to their cognate targets; HIV-1 RT has been crystallized with RPV [22] and, more recently, with DOR [16, 17]. RPV has a pyrimidine core with two aryl groups linked by amines, a benzonitrile group and dimethylphenyl moiety with a cyanovinyl, whereas DOR comprises a trifluoromethyl-pyridone core with a methyl-triazolone and a chlorophenol with a cyano group. The crystal structure of RPV in the NNRTI binding pocket showed that the chlorophenol moiety of RPV has aromatic stacking interactions with Y181 and the appended cyanovinyl extends into a hydrophobic tunnel, located in the upper portion of the NNRTI binding pocket, formed by the sidechains of Y188, F227, W229, and L234. The amine linker nitrogens interact with the main chain carbonyl oxygens of K101 from the p66 subunit and E138 of the p51 subunit through hydrogen bonding. Residues L100, K103, and V179, which are located near the center of the NNRTI binding pocket, interact with the central pyrimidine ring of RPV. The benzonitrile moiety makes contacts with Y318, P225, and P236. The interactions of DOR and the NNRTI binding pocket of RT in the crystal structure showed that the chlorophenol moiety stacks with Y188 and the DOR cyano moiety makes contacts that are similar to those made by RPV within the hydrophobic tunnel. The pyridone core of DOR interacts with V106 and L100 and the methyl triazolone stacks with P236 and interacts with the main chain atoms of K103 [22].
Superimposing the structures of RT with DOR and RPV bound reveals significant differences in their binding (Supp. Fig. 2; Fig. 5) and the residues they contact in the binding pocket, which suggests how the two compounds differ in their susceptibility to NNRTI-resistant mutants. The superimposition shows that DOR resides ~1–2 Å deeper in the NNRTI binding site compared to RPV (Supp. Fig 2). In the DOR structure (Fig. 5, right panel B), Y188 is ~4 Å closer to the middle and rim of the binding pocket to accommodate the chlorophenol moiety of DOR, while Y181 is ~8 Å closer to the rim of the binding pocket compared to the structure with RPV bound (Fig. 5, left panel A). Residue E138 moves ~4.5 Å away from the rim toward the back of the NNRTI binding pocket in the DOR structure as compared to the RPV structure, which prevents DOR from interacting with K101. In addition, the residues in the upper portion of the NNRTI binding pocket, L234, P225, and F227 are shifted by 1.5 Å in the structure with DOR bound compared with RPV RT structure. The substitution of the benzonitrile moiety of RPV with the triazolone of DOR causes a 2–3 Å movement of this substituent towards P236. The differences in the positions of these residues around the NNRTI binding pocket between the two structures matches the residues where mutations are likely to be selected by the two compounds. It appears, based both on the crystal structures and the behavior of the mutants in our antiviral assays, that residues in the upper portion of the binding pocket interact more extensively with DOR and that the residues around the rim of the binding pocket interact more with RPV. RPV is known to change its binding in response to changes in the NNRTI binding pocket that are associated with the presence of drug resistance mutations. We do not yet have access to crystal structures of DOR bound to mutant forms of RT. It is possible that DOR is flexible to some extent [8]; however, it is more broadly susceptible to drug resistance mutations than RPV.
Figure 5. Relative positions of RPV and DOR in the NNRTI binding pocket, showing contacts with specific residues.
