

Abstract
Amyotrophic lateral sclerosis (ALS) is a lethal paralytic disease caused by the degeneration of motor neurons in the spinal cord, brain stem, and motor cortex. Mutations in the gene encoding copper/zinc superoxide dismutase (SOD1) are present in ~20% of familial ALS and ~2% of all ALS cases. The most common SOD1 gene mutation in North America is a missense mutation substituting valine for alanine (A4V). In this study, we analyze sodium channel currents in oocytes expressing either wild-type or mutant (A4V) SOD1 protein. We demonstrate that the A4V mutation confers a propensity to hyperexcitability on a voltage-dependent sodium channel (Nav1.3) mediated by heightened total Na+ conductance and a hyperpolarizing shift in the voltage dependence of Nav1.3 activation. To estimate the impact of these channel effects on excitability in an intact neuron, we simulated these changes in the program NEURON; this shows that the changes induced by mutant SOD1 increase the spontaneous firing frequency of the simulated neuron. These findings are consistent with the view that excessive excitability of neurons is one component in the pathogenesis of this disease.
Electronic supplementary material
The online version of this article (doi:10.1007/s10867-016-9411-x) contains supplementary material, which is available to authorized users.
Keywords: Nav channel, Amyotrophic lateral sclerosis, Hyperexcitability, Superoxide dismutase, Oocyte
Introduction
Amyotrophic lateral sclerosis (ALS) is a lethal paralytic disease caused by neuronal degeneration in the spinal cord, brain stem, and motor cortex [1]. About 10% of ALS cases are familial (FALS) transmitted as dominant traits; to date, mutations in more than 40 different genes have been associated with FALS. A subset of approximately 20% of these cases is a consequence of mutations in the gene encoding cytosolic copper/zinc superoxide dismutase (SOD1) [2]. In North America, by far the most common SOD1 mutation is a substitution of valine for alanine at position 4 (A4V). Regrettably, this mutation is associated with an extremely rapid course, with survival typically less than 1.5 years [3].
Numerous mechanisms underlie the pathophysiology of neuronal degeneration in ALS. Considered together, the diverse set of ALS-associated genes implicates three major categories of cellular pathology in ALS, including (1) instability and misfolding of abundant proteins (such as SOD1), (2) perturbations in RNA biology arising either from aberrant trafficking of nucleic acid binding proteins or from expansions of repeat domains in genomic DNA, and (3) disturbances of cytoskeletal proteins that underlie axonal growth and function. In turn, these three sets of mechanisms lead to multiple adverse downstream events, including: generation of reactive oxygen and nitration species (ROS), mitochondrial degeneration, accumulation of intracellular aggregates of both protein and RNA, induction of ER stress and the unfolded protein response and activation of cell death pathways (both apoptotic and necrotic) [1, 4]. Also evident, both in vivo and in vitro, are data implicating altered neuronal excitability as another key component of the pathophysiology of ALS. Neuronal hyperexcitability has been documented in primary spinal motor neuron cultures [5, 6], in ALS motor neurons derived from induced pluripotential stem cells [7, 8] and in slice preparations from transgenic mice carrying the familial ALS mutation G93A (hSOD1G93A) [9, 10]. In the latter instance, the ALS mice exhibited augmented neuronal excitability and increased persistent sodium current [11]. The hyperexcitability reflects at least three factors: intrinsic hyperexcitability of the neurons themselves, excessive excitatory inputs from interneurons [10], and pro-excitatory effects of soluble factors secreted by astrocytes [9]. Hypoexcitability has also been observed in iPSC-derived ALS motor neurons [12] and is also proposed to be a critical element in the early vulnerability of selected neuronal populations in ALS [13, 14].
Several studies have documented disturbances in voltage-dependent Nav currents and increased persistent sodium current (PICNa) in embryonic neurons from ALS mice. Voltage-gated Na+ channels (Nav), which underlie neuronal excitation and action potential propagation, consist of a 260-kDa α subunit and one or more 30–40-kDa β subunits. Because the α subunits form the actual ion channel as well as sensors of voltage dependence, they are inherently the working cores of the excitation process. β subunits modulate the kinetics of voltage-dependence activation and inactivation as well as channel localization on the cell membrane [15, 16].
In embryonic development, the predominant Na+ isoforms expressed in motor neurons are Nav1.2 and Nav1.3. Expression of Nav1.3 begins very early in embryogenesis and then increases, attaining a maximum level at birth; it then declines after the second postnatal week to very low levels at adulthood [17–19]. Nav1.1 and 1.6 expression levels predominate at later developmental stages [17]. For example, in the rat, Nav1.1, Nav1.2, and Nav1.6 are expressed at robust levels in the adult CNS while Nav1.3 levels are substantially reduced [18]. In the experiments reported here, we elected to focus on Nav1.3 because many of the studies pointing to motor neuron hyperexcitability in ALS are based on embryonic or immediately post-natal motor neurons (particularly in vitro) as discussed below.
To date, there have been no analyses of the impact mutant ALS genes and proteins have on the biophysical properties of individual voltage-gated Na+ channels. In the current study, we show that a common familial ALS mutant, hSOD1A4V, shifts the voltage dependence and augments total Na+ currents of the Nav channel, Nav1.3, an isoform that is characteristic of embryonic motor neurons [17], which is documented to have a persistent inward sodium current and has been implicated in human neurological disease [20, 21]. Our results suggest that changes in the biophysical properties of voltage-gated sodium channels are important in the genesis of mutant SOD1-induced hyperexcitability in ALS.
