

Abstract
The binding profile of a known inhibitor, benzensulfonamide, against a family of carbonic anhydrase isozymes was efficiently enhanced via high-throughput screening of customized combinatorial one-bead-one-compound peptide libraries modified with the inhibitor molecule. The screening of the conjugate libraries recognized subtle variations in the microenvironments of the target enzyme and thus facilitated the identification of short peptide sequences that bind selectively to a close proximity of the active site. The identified peptide portions contributed significantly to the overall binding of the conjugate peptides with greatly enhanced affinity as well as improved specificity towards the target isozyme. The interactions between the inhibitors and the isozymes were validated by surface plasmon resonance (SPR), pull-down assay and enzymatic activity measurement. This high-throughput approach proved useful and efficient to enhance the binding profile of known inhibitors and may apply to developing effective inhibitors for a wide range of isozyme families.
Introduction
Isozymes, known as multiple forms of enzymes, play a major role in many biological processes and often show a great structural homology. Although selective isozyme inhibitors are a key issue in drug development, such a high homology among isozymes has frequently rendered the discovery of selective inhibitors extremely challenging.1 Therefore, tremendous research efforts have been paid to developing effective strategies that allow for enhancing the selective binding of the existing isozyme inhibitors.2-13 Developing allosteric inhibitors is one of the strategies to address such an aim, albeit an uphill challenge.14 Selective inhibition through binding to enzyme active sites is also challenging due to great structural homology among the isozymes.15 The lack of selectivity of conventional inhibitors against isozyme family demonstrates a clear demand for developing efficient strategies to enhance specificity of such inhibitors for diagnostic purposes as well as pharmaceutical application.
Carbonic anhydrases (CAs) is one of the common isozyme families in human body governing inter-conversions between carbon dioxide and bicarbonate with generation of proton. To the best of our knowledge, a total of sixteen human CA isozymes have been identified to date,16,17 including a number of small molecule inhibitors2-7 such as sulfonamides, sulfamates/bis -sulfamates, sulfamides, hydroxamates, sulfocoumarins, and sulfonamide - containing sugar moieties.8,9 These small molecule inhibitors bind to the common zinc ion of the active sites and have structural variations that account for a certain level of selectivity for different isozymes.4,7 These findings suggest possible compositional differences in microenvironments surrounding the isozyme active sites. Benzenesulfonamide is one of the well-known small molecule inhibitors for carbonic anhydrase families, especially with a dissociation constant (KD) of 2.1 - 3.9 μM and 1.3 - 1.5 μM for hCAI and hCAII, respectively.18 It was reported that conjugation of a synthetic polypeptide to this benzenesulfonamide cooperatively enhanced binding affinity as well as specificity.15,19 The enhanced specificity might be attributed to selective binding of the polypeptides in a slightly varied fashion in the proximity to the active site of each isozyme. These findings suggest that the more selective inhibitors can be developed by conjugating a known small molecule inhibitor to second ligand that binds to proximal sites with compositional and/or structural variations in a cooperative fashion. Screening of such conjugate libraries will facilitate cherry-picking of new motifs that bind cooperatively with the existing inhibitor to the active site. Other approaches, for example, phage display, microarray and mechanism-based designation, are also reported to develop such conjugate ligands.15,20
Amongst them, the bead-based combinatorial peptide library approach appears attractive to provide opportunities to disclose such cooperative second binding ligands.21b Peptides have been considered as attractive candidates in development of inhibitors or modulators due to their compositional and structural diversity via the well-established synthetic methods. In addition, peptides can be easily modified to fine-tune the properties such as affinity, stability and solubility.22 One of the most competitive advantages of this method is the capability of robust synthesis and rapid screening of millions of peptide sequences in a reliable fashion. Importantly, the bi-ligand candidates selected from several rounds of screenings would show significantly enhanced binding affinity and specificity against the target enzymes.23
Herein, we investigated whether the subtle variations in isozyme microenvironments can be utilized to enhance the binding profile of a common inhibitor that binds to the active site. Two representative isozymes, hCAII and hCAI were selected as the target molecules as well as hCAIX for a further comparison. With our approach based on conjugating benzenesulfonamide to another peptide ligand as shown in Fig. 1, customized one-bead-one-compound (OBOC) peptide libraries 21 (simply modified with benzenesulfonamide at N-terminus) were constructed, consisting of D-amino acids to circumvent the physiological stability issue in the downstream studies. The conjugate libraries were screened against hCAI and hCAII, respectively, to identify selective peptide sequences. Binding affinity and specificity of the sulfonamide-peptide inhibitor candidates were investigated by surface plasmon resonance (SPR), pull-down assay and enzymatic activity measurement (IC50).