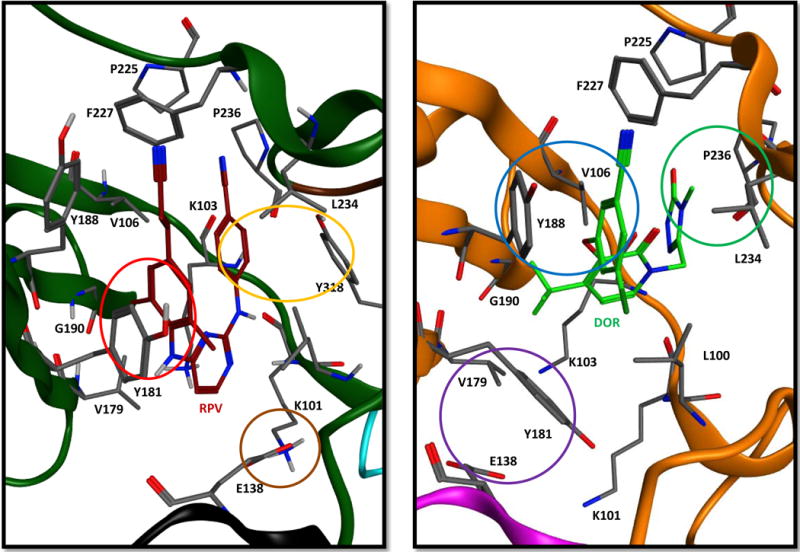
The crystal structures of RPV (left image, A) and DOR (right image, B) are displayed for comparison to illustrate the different contacts made between the inhibitor and the NNRTI binding pocket for each. In the RPV structure, the phenyl moiety of RPV stacks with Y181 (red circle), while E138 interacts through an electrostatic interaction with K101 (brown circle) and the benzonitrile moiety stacks with Y318 (orange circle). In the DOR structure, the chlorophenyl moiety of DOR alternatively stacks with Y188 (blue circle), while the triazolone functionality of DOR interacts with P236 (green circle), and residues E138 and Y181 (purple circle) move toward the rim and back of the NNRTI binding pocket, respectively. In both the RPV and DOR structures, the cyanovinyl and cyano functionalities, respectively, modified on the phenyl moiety extend in the hydrophobic pocket formed by the side chains Y188, F227, W229, and L234. In the figure only E138K is from the RT p51 subunit in the RPV (black) and DOR (magenta) images. The remaining residues are from the RT p66 subunit in the RPV (dark green) and DOR (orange) structures.
Discussion
Current cART regimens use combinations of at least 3 drugs to fully suppress HIV-1 replication. In some cases, drugs in the same class can be used together if they select for non-overlapping resistance mutations, such as the NRTIs tenofovir and FTC [23]. One issue with NNRTIs is their considerable cross-resistance. A new NNRTI, DOR, which is in phase III clinical trials, has potent antiviral activity against WT HIV-1 and some of the well-characterized NNRTI-resistant mutants. Based on analysis of the mutations in RT that were selected by DOR in cell culture, it was suggested that DOR and RPV select for different resistance mutations and that DOR could be a useful new NNRTI used in cART [17]. However, when we analyzed a large panel of NNRTI-resistance mutations, we found that several well-known NNRTI-resistance mutations, either singly, or in combination, caused a considerable loss of DOR potency against HIV-1.
The tripartite structure of RPV is a key to its ability to inhibit a wide range of NNRTI-resistant HIV-1 mutants. The relatively long linker that joins the cyanovinyl group to the pyrimidine scaffold (11.1 Å) allows the cyanovinyl group of RPV to be flexible in response to mutations that change the structure of the binding pocket, allowing the drug to make new contacts with other residues, maintaining its ability to inhibit RT. Increasing the length of the linker groups that join the functional moieties to the pharmacophore is becoming a common theme in the design of HIV-1 inhibitors. Dolutegravir (DTG) is an integrase inhibitor that has a long linker joining its benzyl moiety and tri-cyclic scaffold [24, 25]; this allows DTG to maintain an interaction between the benzyl moiety and penultimate cytosine of the incoming viral DNA [24]. Thus DTG is able to inhibit integration even when resistance mutations alter the structure of the integrase active site. The distances between the pyridone scaffold of DOR and its cyano and triazolone moieties (Supp. Fig. 2) are 7.75 Å and 6.35 Å, respectively, which are considerably shorter than the linkers that join RPV’s cyanovinyl (11.1 Å) and benzonitrile moieties (8.3 Å). This suggests DOR may be more limited in its ability to adapt to the changes in the NNRTI binding pocket caused by some mutations and to overcome resistance. Importantly, classical NNRTI mutants, for example Y188L, caused a substantial drop in DOR susceptibility and other classical NNRTI mutants including K103N and K103N/Y181C caused more modest drops in susceptibility to DOR. Based on the resistance data we present here, we question whether DOR has significant advantages relative to the available NNRTIs. However, it might be possible to develop cART regimens in which DOR was combined with RPV; however, as previously mentioned, E138K mutation could pose a problem for this combined strategy. Although DOR apparently did not select for the E138K mutation in cell culture, we found that this mutation did reduce the susceptibility of HIV-1 to DOR by about 20-fold. As has already been reported, the E138K mutation does not cause a significant reduction in susceptibility to RPV in cell culture assays (including ours). The complication is that, in HIV-infected individuals, RPV selects for the E138K mutation [12, 13]. This apparent paradox has been noted before, by us [20], and by others [10, 15]. However, it is possible that DOR would not be effective in suppressing the selection and replication of E138K mutants that arise during RPV-containing cART.