Materials and methods
Molecular biology
Human Nav1.3 α subunit isoform 2 (NCBI Reference Sequence: NP_001075145.1) DNA was obtained from J.A. Kearney (Vanderbilt University, Nashville, TN, USA). β1 subunit DNA was obtained from C. Ahern (University of British Columbia, Vancouver, Canada). The constructs were linearized with the appropriate restriction enzyme (New England Biolabs) and cRNA was synthesized using in vitro run-off transcription with T7 polymerase (Promega).
SOD1WT/A4V expression and purification
pET3d vectors containing human wild-type (hSOD1WT) and mutant (hSOD1A4V) SOD1 cDNA were expressed in BL21 (DE3) PlysS cells. To express the SOD1 protein, the bacterial culture was incubated with 1 mM isopropyl β-d-thiogalactopyranoside (Sigma) in the presence of 200 μM copper (II) chloride (Sigma) and 200 μM zinc chloride (Sigma) for 3 h at 37 °C (WT) or 30 °C (A4V). The protein was harvested when the culture achieved an optical density (A600) between 0.6 and 0.8; hSOD1WT and hSOD1A4V proteins were harvested and purified from cell pellets as previously described [22, 23].
Immunoblotting
Immunoblots of individuals and groups of oocytes were washed with PBS and lysed in RIPA buffer. Lysates were sonicated and cleared by centrifugation at 12,000 rpm for 5 min. The protein content of the cleared lysate was quantified by BCA colorimetric assay (Thermo, USA). For denaturing Western-blot analyses, 5 μg of total protein was loaded on 12% Tris-glycine gels (Invitrogen, USA) in Tris-Glycine SDS running buffer (Invitrogen, USA) and transferred to nitrocellulose membrane using i-Blot® (Invitrogen, USA). Membranes were blocked for 1 h in blocking solution (LiCor, USA) containing 0.1% Tween-20. Sheep anti-human SOD1 antibody (The Binding Site) was incubated at 4 °C overnight at a 1:2000 dilution. After washing in PBS containing 0.1% Tween-20, blots were incubated for 1 h with fluorophore-conjugated donkey anti-sheep antibody (LiCor, USA) prior to visualization and analysis by the Odyssey infrared imaging system (LiCor, USA).
Electrophysiology
Oocytes were surgically removed from Xenopus laevis as previously described [24]. The extraction procedure and care of Xenopus laevis were approved by the University of Massachusetts Institutional Animal Care and Use Committee. Twenty-four hours after surgery, oocytes were first microinjected with mRNA (6 ng α subunit, 1.2 ng β1 subunit) then subsequently with 25 ng of purified hSOD1 protein. Microinjected oocytes were stored in antibiotic-supplemented ND96 buffer [24].
Currents were measured ~ 18 h after injection using a two-electrode voltage clamp (OC-725; Warner Instrument Corp.); the data were acquired with a Digidata 1322A (Axon Instruments) running pClamp 9 (Axon Instruments) at room temperature. Electrodes were filled with: 3 M KCl, 10 mM HEPES, and 5 mM EGTA, pH 7.6. Currents were measured in ND96 recording buffer (pH 7.6) containing: 96 mM NaCl, 5 mM HEPES, 2 mM KOH, 1 mM MgCl2, and 0.3 mM CaCl2.
Total current
The quantification of total current through the oocytes, the voltage dependence of activation, and the kinetics of fast inactivation were evaluated with a protocol in which oocytes were depolarized from a holding potential of –100 mV to a range of potentials from –80 to +60 mV in 10 mV increments for 50 ms. Voltage step depolarizations were delivered every 5 s. Data analysis was performed with Clampfit 9 (Axon Instruments) and Prism 5 software (GraphPad). The amplitude of the current was measured at each potential and normalized such that the maximal current in oocytes injected with mRNA alone was equal to 1. The currents from potentials –80 mV to +60 mV of each oocyte from the protein injected group (WT or mutant) were normalized to the mRNA control group. For this and all other electrophysiology experiments, the Grubbs test was used to exclude any outliers. Data are presented as mean ± SEM. Differences between values were examined by ANOVA followed by Tukey’s multiple comparison test with statistical significance established at p < 0.05.
Voltage dependence of activation
Current–voltage relationships were measured by holding at –100 mV and stepping to a series of test potentials for 50 ms in 10 mV increments, followed by a tail pulse at –100 mV. Voltage step depolarizations were delivered every 5 s to allow for channels closing between depolarizations. To measure the voltage dependence of activation, maximum currents at each potential were normalized to the maximum peak current. Normalized tail currents were plotted versus the test potential to produce activation curves and fit to the Boltzmann equation:
where V is the applied pulse potential and V1/2 is the voltage of half-maximal activation. This was calculated for each individual experiment. Within each experiment set (control, hSOD1WT, hSOD1A4V) the V1/2 values were then averaged to obtain the mean V1/2 for that set.
Voltage dependence of inactivation
Oocytes were held at –100 mV. A conditioning pulse was applied to a range of potentials from –120 to 0 mV in 20-mV increments for the duration of 500 ms, followed by a test pulse to –30 mV for 50 ms. Voltage step depolarizations were delivered every 5 s to allow for channel closing. The peak current at each test pulse was normalized to the current of the conditioning pulse. Normalized values were plotted against the test pulse voltage and the data were fit with a Boltzmann sigmoidal function:
where V is the applied pulse potential and V1/2 is the half maximal inactivation.
Kinetics of fast inactivation
Oocytes were held at a holding potential of –100 mV. Oocytes were depolarized to a range of potentials from –80 to +60 mV in 10 mV increments for 50 ms. Voltage step depolarizations were delivered every 5 s to allow for channel closing between depolarizations. To evaluate the kinetics of fast inactivation, the decay phase of current traces acquired by a depolarizing step to –30, –20, –10, 0, and 10 mV from a holding potential of –100 mV were fitted best by the sum of two exponential standard functions:
where A1 and A2 are the approximate portions of the inactivated currents and τ1 and τ2 the respective time constants. The function consisted of a fast component, which forms the transient Na+ current and a long-lasting, slow, component, which forms PICNa.