Fig. 1.

Approaches based on conjugation of benzenesulf onamide to a selective peptide that binds to a close proximity of the active site of the two isozymes (blue: N, red: O, yellow: S).
Results and Discussion
Previously, we introduced an efficient platform for high-throughput screening of combinatorial peptide libraries by tackling several unsolved problems including the undesired high background interactions stemming from the charged fluorescent dyes used for labelling the target enzymes.23 With an aim to apply this versatile tool to identifying selective peptide ligands, a comprehensive pentamer peptide library consisting of 18 unnatural (D-form) amino acids, excluding methionine and cysteine, was conjugated to 4-carboxybenzylsulfonamide (4-CBS) with (Lib 1) and without a spacer (Lib 2), as shown in Fig. 2(a). Two representative spacer groups (i.e., PEG2 [20 atm] and nil) were adopted although they might be optimized for further enhanced bindings afterwards . A small amount of beads with a dummy sequence (GGGGG) conjugated to 4-CBS with/without a spacer (Con 1 and Con 2) were also prepared for control experiments, thereby to observe the magnitude of contribution from the peptide region in the binding to the target enzymes.
Fig. 2.

(a) Structures of the combinatorial library beads (Lib 1 and Lib 2) and the control beads (Con 1 and Con 2). (PEG)2: 20 atoms; aa: 18 D-amino acids; R: L-arginine; M: L-methionine. (b) Sorting images by COPAS from screening against hCAI and hCAII [X-axis = time of flight (TOF); Y-axis = fluorescence intensity (a.u.)]. Excitation: 640 nm solid-state laser; Detection conditions: Gain = 4, PMT = 750.
The designed combinatorial libraries were screened against the target isozymes (hCAII and hCAI) labelled with a zwitterionic near-infrared fluorescent dye (ZW700-1c).23,24 Based on fluorescence intensity, bright positive beads were sorted out into 96-well plates by an automatic bead sorter (COPAS™ PLUS, Union Biometrica, Inc.) as demonstrated in our previous work.23 The fluorescence intensity in the screening of Lib 1 against hCAII was rather comparable to that of the control beads under the identical conditions (Fig. 2b). These phenomena illustrate that the peptide region in Lib 1 does not contribute significantly to enhancing the binding affinity of 4-CBS. Besides, the collected positive peptides disclosed a random distribution of amino acids at each position (see SI, Fig. S1). Meanwhile, Lib 2 exhibited distinct screening and sequencing results for both target enzymes. Fluorescence level in the screening of Lib 2 against hCAII turned out considerably higher than that of the control beads under identical conditions, elucidating that the contribution of the peptide region of Lib 2 towards the bindings is significant (Fig. 2b). These libraries and controls were also screened against hCAI to further compare the fluorescence intensity associated with the contribution of the peptide region (Fig. 2c). Neither peptide motif nor spacer group noticeably enhances the binding affinity against hCAI based on fluorescence intensity. According to the sorting campaign by COPAS, enhancement of binding to hCAII by the peptides of Lib2 is greater than that to hCAI (Fig. 2b and 2c). Positive beads from both screenings contain dominantly negatively charged and hydrophobic amino acids, thereby to show a similar trend (see SI, Fig. S2). These results presumably stem from interactions with the positively charged and hydrophobic residues in the vicinity of the active site of the target enzymes.4b The results from a series of screenings suggest that Lib 2 is more promising for enhancing the binding properties of 4-CBS through further development.
The screening of Lib 2 against the two isozymes was repeated twice in order to accumulate more data in a reproducible fashion and thereby to propose a common focused library based on the occurrence of amino acids (see SI, Table S1). The focused library was screened against both hCAI and hCAII, respectively. These results contained highly homologous positive hit sequences with several conserved motifs such as d-v-d-d-x for hCAII and d-v-d-v-v for hCAI, respectively, where d represents the negatively charged and v for the hydrophobic D-amino acids, whilst x indicates all entities (see SI, Fig. S3 and S4). With two screening campaigns of the focused library completed, several repeating peptide sequences were short-listed as candidates for validation by SPR to measure their binding affinities. Table 1 depicts the selected peptides, i.e., seven for hCAII (Entries 2 – 8) and three for hCAI (Entries 9 – 11).