We superimposed the structures of HIV-1 RT with RPV or DOR to determine if differences in the binding of the compounds can explain the differences in their resistance susceptibility profiles. Based on the structure of RPV bound to RT, it is not surprising that the K101P caused a substantial decrease in susceptibility to RPV. K101P would appear to cause a steric clash with RPV in the binding pocket and the interaction of E138 with K101 would be lost [21]. Mutating these residues likely affects the rates of association/dissociation of RPV within the NNRTI binding pocket. The K101P/V179I mutant caused a large decrease in RPV susceptibility because it affects both the contacts between the amine linkers and pyrimidine core with the residues of the binding pocket by introducing steric clash through the branched isoleucine and the cyclic proline. The Y181I mutant also displayed a decrease in susceptibility to RPV, unlike the other mutant at this position, Y181C, likely because of the unavoidable steric clash from beta-branched isoleucine that prevents RPV from recruiting Y183 toward the NNRTI binding pocket to compensate for the loss of interaction between Y181 and its phenyl moiety [22]. However, RPV did not lose efficacy against viruses with mutations in the upper portion of the NNRTI binding pocket (including the DOR-associated mutations), suggesting that residues on the rim of the NNRTI binding pocket are much more important for the binding of RPV. Conversely, DOR interacts primarily with residues in the upper portion of the NNRTI binding pocket, making important contacts with K103, V106, Y188, and P236. Mutations at V106, with or without additional secondary mutations (F227L, L234I, and F227L/L234I), caused substantial decreases in susceptibility to DOR. The branched, hydrophobic side chain of V106 interacts with the pyridone core of DOR; in the presence of the V106A mutation, this important contact is lost. The Y188L mutation causes the loss of the π-π stacking interaction, which in turn reduces the potency of DOR inhibition, while the lack of interaction with Y181 explains the fact that the Y181C and Y181I mutants are susceptible to DOR. The P236L mutation causes a modest decrease in DOR susceptibility by introducing clash and preventing the CH-π interaction between the triazolone of DOR and P236. Interestingly, the E138K mutant caused a 20-fold drop in susceptibility to DOR. Although the E138 residue does not make any direct contacts with DOR, the binding of DOR within the pocket caused the position of this residue to shift substantially, which could prevent the interaction between E138K of the p51 subunit and K101 of the p66 subunit.
We suggest that there might be advantages to a 4 drug regimen that would include one or two NRTIs or an NRTI and an integrase inhibitor, in combination with 2 NNRTIs. If the NNRTIs are carefully chosen, it should be difficult for the virus to develop resistance, and the low toxicity of the NNRTIs would be an advantage for patients [26, 27]. Whether DOR and RPV represent a clinically useful pair of NNRTIs that could be used together in cART remains to be seen; however, it would be useful to find additional pairs of NNRTIs whose resistance profiles do not overlap. Thus there is a need to develop new NNRTIs that are broadly effective against known NNRTI-resistant mutants and the development of new NNRTIs should make use of the available structural data, in relation to the resistance susceptibility profiles of the compounds, as a guide to develop compounds (or combinations of compounds) that can overcome known NNRTI-resistant mutants.