Recovery from fast inactivation
Oocytes were held at –100 mV. A depolarization pulse was applied from the holding potential to a step potential of 0 mV for 50 ms to induce inactivation. A second depolarizing pulse (also from –100 to 0 mV) was applied to activate the Na+ current. The inter-pulse interval was initially 50 ms and was increased by 1-ms steps to define the rate of recovery. The entire protocol entailed 40 sweeps. The time courses for recovery from fast inactivation data were evaluated by normalizing the current amplitude during the second depolarization test pulse to that of the first depolarization test pulse. The data were fit to the one phase decay function:
where K is the rate constant.
All experiments were conducted using three different batches of oocytes; we typically tested three conditions (control, hSOD1WT and hSOD1A4V) on subsets of equivalent numbers of oocytes from each batch.
NEURON program simulations
The open-source program NEURON [25] was used to model the effects on firing frequency of hSOD1WT and hSOD1A4V proteins. This model uses conventional Hodgkin and Huxley simulations [26] involving the neuronal soma and two passive membrane dendrites in order to keep the simulation environment simple, reflecting our oocyte conditions. The basic equations used for our simulations were taken from [27]:
| 1 |
| 2 |
| 3 |
| 4 |
| 5 |
| 6 |
| 7 |
| 8 |
The equations used for the ‘m’ sodium
activation system were
| 9 |
| 10 |
and for the ‘h’ sodium inactivation system we have
| 11 |
| 12 |
Our simulations of excitability in the presence of hSOD1WT protein used the baseline parameters listed in Table 1a, b; these were derived from the following file: (http://neuron.yale.edu/hg/neuron/nrn/file/tip/src/nrnoc/hh.mod). We introduced our experimental data by varying first independently and then simultaneously the two parameters that we observed to be experimentally significant: (1) the peak conductance of the Nav channel (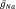 ) and (2) the voltage dependence of activation.
) and (2) the voltage dependence of activation.
Table 1.
Parameters for simulations*
| A. | ||||
| nseg | diam | L | Ra | |
| Soma | 1 | 18.8 | 18.8 | 123 |
| Dendrite #1 | 5 | 3.18 | 701.9 | 123 |
| Dendrite #2 | 5 | 2 | 549.1 | 123 |
| B. | ||||
| (S/cm2) | ||||
|
(S/cm2) |
0.25 | |||
| gI (S/cm2) | 0.0001666 | |||
| gKbar (S/cm2) | 0.036 | |||
| el (mV) | −60 | |||
*
the maximum specific sodium channel conductance, gKbar the maximum specific potassium channel conductance, gl the maximum specific leakage conductance, E
L the reversal potential for the leakage channel, nseg is the number
The peak conductance of Nav channel () simulations
The conductance gnabar (the maximum specific sodium channel conductance) was set at 0.25 S/cm2 at the baseline, reflecting conditions of electrical silence when wild-type SOD1 was present. To model the impact of mutant SOD1, we increased this conductance by 0.04 to 0.29 S/cm2. This initiated spontaneous generation of action potentials.
The voltage dependence of activation of the Na+ current
The baseline activation parameters for the midpoint of m gating (activation) included αm = –40 mV and βm = –65 mV (Eqs. 9 and 10); this baseline simulated both the control situation and the experiments in which WT SOD1 was present; in this circumstance, the model did not demonstrate spontaneous generation of action potentials. To simulate the impact of mutant SOD1, the observed hyperpolarizing change in the voltage dependence of activation (V1/2) was modeled by shifting the gating constant for activation (alpha and beta) by 1 mV hyperpolarizing increments. In each instance, the key output was the frequency of spontaneous firing. A shift of 3 mV (αm = –43 mV; βm = –68 mV), which was observed experimentally (see Results below), was associated with activation of repetitive firing. In these simulations, gating of the potassium currents was not changed.
Additionally, we explored the impact of altering the voltage dependence of inactivation. At the baseline, steady-state half-inactivation, αh = –65 mV and βh = –35 mV, (Eqs. 11, 12). These constants were left at their default value for our control and wild-type protein experimental conditions. To simulate the effect of a depolarizing shift in the voltage dependence of inactivation, alpha and beta were in the depolarizing direction; experimentally, the shift was –3 mV (αh = –62 mV; βh = –32 mV). Gating for potassium activations were left at their default values.
Results
To study the impact of WT and mutant (A4V) SOD1 protein on the properties of Nav1.3, we used an oocyte expression system. We elected to study the A4V variant of SOD1 because this is the most common SOD1 gene mutation in North America and because it has an aggressive clinical course, with survival typically about 1 year [3]. As described above, electrophysiological studies were performed approximately 18 h after injecting purified SOD1 protein together with the cDNA expressing the α and β subunits of Nav1.3. We first documented that hSOD1 protein (both WT and A4V) could be detected by Western blotting in oocyte extracts not only immediately after injection but also at 18 h post-injection (Fig. S1a, c). We confirmed expression and trafficking of Nav1.3-β1 complexes by measuring the current (Fig. S1b); uninjected oocytes have negligible sodium currents. We first compared the influence of hSOD1WT vs. hSOD1A4V on peak Nav1.3 currents elicited by –100 mV to a range of test potentials. Figure 1 shows a normalized Nav1.3-β1 channel current elicited by 50-ms depolarizations in standard ND96 buffer. For all but two test voltages (–40 and 10 mV), the peak Na+ current in the presence of hSOD1A4V exceeded that seen with hSOD1WT (Table 2).
Fig. 1.