Table 1.
Summarized dissociation constants (KD) by SPRa
| Peptides | hCAII | hCAI | |
|---|---|---|---|
| KD, nM | KD, nM | ||
| 1 |
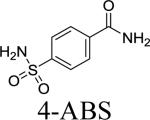
|
959 | 1220 |
| 2 | 4-CBS-dlddy | 49 | 89 |
| 3 | 4-CBS-dvdey | 63 | 55 |
| 4 | 4-CBS-dveye | 97 | 121 |
| 5 | 4-CBS-dleey | 88 | 116 |
| 6 | 4-CBS-dvddy | 82 | 102 |
| 7 | 4-CBS-dlddw | 22 | 25 |
| 8 | 4-CBS-dvddl | 156 | 137 |
| 9 | 4-CBS-dvdvl | 82 | 83 |
| 10 | 4- CBS-dvevl | 82 | 151 |
| 11 | 4- CBS-dadvl | 185 | 130 |
| 12 | 4- CBS-LKEYD | 59 | 81 |
KD values were obtained from a steady state fitting.
To investigate the influence of chirality, an analogous comprehensive library to Lib 2 was constructed with 18 natural (L-form) amino acids as the diversity elements (Lib 3). The positive hits were identified under identical screening conditions. Similar to the screening results from Lib 2, negatively charged and hydrophobic amino acids were dominant in the hit list from both the comprehensive and the focused libraries (see SI, Figure S5 and S6). However, individual positive sequences differ slightly from the case of Lib 2. One of the L-peptide identified from screening Lib 3 was included in the SPR experiments (Entry 12 in Table 1).
Association and dissociation kinetic constants of the 4-CBS-peptide conjugates were measured by SPR (see SI, Fig. S8 for representative sensograms). The results were compared with 4-amidobenzylsulfonamide (4-ABS). Although the peptide itself did not interact strongly with the target enzymes, the conjugated derivatives enhanced the binding affinity of 4-ABS up to 40 times in a synergistic fashion. All the selected sulfonamide-peptide conjugate candidates showed reasonably high degree of binding affinities with KD values between 22 and 156 nM for hCAII and between 25 and 151 nM for hCAI, respectively (Table 1). Based on the obtained KD values, the peptides 1, 2, 7 and 9 were selected for further validations by both pull-down assay and enzymatic activity measurements. It is noteworthy that the most frequently occurring hit peptides for each enzyme target during the screenings displayed the highest binding affinity for the respective enzymes in the pull-down experiments (see Fig. 3).
Fig. 3.

Pull-down experiments using the conjugated peptides 1, 2, 7 and 9, (a) with hCAII and hCAI, [hCA] = 0.57 μM, C1: control, mixture of hCAII and hCAI, (b) and (c) with a mixture of ZW700-hCAII and CY3-hCAI, C2: control, mixture of dye-labeled hCAII and hCAI. Visualization of the SDS gel under irradiation at (b) 645 nm, and (c) 550 nm.
The bindings of 1, 2, 7 and 9 towards hCAII and hCAI were investigated by pull-down experiments. Since these two isozymes have nearly identical molecular weights, they are inseparable by SDS-PAGE.15 Therefore, bindings between the conjugates and the target enzymes were investigated separately as shown in Fig. 3a. All of 2, 7 and 9 showed improved selectivity for hCAII compared to 4-ABS (1). In particular, 9 demonstrated the greatest capturing ability for hCAI than 1, 2 and 7. Considering that both isozymes bind to the benzenesulfonamide in a similar manner with KD values of 1 – 4 μM,18 conjugation to the peptides has increased the binding affinity considerably, consistent with the SPR results to some extent. To study the selectivity of the conjugates, a series of pull-down experiments were carried out with a mixture of ZW700-hCAII (red fluorescent dye)23 and CY3-hCAI (CY3: Cyanine3, green fluorescent dye), which enabled separate visualization of each enzyme on SDS-PAGE. The results show that 9 binds strongly to both targets although 2 and 7 have significantly increased binding selectively to hCAII (see Fig. 3b and 3c). It is noteworthy that a small variation in peptide sequence led to such a significant shift in the binding affinity between the isozymes. The fact that 9 appeared most frequently in the screening of the focused libraries against hCAI clearly addresses that our high-throughput screening platform worked efficiently to identify the optimal peptide sequences that bind strongly to the target enzyme (see SI, Table S7). To examine the specificity of the conjugates towards the CA family, pull-down experiments were carried out with a mixture of CA isozymes including a cytosolic protein (hCAII), trans membrane-associated proteins (hCAIX and hCAXII), and a mitochondria protein (hCAVB). All of 2, 7 and 9 showed selective capturing of hCAII on a SDS gel to a certain extent as shown in Fig. 4. These conjugated peptides exhibited good specificity towards hCAII, to meet our expectation.