Supplementary Material
Acknowledgments
The authors would like to thank Teresa Burdette for help with the manuscript and Joseph Meyer for assisting with the figures. This research was supported by the Intramural Research Programs of the National Cancer Institute and the Intramural AIDS Targeted Antiviral Program (IATAP) and NIH grants R01 AI080290 (ZA) and T32 AI065380 (KM).
Footnotes
This work has not been presented at meetings
RPV and DOR have complementary efficacies
References
- 1.Das K, Arnold E. HIV-1 reverse transcriptase and antiviral drug resistance. Part 1. Curr Opin Virol. 2013;3(2):111–8. doi: 10.1016/j.coviro.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Das K, Arnold E. HIV-1 reverse transcriptase and antiviral drug resistance. Part 2. Curr Opin Virol. 2013;3(2):119–28. doi: 10.1016/j.coviro.2013.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spence RA, et al. Mechanism of inhibition of HIV-1 reverse transcriptase by nonnucleoside inhibitors. Science. 1995;267(5200):988–93. doi: 10.1126/science.7532321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rittinger K, Divita G, Goody RS. Human immunodeficiency virus reverse transcriptase substrate-induced conformational changes and the mechanism of inhibition by nonnucleoside inhibitors. Proc Natl Acad Sci U S A. 1995;92(17):8046–9. doi: 10.1073/pnas.92.17.8046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Das K, et al. HIV-1 reverse transcriptase complex with DNA and nevirapine reveals non-nucleoside inhibition mechanism. Nat Struct Mol Biol. 2012;19(2):253–9. doi: 10.1038/nsmb.2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shirasaka T, et al. Emergence of human immunodeficiency virus type 1 variants with resistance to multiple dideoxynucleosides in patients receiving therapy with dideoxynucleosides. Proc Natl Acad Sci U S A. 1995;92(6):2398–402. doi: 10.1073/pnas.92.6.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Das K, et al. Crystallography and the design of anti-AIDS drugs: conformational flexibility and positional adaptability are important in the design of non-nucleoside HIV-1 reverse transcriptase inhibitors. Prog Biophys Mol Biol. 2005;88(2):209–31. doi: 10.1016/j.pbiomolbio.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 8.Das K, et al. Roles of conformational and positional adaptability in structure-based design of TMC125-R165335 (etravirine) and related non-nucleoside reverse transcriptase inhibitors that are highly potent and effective against wild-type and drug-resistant HIV-1 variants. J Med Chem. 2004;47(10):2550–60. doi: 10.1021/jm030558s. [DOI] [PubMed] [Google Scholar]
- 9.Janssen PA, et al. In search of a novel anti-HIV drug: multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1E)-2-cyanoethenyl]-2,6-dimethylphenyl]amino]-2-pyrimidinyl]amino]benzonitrile (R278474, rilpivirine) J Med Chem. 2005;48(6):1901–9. doi: 10.1021/jm040840e. [DOI] [PubMed] [Google Scholar]
- 10.Azijn H, et al. TMC278, a next-generation nonnucleoside reverse transcriptase inhibitor (NNRTI), active against wild-type and NNRTI-resistant HIV-1. Antimicrob Agents Chemother. 2010;54(2):718–27. doi: 10.1128/AAC.00986-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lazzarin A, et al. Efficacy and safety of TMC125 (etravirine) in treatment-experienced HIV-1-infected patients in DUET-2: 24-week results from a randomised, double-blind, placebo-controlled trial. Lancet. 2007;370(9581):39–48. doi: 10.1016/S0140-6736(07)61048-4. [DOI] [PubMed] [Google Scholar]
- 12.Cohen CJ, et al. Efficacy and safety of rilpivirine (TMC278) versus efavirenz at 48 weeks in treatment-naive HIV-1-infected patients: pooled results from the phase 3 double-blind randomized ECHO and THRIVE Trials. J Acquir Immune Defic Syndr. 2012;60(1):33–42. doi: 10.1097/QAI.0b013e31824d006e. [DOI] [PubMed] [Google Scholar]