SOD1 protein increases NaV1.3-β1 channel current. Quantification of Nav1.3 current levels from oocytes co-injected with different hSOD1 proteins. The effect of hSOD1WT vs. hSOD1A4V protein on peak NaV1.3-β1 channel currents was compared for depolarizations from –100 mV to a range of test potentials. Values are normalized to oocytes injected with only α and β subunit mRNA (without exogenous hSOD1). For all but two test voltages (–40 and 10 mV), the peak Na+ current in the presence of hSOD1A4V exceeded that seen with hSOD1WT. Data are presented as the mean ± SEM from multiple oocytes (Table 2). Asterisks indicate significant difference between data using hSOD1A4V and either hSOD1WT or no hSOD1. (ANOVA with Tukey’s post test ***p ≤ 0.001, **p ≤ 0.01, *p ≤ 0.05)
Table 2.
Normalized current and standard errors of the mean, Fig. 1
| Voltage (mV) | Control (n = 26) | hSOD1WT (n = 23) | hSOD1A4V (n = 22) |
|---|---|---|---|
| −40 | 1 ± 0.29 | 4.02 ± 1.78 | 6.23 ± 2.68 |
| −30 | 1 ± 0.15 | 1.42 ± 0.37 | 2.36 ± 0.36 |
| −20 | 1 ± 0.12 | 1.34 ± 0.23 | 1.82 ± 0.24 |
| −10 | 1 ± 0.12 | 1.19 ± 0.13 | 1.73 ± 0.22 |
| 0 | 1 ± 0.12 | 1.35 ± 0.18 | 2.01 ± 0.27 |
| 10 | 1 ± 0.14 | 1.43 ± 0.20 | 2.22 ± 0.35 |
| 20 | 1 ± 0.18 | 1.44 ± 0.22 | 2.63 ± 0.48 |
| 30 | 1 ± 0.23 | 1.44 ± 0.29 | 3.20 ± 0.69 |
To determine whether this increase in current was due to a shift in the voltage sensitivity of NaV1.3-β1 channel complexes, we measured the channel current at different voltages (Fig. 2a). The threshold and voltage dependence of activation for the peak sodium current were similar for control and complexes injected with hSOD1WT (Fig. 2b). By contrast, in the presence of the mutant hSOD1A4V protein, there was a significant hyperpolarizing shift of the midpoint of activation (V1/2). There was also a slight decrease in the voltage dependence (slope) of NaV1.3-β1 complexes (Fig. 2c) that was not significant.
Fig. 2.

SOD1 protein induces a hyperpolarizing shift in the voltage dependence of NaV1.3-β1 channel activation. a Time course of Nav1.3 currents. Nav1.3 currents elicited from a holding potential of –100 mV by 50-ms steps to potentials of –80 to 60 mV are displayed for oocytes injected with only α and β subunit channel mRNA, or α and β subunit mRNA combined with either hSOD1WT or hSOD1A4V protein (scale bar x = 0.2 ms, y = 2 μA). b Current–voltage curves for NaV1.3-β1 complexes. Current tracings over time are normalized to the maximum current. The threshold and voltage dependence of activation for the peak sodium current were similar for complexes injected with hSOD1WT or mutant hSOD1A4V. c Voltage-activation curves calculated from the analyses of dependence of activation on voltage. The solid curves illustrate the Boltzmann curves corresponding to the data. Data are presented as the mean ± SEM (n = 14–20 oocytes). The average values of the midpoint of activation (V1/2) were: control (–30.89 ± 0.44, n = 20); hSOD1WT (–30.64 ± 0.49, n = 18) and hSOD1A4V (–33.79 ± 1.21, n = 14); V½ for hSOD1A4V differed significantly from V1/2 for either hSOD1WT or control (p ≤ 0.05). The average values of the activation curve slopes did not differ significantly among control (2.14 ± 0.13, n = 20), hSOD1WT (2.19 ± 0.17, n = 19) and hSOD1A4V (1.85 ± 0.13, n = 13) groups (p > 0.05, ANOVA with Tukey’s post test)
We next measured the voltage dependence of inactivation of the Nav1.3-β1 channel complex (Fig. 3). Figure 3a shows the traces recorded by step depolarizations to –30 mV from a conditioning test potential between –120 and 0 mV. The resultant currents were normalized to the first current elicited during the protocol, graphed as an exponential function and fit to a Boltzmann equation. Neither the average midpoint of inactivation (V1/2) nor the voltage dependence (slope) differed significantly between the three groups (Fig. 3b).
Fig. 3.

SOD1 protein does not affect the voltage dependence of inactivation of NaV1.3-β1 channel complexes. a Nav1.3 current was recorded by step depolarizations to –30 mV from a conditioning test potential between –120 and 0 mV, in 20 mV steps (scale bar x = 0.2 ms, y = 1 μA). b Inactivation curves were calculated from the data in a. The solid lines represent Boltzmann fits to the data. The average midpoint of inactivation (V1/2) for control (–61.55 ± 0.82, n = 19), hSOD1WT (–64.24 ± 1.82, n = 17) and hSOD1A4V (–61.89 ± 0.95, n = 13) and the voltage dependency (slope) of inactivation curves for control (–4.88 ± 0.39, n = 19), hSOD1WT (–5.00 ± 0.31, n = 17) and hSOD1A4 (–5.32 ± 0.40, n = 13) were similar (p > 0.05, ANOVA with Tukey’s post test)
It has previously been observed in mice expressing hSOD1G93A that cultured neurons demonstrate an increase in the rate of recovery from inactivation [28]. For this reason, we next determined whether the increase in current caused by hSOD1A4V was due to changes in the inactivation kinetics of Nav1.3-β1 channel complexes. To determine the effect hSOD1 had on the kinetics of Nav1.3-β1 channel complexes inactivation, the decay phases of activation current traces were fit to a two-phase exponential decay function. As seen in Fig. 4a, these currents had a noticeable fast transient component and a slow persistent component. The fast and slow time constants for all three groups were similar (Table 3, Fig. 4b). We also used a two-step protocol to measure the effect of hSOD1WT vs. hSOD1A4V on the NaV1.3-β1 channel complex recovery from fast inactivation (Fig. 5a). The resultant traces were fit to a single-phase exponential decay function (Fig. 5b). Although mutant hSOD1A4V did decrease the rate of recovery from fast inactivation, this difference was not significant (Fig. 5c).