Fig. 4.

SDS-PAGE analy sis of the isozy mes captured by 1, 2, 7 and 9 in the pull-down experiments with a mixture of hCAIX, hCAXII, hCAVB and hCAII. [CA] = 1.00 μM.
In addition, we measured the ability of 1, 2, 7 and 9 to inhibit in vitro esterase activity of the three CAs (Fig. 5, Table 2 and see SI, Figure S9). The obtained IC50 values of 4-ABS against hCAI, II and IX are similar to those in the reported values.7 Compared to 4-ABS, the inhibitory activity of 2 and 7 against hCAII resulted in ca. 17-fold increase. That of 9 against hCAI was particularly boosted by 30-fold, while the inhibitory activity against hCAII increased merely by 2.6-fold.
Fig. 5.

IC50 analysis of the conjugates for a) hCAII and b) hCAI. [hCA] = 200 nM
Table 2.
Average IC50 values of representative conjugated peptides
| Compound | hCAII | hCAI | hCAIXa |
|---|---|---|---|
| IC50 (nM) | IC50 (nM) | IC50 (nM) | |
| 1 | 1951 ± 4 | 3526 ± 6 | 3240 ± 4 |
| 2 | 112 ± 3 | 810 ± 3 | 1052 ± 4 |
| 7 | 104 ± 3 | 430 ± 3 | 963 ± 4 |
| 9 | 764 ± 2 | 120 ± 3 | 981 ± 3 |
| 12 | 210 ± 3 | 755 ± 2 | 880 ± 5 |
see SI, Fig. S9
These candidates were also tested for inhibition of the esterase activity of hCAIX (see SI, Fig. S9(c)). The obtained IC50 value was 10-fold higher than those values against hCAII and hCAI. These results highly comply with the pull-down experiments, but slightly differ from the SPR measurements. The consolidated results clearly address that the binding affinity and specificity of a common inhibitor can be significantly enhanced by our approach. In particular, 9 showed a good specificity for hCAI in terms of enzymatic inhibitory activity (IC50).
Conclusions
In this work, we successfully demonstrated that slight variations in microenvironments surrounding the enzyme active sites could be still distinguished as a way to enhance the binding profile of known inhibitors that commonly bind to the active sites of a series of isozymes. A combinatorial peptide library conjugated to benzenesulfonamide at N-terminus was constructed and screened by using our well-established high-throughput screening platform to efficiently identify the peptides with improved selectivity that bind to the close proximity of the active sites of hCAII and hCAI, respectively. Several peptide-benzenesulfonamide conjugates obtained from the screenings showed significantly higher binding affinity (KD: 20 – 180 nM) than benzenesulfonamide alone (KD: 1.3 – 1.5 μM). Although the potential conjugate inhibitors showed similar binding affinity for both isozymes by SPR measurements, their IC50 values against each isozyme and the results from pull-down experiments confirmed that specificity of the original small inhibitor can be improved by our approach based upon the robust high-throughput screening platform. Particularly, our screening platform allowed for discriminating only subtle variations in the binding sites, beneficially with a great reliability. These outcomes shed light upon developing useful applications for improving binding properties of existing inhibitors or modulators. Currently, we endeavor to apply this versatile approach to developing effective inhibitors for other isozyme families.2,25,26
Experimental Section
Materials and characterization
N-methylpyrrolidone (NMP), diethylether, dichloromethane (DCM) and Fmoc-PEG1-OH were purchased from Merck. Fmoc-protected D-amino acids (Fmoc-AAs) were purchased from GL Biochem Ltd (Shanghai, China). TentaGel S amino resin was purchased from Rapp Polymere. α-Cyano-4-hydroxycinnamic acid (CHCA) was purchased from Bruker. EZ-Link NHS-Biotin reagent was purchased from Thermo Scientific. hCAII (Aldrich), hCAI (Aldrich), hCAVB, hCAIX and hCAXII (Sinobiological Inc.) were purchased from commercial sources. Unless otherwise specified, chemicals were purchased from Aldrich. MALDI-MS and MS/MS spectra were obtained using ultrafleXtreme™ TOF/TOF (Bruker). Microwave-assisted CNBr-based cleavage reactions were performed by a household microwave oven (model R-248J, 800 W, 2450 MHz) from Sharp Inc. The PEAKS software was purchased from Bioinformatics Solutions Inc. The purification of bulk peptides was done by a preparative HPLC system from Gilson on a C18 reversed phase preparative column (Kromasil® from AkzoNobel, 5 μm, 250 × 30 mm).