- 13.Rimsky L, et al. Genotypic and phenotypic characterization of HIV-1 isolates obtained from patients on rilpivirine therapy experiencing virologic failure in the phase 3 ECHO and THRIVE studies: 48-week analysis. J Acquir Immune Defic Syndr. 2012;59(1):39–46. doi: 10.1097/QAI.0b013e31823df4da. [DOI] [PubMed] [Google Scholar]
- 14.Cohen CJ, et al. Rilpivirine versus efavirenz with two background nucleoside or nucleotide reverse transcriptase inhibitors in treatment-naive adults infected with HIV-1 (THRIVE): a phase 3, randomised, non-inferiority trial. Lancet. 2011;378(9787):229–37. doi: 10.1016/S0140-6736(11)60983-5. [DOI] [PubMed] [Google Scholar]
- 15.Xu HT, et al. Compensation by the E138K mutation in HIV-1 reverse transcriptase for deficits in viral replication capacity and enzyme processivity associated with the M184I/V mutations. J Virol. 2011;85(21):11300–8. doi: 10.1128/JVI.05584-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lai MT, et al. In vitro characterization of MK-1439, a novel HIV-1 nonnucleoside reverse transcriptase inhibitor. Antimicrob Agents Chemother. 2014;58(3):1652–63. doi: 10.1128/AAC.02403-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng M, et al. In vitro resistance selection with doravirine (MK-1439), a novel nonnucleoside reverse transcriptase inhibitor with distinct mutation development pathways. Antimicrob Agents Chemother. 2015;59(1):590–8. doi: 10.1128/AAC.04201-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marinello J, et al. Comparison of raltegravir and elvitegravir on HIV-1 integrase catalytic reactions and on a series of drug-resistant integrase mutants. Biochemistry. 2008;47(36):9345–54. doi: 10.1021/bi800791q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith SJ, Hughes SH. Rapid screening of HIV reverse transcriptase and integrase inhibitors. J Vis Exp. 2014;(86) doi: 10.3791/51400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson BC, et al. A comparison of the ability of rilpivirine (TMC278) and selected analogues to inhibit clinically relevant HIV-1 reverse transcriptase mutants. Retrovirology. 2012;9:99. doi: 10.1186/1742-4690-9-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith SJ, et al. Rilpivirine analogs potently inhibit drug-resistant HIV-1 mutants. Retrovirology. 2016 doi: 10.1186/s12977-016-0244-2. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Das K, et al. High-resolution structures of HIV-1 reverse transcriptase/TMC278 complexes: strategic flexibility explains potency against resistance mutations. Proc Natl Acad Sci U S A. 2008;105(5):1466–71. doi: 10.1073/pnas.0711209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Das K, et al. Structural basis for the role of the K65R mutation in HIV-1 reverse transcriptase polymerization, excision antagonism, and tenofovir resistance. J Biol Chem. 2009;284(50):35092–100. doi: 10.1074/jbc.M109.022525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hare S, et al. Structural and functional analyses of the second-generation integrase strand transfer inhibitor dolutegravir (S/GSK1349572) Mol Pharmacol. 2011;80(4):565–72. doi: 10.1124/mol.111.073189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kobayashi M, et al. In Vitro antiretroviral properties of S/GSK1349572, a next-generation HIV integrase inhibitor. Antimicrob Agents Chemother. 2011;55(2):813–21. doi: 10.1128/AAC.01209-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Margolis AM, et al. A review of the toxicity of HIV medications. J Med Toxicol. 2014;10(1):26–39. doi: 10.1007/s13181-013-0325-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stolbach A, et al. A Review of the Toxicity of HIV Medications II: Interactions with Drugs and Complementary and Alternative Medicine Products. J Med Toxicol. 2015;11(3):326–41. doi: 10.1007/s13181-015-0465-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.