Fig. 4.

hSOD1 does not affect the inactivation kinetics of NaV1.3–β1 channel complexes. a Representative Nav1.3 current traces for control, hSOD1WT, and mutant hSOD1A4V obtained following depolarization from a holding potential of –100 mV to –20 mV. These currents demonstrated a fast transient component and a slow component (scale bar x = 0.2 ms, y = 2 μA). b The decay phases of activation current traces were fit to a two-phase exponential decay function. Bar graphs display the mean ± SEM of the τ values of fast inactivation of three groups. Averages of both the fast and slow time constants for all three groups were similar (p > 0.05)
Table 3.
The kinetics of fast inactivation is similar for oocytes injected with WT and mutant hSOD1*
| Control τ1 |
hSOD1WT
τ1 |
hSOD1A4V
τ1 |
CONTROL τ2 |
hSOD1WT
τ2 |
hSOD1A4V
τ2 |
|
|---|---|---|---|---|---|---|
| −30 mV | 1.82 ± 0.15 | 1.60 ± 0.12 | 1.97 ± 0.34 | 0.69 ± 0.07 | 0.53 ± 0.06 | 0.77 ± 0.08 |
| −20 mV | 2.25 ± 0.18 | 1.86 ± 0.17 | 2.22 ± 0.21 | 0.67 ± 0.07 | 0.36 ± 0.07 | 0.60 ± 0.15 |
| −10 mV | 2.73 ± 0.10 | 2.27 ± 0.17 | 2.69 ± 0.10 | 0.58 ± 0.04 | 0.49 ± 0.03 | 0.49 ± 0.03 |
| 0 mV | 2.46 ± 0.12 | 2.19 ± 0.13 | 2.46 ± 0.09 | 0.63 ± 0.09 | 0.46 ± 0.06 | 0.42 ± 0.04 |
| 10 mV | 2.25 ± 0.10 | 1.99 ± 0.09 | 2.08 ± 0.09 | 0.57 ± 0.05 | 0.40 ± 0.06 | 0.43 ± 0.07 |
*The rates of inactivation were measured by fitting currents to a double exponential. Data are represented as the mean ± SEM (–30 mV: n = 23 (control), n = 15 (hSOD1WT), n = 15 (Mutant hSOD1A4V); –20 mV: n = 20 (control), n = 15(hSOD1WT), n = 13(hSOD1A4V); –10 mV: n = 15 (control), n = 11 (hSOD1WT), n = 13 (hSOD1A4V); 0 mV: n = 20 (control), n = 17(hSOD1WT), n = 16 (hSOD1A4V); 10 mV: n = 13 (control), n = 14 (hSOD1WT), n = 15 (hSOD1A4V)
Fig. 5.

A two-step protocol to measure the effect of hSOD1 on the recovery of NaV1.3 current from fast inactivation. a Current traces for the recovery from fast inactivation in control oocytes and in the presence of either hSOD1WT or hSOD1A4V protein (scale bar x = 10 ms, y = 2 μA). See Methods for details. b Profile of the peak current traces from a expressed as a single-phase exponential decay function. c Comparison of decay constants from b. Current amplitude during the second depolarization test pulse was normalized to the current amplitude during the first depolarization test pulse and a recovery value was found for every sweep. The SOD1A4V protein slightly decreased the rate of recovery from fast inactivation (hSOD1A4V 3.91 ± 0.22, n = 19) but this was not significant compared to control (4.52 ± 0.57, n = 19) or hSOD1WT (4.50 ± 0.32, n = 12) (p > 0.05 ANOVA with Tukey’s post test)
These studies define two specific effects of hSOD1A4V on the Nav1.3-β1 complex: (1) an increase in peak Na+ current after depolarization and (2) a shift in the voltage dependence of activation in a hyperpolarizing direction. We next undertook computational studies to model the impact of these changes, considered singly and together, on the overall firing frequency of a mammalian neuron. For this purpose, we ran calculations using the open-source program NEURON [25], which incorporates mammalian neuronal channel parameters (see Methods). We first estimated the impact of the increase in peak Nav1.3 current on firing frequency. At the baseline, which was 0.25 Siemens/cm2 conductance, the model did not generate action potentials spontaneously (resting potential –62 mV, Fig. 6a, left). However, spontaneous electrical firing was elicited when the conductance was increased to 0.29 Siemens/cm2 (see Fig. 6). Moreover, the firing frequency increased progressively with further increases in Na+ conductance until an increase of 134%, at which point the membrane became depolarized (–27 mV) and unexcitable (Fig. 6b, right). Because the range of conductance changes induced by hSOD1A4V were on the order of 50–200% (Fig. 1), we conclude that hSOD1A4V can exert a pro-excitatory influence on Nav1.3- β1 conductance and that this mutant protein might ultimately lead to depolarization arrest of excitability.
Fig. 6.