Construction of peptide libraries
Random OBOC peptide libraries were synthesized using our reported method.27,28 For the solutions of Fmoc-isoleucine and Fmoc-glutamine, it contained 10 mol% of glycine each for discrimination of isobaric residues in the semi-automatic peptide sequencing. Peptide density was reduced to quarter loading to minimize the non-specific binding of protein containing of a negatively charged surface.29

Labeling of protein with dye
To label hCAII and hCAI with ZW700-1c,24 100 μl of a hCAII solution (2 mg/mL) in PBS (pH 8.0) was mixed with 4 molar equivalents of NHS-activated dye dissolved in DMSO (10 mg/ml). The mixture was incubated for 1 h at room temperature under dark conditions. The dye-labeled protein was purified by using size exclusion chromatography. Upon purification the resulting dye-labeled protein was characterized by UV-vis spectroscopy and SDS-PAGE. Protein concentration was determined by UV absorbance at 280 nm.
Library screening
For the screen, 100 mg of library resin was transferred into an Alltech vessel (8 ml, equipped with a filer) and pre-incubated in a blocking solution, comprising of 0.05% NaN3, 0.05% Tween 20, and 1% BSA in PBS buffer (pH 7.4), for 1 h on a 360° shaker at 25 °C. Dye-labeled protein was added to a final concentration of 200 nM and the library was incubated for overnight on a 360° shaker at 4 °C. The liquid was drained and the resulting beads were washed three times with the blocking solution and three times with 0.05% Tween 20 in PBS buffer sequentially. After washing, the beads were transferred into a sample vessel of COPAS Plus (Union Biometrica) and diluted with 200 mL of PBS buffer with 0.05% Tween 20 (pH 7.4). Two-step sorting was carried out to sort out positive beads. In the second sort, positive beads were directly sorted into a 96 titer well plate with cone-shaped wells.
CNBr-based cleavage of peptides from single beads
The 96 well plate with sorted beads was purged by argon for 15 min and then CNBr (10 μL, 0.50 M in 0.2 N HCl solution) was added into each well. After additional purging by argon/nitrogen for 15 min, the 96 well plate was sealed and placed under microwave radiation for 1 min. The resulting solution was concentrated under centrifugal vacuum for 2.5 h at 45 °C.30
MALDI-MS and MS/MS analysis of peptides cleaved from single beads
To each well were added CHCA (7 μL, 0.4% solution in acetonitrile/water (1:1)) and then acetonitrile/ water (7 μL, 1:1 with 0.1% trifluoroacetic acid (v/v)). 2.5 μL of the mixture was spotted onto a 384-well MALDI plate, which was allowed to stand for 15 min to dry naturally. MS and MS/MS acquisition was conducted with ultrafleXtreme™ MALDI-TOF/TOF mass spectrometer from Bruker Daltonics.
Synthesis of individual peptides
Each peptide was synthesized on Rink amide resins (0.63 mmol/g) on a typical resin scale of 20 mg per sequence in bulk for affinity measurements . With the desired sequence of peptide attained, the resin was treated in trifluoroacetic acid (95%), water (2.5%), and triisopropylsilane (2.5%) for 2 h. The cleavage cocktail was concentrated in a continuous flow of nitrogen, and the crude peptides were precipitated in diethyl ether. The resulting white solid was then purified to >95% in purity by HPLC bearing a C18 reversed-phase preparative column. The purified peptides were used for validation, for example, affinity measurements via surface plasmon resonance (SPR).
Bulk synthesis of biotinylated peptides
Bulk synthesis of biotinylated peptides was performed on Rink amide resins (0.31 mmol/g) on a typical resin scale of 50 mg per sequence. First, Fmoc-Lys(Mtt)-OH was coupled to the resin. Then, 4-methyltrityl (Mtt) group was deprotected selectively by reacting with TFA/TIS/DCM (3/3/94) for 2 min, 5 min and 30 min successively, using a fresh aliquot each time. At the following step, biotin-NHS in NMP with DIEA was added and the mixture was vortexed for 30 min. With all amino acids coupled, the resin was treated in trifluoroacetic acid (95%), water (2.5%), and triisopropylsilane (2.5%) for 2 h. The cleavage cocktail was concentrated in a continuous flow of nitrogen, and the crude peptides were precipitated in diethyl ether. The resulting white solid then purified to >95% in purity by HPLC bearing a C18 reversed-phase preparative column. The purified peptides were used for validation, specificity test.
Affinity measurements
Affinity measurements were carried out on a Biacore T100 instrument (GE Healthcare). The CM5 sensor chip was used for all measurements. The sensor was primed with HBSEP+ (10 mM HEPES pH 7.4, 150 mM NaCl, 3.4 mM EDTA, 0.005% P20, GE Healthcare) buffer and was activated using amine coupling through 1:1 mixture of 0.4 M EDC and 0.1M NHS and remaining activated groups was blocked with 1 M solution of ethanolamine (pH 8.5). Either hCAII or hCAI was immobilized onto the sensor chip surface by approximately 5500 response units (RU). A blank flow cell (no immobilized protein) was used as a reference to subtract nonspecific binding, drift, and the bulk refractive index. Next, varying concentrations of peptide (31-500 nM) were passed over the chip for 25 min at a flow rate of 50 μL/min. Association (ka) and dissociation (kd) rate constants were calculated with a 1:1 binding model using Biacore evaluation software, and KD values were calculated from the ratio of kd to ka. Kinetic parameters were obtained by fitting curves to a 1:1 Langmuir model with baseline correction.
Pull-down experiment
Add 70 μL of streptavidin agarose resin (Thermo Scientific) to 1ml PBS and drain all solution. Excess of biotinylated peptides was added into streptavidin agarose resin in 300 μL PBS, then was incubated for 2 h. Resin was washed using PBS and PBST 0.05% for 3-4 times to remove the excess of peptide. It was incubated with target proteins, hCAII and hCAI, respectively at 4°C for overnight then was washed. 4× SDS loading buffer, reducing agent were added to the resins and then boiled for 10 min at 99 °C for denaturation. For the fluorescence image, dye labelled proteins were used, ZW700-hCAII and CY3-hCAI, respectively.
Esterase activity
Carbonic anhydrase catalyzed the hydrolysis of 4-nitrophenyl acetate (4-NPA) to nitrophenol, whose appearance was monitored by absorbance at 400 nm.31 The solution assay includes 0.2 μM CA, appropriate amount of conjugates as a blocker and 50 μM 4-NPA in 200 μL in tris buffer composed of 9 mM Tris-HCl, 81 mM NaCl, 10% acetonitriles (v/v) and 1% DMSO (v/v). All experiments were performed at least in triplicates.
Supplementary Material
Acknowledgements
This work was supported by the Institute of Bioengineering and Nanotechnology (Biomedical Research Council (BMRC), Agency for science, Technology and Research (A*STAR), BMRC-Science and Engineering Research Council Diagnostics Grant (A*STAR), and the National Institutes of Health: NIBIB grant #R01-EB-011523 (HSC).
Footnotes
† Footnotes relating to the title and/or authors should appear here.