Firing patterns of a mammalian neuron in response to changes in Nav conductance induced by hSOD1A4V. a At the baseline (see Methods), there is no predicted spontaneous firing as modeled using NEURON software (left; resting membrane potential –62 mV). Transition from silence to firing was observed first at a conductance of 0.29 Siemens/cm2 (right). b When the Nav conductance in the model is increased by 90% (from 0.25 to 0.475 Siemens/cm2, which is the mean increment in conductance recorded experimentally), the model fires spontaneously (left). When this conductance is further increased to 1.34 Siemens/cm2 (a 4.4-fold increase), the model is depolarized to –27 mV and becomes unexcitable (right). c This demonstrates dependence of firing frequency on Nav channel conductance, indicating that as the conductance increases there is a progressive increase in frequency until depolarization
Analogously, we modeled the impact on the firing frequency of hyperpolarizing shifts in the voltage dependence of activation of the Nav1.3 current. The conductance was left at the 0.25 baseline value at all conditions in this model and the baseline V1/2 for the steady-state activation curve for the sodium current was –40 mV (αm), –65 mV (βm). The neuron was electrically quiescent at the baseline V1/2 (Fig. 7a, left) but demonstrated spontaneous generation of trains of action potentials with a 0.6-mV hyperpolarization (Fig.7a, right). As documented in Fig. 7b left, c, the model shows induction of repetitive firing with hyperpolarization as small as 1 mV from this baseline. The firing frequency further increased with additional hyperpolarizing shifts up to 9 mV. With a 10-mV hyperpolarizing shift, the membrane became unexcitable and depolarized (–37 mV) (Fig. 7b, right).
Fig. 7.

Firing patterns of a mammalian neuron in response to changes in voltage dependence of activation induced by hSOD1A4V. The conductance was set at the baseline level of 0.25 S/cm2 at all conditions in this model; the baseline V1/2 for the steady-state activation curve for the sodium current was –40 mV (alpha), –65 mV (beta). a The neuron is electrically at rest at the baseline V1/2 (left). A single spontaneous action potential is initiated with a 0.6-mV hyperpolarization (right). b When the voltage dependence of Nav channel activation is shifted 3 mV in the hyperpolarizing direction (as observed experimentally), continuous spontaneous firing is triggered (left). When the hyperpolarizing shift exceeds 10 mV, the model becomes depolarized to –37 mV and inexcitable (right). Panel c is an equivalent relationship for the dependence of firing frequency on the voltage dependence of Nav channel activation
Because hSOD1A4V both augments the peak Na+ current and hyperpolarizes the activation process, we also modeled the impact on the firing frequency of simultaneous changes in these parameters. As shown in Fig. 8, the prediction of this model is that the simultaneous impact of a 90% increase in and a 3-mV hyperpolarizing shift in m∞ is a state of stable, repetitively firing action potentials at 0.066/ms (66/s).
Fig. 8.

Dependence of firing frequency on simultaneous changes induced by hSOD1A4V on Na+ conductance and voltage dependence of activation. The modeled conductance and voltage dependences of Nav channel activation are indicated in the x and y axes, respectively. The firing frequency is indicated in the z-axis. The yellow dot indicates the estimated position of the frequency in the presence of hSOD1A4V-induced changes (0.475 siemens/cm2, –43 mV). This predicts that under the experimental conditions induced by hSOD1A4V the model will fire spontaneously and thus is hyperexcitable as compared to the baseline in the presence of hSOD1WT (green dot)
Although the voltage dependence of inactivation did not significantly change with the mutant protein injection, we also wondered if the ~3-mV depolarization shift in the inactivation V1/2 (Fig. 3) would induce neuronal firing. Analogously to voltage dependence of activation simulations, the conductance was left at the 0.25 baseline value and the values for alpha and beta were shifted from 65 mV (alpha), –35 mV (beta) in a depolarizing direction. There were no spontaneous action potential trains at the baseline but firing of single action potentials was triggered with a 2.6-mV depolarization (Fig. 9). When the depolarizing shift exceeded 5 mV, spontaneous firing was triggered (c, left). When the depolarizing shift exceeded 19 mV, the model became depolarized to –25 mV and inexcitable (c, right).
Fig. 9.

Firing patterns of a mammalian neuron in response to changes in voltage dependence of inactivation induced by hSOD1A4V. The conductance was at the 0.25 baseline value at all conditions in this model; the baseline V1/2 for the steady-state inactivation curve for the sodium current was –65 mV (αh), –35 mV (βh). a The neuron was electrically silent at the baseline V1/2 (left); an initial, single action potential was seen with a 2.6-mV depolarization (right). b When the voltage dependence of the Nav channel inactivation was shifted 3 mV in the depolarizing direction (as observed experimentally), no spontaneous firing was observed (resting membrane potential –40 mV). c When the depolarizing shift exceeds 5 mV, repetitive spontaneous firing is triggered (left). When the depolarizing shift exceeds 19 mV, the model becomes depolarized to –25 mV and inexcitable (right)
Discussion
Using an oocyte expression system, we examined the impact of hSODA4V protein on the physiological parameters of a complex of Nav1.3 consisting in its α and β subunits; to our knowledge, this is the first report to examine the impact of mutant SOD1 protein at the level of a specific Na (V) channel complex. As noted above, in this study we focused on Nav1.3, an embryonically expressed isoform, because many of the studies pointing to motor neuron hyperexcitability in ALS are based on embryonic or immediately post-natal motor neurons (particularly in vitro) e.g., [29]. Thus, in transgenic hSOD1G93A ALS mice, heightened excitability is evident before or shortly after birth [30–32]. Also, embryonic spinal cord cultures of 4–7 DIV have elevated persistent inward Na+ currents when treated with astrocyte conditioned media from SOD1G93A mice [33]; this study showed that astrocyte conditioned media from hSOD1G93A mice target Nav1.2 and Nav1.3.