Electronic Supplementary Information (ESI) available: Detailed experimental section, additional results, SPR data and peptide sequences. See DOI: 10.1039/b000000x/
References
- 1.Supuran CT. Nat. Rev. Drug Discovery. 2008;7:168–181. doi: 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- 2.Tanc M, Carta F, Bozdag M, Scozzafava A, Supuran CT. Bioorg. & Med. Chem. 2013;21:4502–4510. doi: 10.1016/j.bmc.2013.05.032. [DOI] [PubMed] [Google Scholar]
- 3.Addy PS, Saha B, Singh NDP, Das AK, Bush JT, Lejeune C, Schofield CJ, Basak A. Chem. Commun. 2013;49:1930–1932. doi: 10.1039/c3cc38251f. [DOI] [PubMed] [Google Scholar]
- 4.a Bozdag M, Ferra roni M, Carta F, Vullo D, Lucarini L, Orlandini E, Rossello A, Nuti E, Scozzafava A, Masini E, Supuran CT. J. Med. Chem. 2014;57:9152–9167. doi: 10.1021/jm501314c. [DOI] [PubMed] [Google Scholar]; b Alterio V, Fiore AD, D'Ambrosio K, Supuran CT, Simone GD. Chem. Rev. 2012;112:4421–4468. doi: 10.1021/cr200176r. [DOI] [PubMed] [Google Scholar]; c Vitale RM, Alterio V, Innocenti A, Winum J-Y, Monti SM, De Simone G, Supuran CT. J. Med. Chem. 2009;52:5990–5998. doi: 10.1021/jm900641r. [DOI] [PubMed] [Google Scholar]
- 5.Gavernet L, Funes JLG, Blanch LB, Estiu G, Maresca A, Supuran CT. J. Chem. Inf. Model. 2010;50:1113–1122. doi: 10.1021/ci100112s. [DOI] [PubMed] [Google Scholar]
- 6.Scolnick LR, Clements AM, Liao J, Crenshaw, Hellberg M, May J, Dean TR, Christianson DW. J. Am. Chem. Soc. 1997;119:850–851. [Google Scholar]
- 7.Lyer R, Barrese AA, III, Parakh S, Oarker CN, Tripp BC. J. Biomol. Screen. 2006;11:782–791. doi: 10.1177/1087057106289403. [DOI] [PubMed] [Google Scholar]
- 8.Ombouma J, Vullo D, Dumy P, Supuran CT, Winum J-Y. ACS Med. Chem. Lett. 2015;6:819–821. doi: 10.1021/acsmedchemlett.5b00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lopez M, Bornaghi LF, Innocenti A, Vullo D, Charman SA, Supuran CT, Poulsen S-A. J. Med. Chem. 2010;53:2913–2926. doi: 10.1021/jm901888x. [DOI] [PubMed] [Google Scholar]
- 10.a Krall N, Pretto F, Decurtins W, Bernardes GJL, Supuran CT, Neri D. Angew. Chem. Int. Ed. 2014;53:4231–4235. doi: 10.1002/anie.201310709. [DOI] [PubMed] [Google Scholar]; b Krall N, Pretto F, Neri D. Chem. Sci. 2014;5:3640–3644. [Google Scholar]
- 11.Touisni N, Kanfar N, Ulrich S, Dumy P, Supuran CT, Mehdi A, Winum J-Y. Chem. Eur. J. 2015;21:10306–10309. doi: 10.1002/chem.201501037. [DOI] [PubMed] [Google Scholar]
- 12.Tanpure RP, Ren B, Peat TS, Bornaghi LF, Vullo D, Supuran CT, Poulsen S-A. J. Med. Chem. 2015;58:1494–1501. doi: 10.1021/jm501798g. [DOI] [PubMed] [Google Scholar]
- 13.a Mack ET, Snyder PW, Perez-Castillejos R, Bilgiçer B, Moustakas DT, Butte MJ, Whitesides GM. J. Am. Chem. Soc. 2012;134:333–345. doi: 10.1021/ja2073033. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Banerjee AL, Eiler D, Roy BC, Jia X, Haldar MK, Mallik S, Srivastava DK. Biochemistry. 2005;44:3211–3224. doi: 10.1021/bi047737b. [DOI] [PubMed] [Google Scholar]
- 14.a Hardy JA, Wells JA. Curr. Opin. Struct. Biol. 2004;14:706–715. doi: 10.1016/j.sbi.2004.10.009. [DOI] [PubMed] [Google Scholar]; b Laskowski RA, Gerick F, Thornton JM. FEBS Lett. 2009;583:1692–1698. doi: 10.1016/j.febslet.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 15.Tegler LT, Fromell K, Jonsson B-H, Viljanen J, Winander C, Carlsson J, Baltzer L. Chembiochem. 2011;12:559–566. doi: 10.1002/cbic.201000556. [DOI] [PubMed] [Google Scholar]
- 16.Frost SC. Subcell. Biochem. 2014;75:9–30. doi: 10.1007/978-94-007-7359-2_2. [DOI] [PubMed] [Google Scholar]