We elected to study the missense mutant protein SOD1A4V because the A4V substitution is the most common and clinically most severe SOD1 mutation in North America [3]. We observed that, compared to hSOD1WT, the mutant SOD1 protein induced two significant changes in the properties of the Nav1.3 complex: an increase in the peak Na+ current and a hyperpolarizing shift in the voltage dependence of Nav channel activation. Using the software program NEURON, we documented that these changes are predicted to increase the firing frequency of a simulated model of a mammalian neuron. We selected NEURON as our model primarily because it has been used extensively to model the behavior of a mammalian neuron [25].
Our observations are consistent with several lines of experimental data that implicate excessive firing of motor neurons as a central element in the pathogenesis of ALS. These studies included investigations of human motor cortex [34], murine cell culture and slice preparations of motor cortex [11, 35], spinal cord [5, 28, 30, 36–41], and brainstem [10, 42]. Most recently, in vitro studies have also shown hyperexcitability of cultured iPSC-derived motor neurons from individuals with SOD1, FUS, and C9orf72 gene mutations [7, 8]. The mechanisms whereby neurons in ALS may become hyperexcitable are multiple, with data implicating both endogenous membrane changes as well as excessively excitatory synaptic inputs [10, 42, 43] and excitatory stimuli from an astrocyte conditioned medium [9]. However, as indicated above, recent data have raised the important possibility that early motor neuronal hypoexcitability may be pivotal in ALS and other neurodegenerative disorders, initiating a cascade of events that compromise neuronal viability with hyperexcitability as a compensatory rather than a primary phenomenon [13, 14]. Resolution of these two viewpoints will await further studies, although we note that the observation that mutant SOD1 directly augments sodium channel excitability is consistent with a primary, upstream role for hyperexcitability in neuronal pathology in ALS.
It is likely that excessive firing will be detrimental to motor neurons in two ways. In our view, the primary adverse impact of heightened electrical activity is likely to be mediated by a downstream effect, such as elevations in cytosolic calcium, with subsequent activation of calcium-dependent enzymes (e.g., proteases, lipases). These changes will lead to cytotoxicity, cell death, muscle denervation, and paralysis. In addition, it is possible that excessive firing will be adverse at least transiently because it can lead to depolarization arrest, which will prevent motor neuron activation of contraction, even if the neuromuscular junction is intact. The possibility of depolarization arrest of motor neurons (presumably early in the illness) has not been studied. The potentially important implication is that in ALS cases there may be populations of neurons that can be rapidly rendered functional by interventions that repolarize the electrically silent cells. Such reagents might include not only Nav channel blocking compounds (such as mexiletine) but also compounds that augment K+ channel activity (e.g., retigabine).
We do not know the mechanism(s) whereby mutant SOD1 protein affects the biophysical properties of the Nav1.3 complex. Potential mechanisms are numerous and, at this point, speculative. There may be a direct effect on the channel proteins, perhaps mediated via channel oxidation or phosphorylation. We note, for example, that oxidative stress can activate protein kinases such as protein kinase C (PKC). This is of potential interest in the context of our studies because levels of PKC activity are elevated in hSOD1G93A mice and human ALS patients. Moreover, PKC is known to phosphorylate the α subunit of some Nav channels [44]. That there might be a direct effect of mutant SOD1 on the channel is further suggested by the observation that mutant SOD1 can interact directly with the cytoplasmic face of the voltage-dependent anion channel, VDAC1 [45].
Alternatively, it is conceivable that mutant SOD1 has an indirect effect on the Nav1.3 complex by a mechanism such as increased channel trafficking to the membrane. For example, in the study done by Van Zundert et. al. [33], conditioned media from astrocytes expressing mutant SOD1 selectively lead to motoneuron death through voltage-gated sodium channels. The authors of that study also indicated that incubation of the motoneuron cultures in mutant SOD1 and TDP 43 astrocyte conditioned medium with sodium channel blockers such as mexiletine (antiarrhythmic drug targeting Nav channel receptor site, [46], spermidine (a polyamine acting as an activity-dependent Nav channel blocker [47]) or a riluzole neural excitability suppressor via Nav channels [5, 9]) inhibit both motoneuron death and nitro-oxidative stress. This study indicates that mutant SOD1 or TDP43 proteins mediate secretion of soluble factors leading to motoneuron cell death via Nav channels.
Finally, our findings are consistent with the view that excessive neuronal excitability is a potential target for therapy in ALS. A compelling point supporting this view is the fact that riluzole, a compound that blocks neuronal firing (presumably via blockage of Nav channels) [42], prolongs survival in ALS and is an FDA-approved ALS therapy [40, 48]. Conceivably, other Nav channel blocking compounds may also be beneficial in treating ALS.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
Exogenous hSOD1 and hNav1.3 are expressed in Xenopus oocytes. a hSOD1 protein was identified in extracts from injected oocytes by Western-blot analysis. The first three lanes represent 5 μg total protein from 23 oocyte lysates whereas the following two lanes represent 5 μg total protein from a single oocyte lysate. There is a non-specific band at ~15 kD that is present in both injected and uninjected oocytes. b Representative traces from oocytes that were uninjected or injected with Nav1.3 mRNA. Proper assembly and trafficking of Nav1.3 channels was confirmed by the presence of Na+ current elicited from a single pulse to 0 mV from a holding potential of –100 mV. The dotted line indicates zero current; scale bar is 1 μA and 10 ms. c hSOD1 protein was identified in oocyte extracts immediately after injection and up to 24 h post-injection. Each lane contains 5 μg total protein from a single oocyte lysate (GIF 55 kb)
Acknowledgments
This study was supported by the Angel Fund, the ALS Therapy, Project ALS, P2ALS, the Pierre L. De Bourgknecht ALS Research Fund, and the National Institute of Neurological Disorders and Stroke (NINDS) awards 1RC2NS070342-01, 1RC1NS068391-01, R01NS050557-05, and R01NS065847-01A1. We thank David Paydarfar for comments on the manuscript, Daryl Bosco for providing purified WT and mutant (A4V) human SOD1 protein, and Nick Wightman for assistance with Western immunoblotting.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no competing interests.