- 17.Supuran CT. Curr. Pharm. Des. 2008;14:603–614. doi: 10.2174/138161208783877884. [DOI] [PubMed] [Google Scholar]
- 18.a Winum J-Y, Vullo D, Casini A, Montero J-L, Scozafava A, Supuran CT. J. Med. Chem. 2003;46:2197–2204. doi: 10.1021/jm021124k. [DOI] [PubMed] [Google Scholar]; b Papalia GA, Leavitt S, Bynum MA, Katsamba PS, Wilton R, Qiu H, Steukers M, Wang S, Bindu L, Phogat S, Giannetti AM, Ryan TE, Pudlak VA, Matusiewicz K, Michelson KM, Nowakowski A, Pham-Baginski A, Cho YH, Wit MD, Smets A, Vandersmissen J, Michiels L, Myszka DG. Anal Biochem. 2006;359:94–105. doi: 10.1016/j.ab.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 19.Baltzer L. Anal. Bioanal. Chem. 2011;400:1653–1664. doi: 10.1007/s00216-011-4905-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a Morimoto J, Hayashi Y, Suga H. Angew. Chem. Int. Ed. 2012;51:3423–3427. doi: 10.1002/anie.201108118. [DOI] [PubMed] [Google Scholar]; b Shomin CD, Meyer SC, Ghosh I. Bioorg. Med. Chem. 2009;17:6196–6202. doi: 10.1016/j.bmc.2009.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lim H-S, Reddy MM, Xiao X, Wilson J, Wilson R, Connell S, Kodadek T. Bioorg. Med. Chem. Lett. 2009;19:3866–3869. doi: 10.1016/j.bmcl.2009.03.153. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Meyer SC, Shomin CD, Gaj T, Ghosh I. J. Am. Chem. Soc. 2007;129:13812–13813. doi: 10.1021/ja076197d. [DOI] [PubMed] [Google Scholar]
- 21.a Lam KS, Salmon SE, Hersh EM, Hruby VJ, Kazmierski WM, Knapp RJ. Nature. 1991;354:82–84. doi: 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]; b Lam KS, Lebl M, Krchňák V. Chem. Rev. 1997;97:411–448. doi: 10.1021/cr9600114. [DOI] [PubMed] [Google Scholar]
- 22.Gray BP, Brown KC. Chem. Rev. 2014;114:1020–1081. doi: 10.1021/cr400166n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jee J-E, Lim J, Hyun H, Oon J, Ong YS, Massif C, Chang Y-C, Choi HS, Lee SS. Chem Commun. 2014;50:15220–15223. doi: 10.1039/c4cc07008a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hyun H, Henary M, Gao T, Narayana L, Owens EA, Lee JH, Park G, Wada H, Ashitate Y, Frangioni JV, Choi HS. Mol Imaging Biol. 2016;18:52–61. doi: 10.1007/s11307-015-0870-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Winum J-Y, Rami M, Scozzafava A, Montero JL, Supuran CT. Med. Res. Rev. 2008;28:445–463. doi: 10.1002/med.20112. [DOI] [PubMed] [Google Scholar]
- 26.Mahon BP, Lomelino CL, Ladwig J, Rankin GM, Driscoll JM, Salguero AL, Pinard MA, Vullo D, Supuran CT, Poulsen S-A, Mckenna R. J. Med. Chem. 2015;58:6630–6638. doi: 10.1021/acs.jmedchem.5b00845. [DOI] [PubMed] [Google Scholar]
- 27.Lee SS, Lim J, Tan S, Cha J, Yeo SY, Agnew HD, Heath JR. Anal. Chem. 2010;82:672–679. doi: 10.1021/ac902195y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cha J, Lim J, Zheng Y, Tan S, Ang YL, Oon J, Ang MW, Ling J, Bode M, Lee SS. J. Lab. Autom. 2012;17:186–200. doi: 10.1177/2211068211433503. [DOI] [PubMed] [Google Scholar]
- 29.Chen X, Tan PH, Zhang Y, Pei D. J. Comb. Chem. 2009;11:604–611. doi: 10.1021/cc9000168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee SS, Lim J, Cha J, Tan S, Heath JR. J. Comb. Chem. 2008;10:807–809. doi: 10.1021/cc800113d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agnew HD, Rohde RD, Millward SW, Nag A, Yeo W-S, Hein JE, Pitram SM, Tariq AA, Burns VM, Krom RJ, Fokin VV, Sharpless KB, Heath JR. Angew. Chem. Int. Ed. 2009;48:4944–4948. doi: 10.1002/anie.200900488. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.