References
- 1.Sreedharan J, Brown RH., Jr Amyotrophic lateral sclerosis: problems and prospects. Ann. Neurol. 2013;74(3):309–316. doi: 10.1002/ana.24012. [DOI] [PubMed] [Google Scholar]
- 2.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 3.Cudkowicz, M.E., McKenna-Yasek, D., Sapp, P.E., Chin, W., Geller, B., Hayden, D.L., Schoenfeld, D.A., Hosler, B.A., Horvitz, H.R., Brown, R.H.: Epidemiology of mutations in superoxide dismutase in amyotrophic lateral sclerosis. Ann. Neurol. 41(2), 210–221 (1997) [DOI] [PubMed]
- 4.Al-Chalabi A, Hardiman O. The epidemiology of ALS: a conspiracy of genes, environment and time. Nat. Rev. Neurol. 2013;9(11):617–628. doi: 10.1038/nrneurol.2013.203. [DOI] [PubMed] [Google Scholar]
- 5.Kuo JJ, Siddique T, Fu R, Heckman CJ. Increased persistent Na(+) current and its effect on excitability in motoneurones cultured from mutant SOD1 mice. J. Physiol. 2005;563(Pt 3):843–854. doi: 10.1113/jphysiol.2004.074138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuo JJ, Schonewille M, Siddique T, Schults AN, Fu R, Bar PR, Anelli R, Heckman CJ, Kroese AB. Hyperexcitability of cultured spinal motoneurons from presymptomatic ALS mice. J. Neurophysiol. 2004;91(1):571–575. doi: 10.1152/jn.00665.2003. [DOI] [PubMed] [Google Scholar]
- 7.Donnelly CJ, Zhang PW, Pham JT, Heusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, Maragakis N, Tienari PJ, Petrucelli L, Traynor BJ, Wang J, Rigo F, Bennett CF, Blackshaw S, Sattler R, Rothstein JD. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80(2):415–428. doi: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wainger, B.J., Kiskinis, E., Mellin, C., Wiskow, O., Han, S.S., Sandoe, J., Perez, N.P., Williams, L.A., Lee, S., Boulting, G., Berry, J.D., Brown Jr., R.H., Cudkowicz, M.E., Bean, B.P., Eggan, K., Woolf, C.J.: Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell. Rep. 7(1), 1–11 (2014) [DOI] [PMC free article] [PubMed]
- 9.Fritz E, Izaurieta P, Weiss A, Mir FR, Rojas P, Gonzalez D, Rojas F, Brown RH, Jr, Madrid R, Zundert B. Mutant SOD1-expressing astrocytes release toxic factors that trigger motoneuron death by inducing hyperexcitability. J. Neurophysiol. 2013;109(11):2803–2814. doi: 10.1152/jn.00500.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Zundert B, Peuscher MH, Hynynen M, Chen A, Neve RL, Brown RH, Jr, Constantine-Paton M, Bellingham MC. Neonatal neuronal circuitry shows hyperexcitable disturbance in a mouse model of the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J. Neurosci. 2008;28(43):10864–10874. doi: 10.1523/JNEUROSCI.1340-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pieri M, Carunchio I, Curcio L, Mercuri NB, Zona C. Increased persistent sodium current determines cortical hyperexcitability in a genetic model of amyotrophic lateral sclerosis. Exp. Neurol. 2009;215(2):368–379. doi: 10.1016/j.expneurol.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 12.Sareen D, O'Rourke JG, Meera P, Muhammad AK, Grant S, Simpkinson M, Bell S, Carmona S, Ornelas L, Sahabian A, Gendron T, Petrucelli L, Baughn M, Ravits J, Harms MB, Rigo F, Bennett CF, Otis TS, Svendsen CN, Baloh RH. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 2013;5(208):208ra149. doi: 10.1126/scitranslmed.3007529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roselli F, Caroni P. From intrinsic firing properties to selective neuronal vulnerability in neurodegenerative diseases. Neuron. 2015;85(5):901–910. doi: 10.1016/j.neuron.2014.12.063. [DOI] [PubMed] [Google Scholar]
- 14.Saxena S, Roselli F, Singh K, Leptien K, Julien JP, Gros-Louis F, Caroni P. Neuroprotection through excitability and mTOR required in ALS motoneurons to delay disease and extend survival. Neuron. 2013;80(1):80–96. doi: 10.1016/j.neuron.2013.07.027. [DOI] [PubMed] [Google Scholar]
- 15.Chen-Izu, Y., Shaw, R.M., Pitt, G.S., Yarov-Yarovoy, V., Sack, J.T., Abriel, H., Aldrich, R.W., Belardinelli, L., Cannell, M.B., Catterall, W.A., Chazin, W.J., Chiamvimonvat, N., Deschenes, I., Grandi, E., Hund, T.J., Izu, L.T., Maier, L.S., Maltsev, V.A., Marionneau, C., Mohler, P.J., Rajamani, S., Rasmusson, R.L., Sobie, E.A., Clancy, C.E., Bers, D.M.: Na(+) channel function, regulation, structure, trafficking and sequestration. J. Physiol. 593(6), 1347–1360 (2015) [DOI] [PMC free article] [PubMed]
- 16.Catterall, W.A.: Structure and function of voltage-gated sodium channels at atomic resolution. Exp. Physiol. 99(1), 35–51 (2014) [DOI] [PMC free article] [PubMed]
- 17.Alessandri-Haber, N., Alcaraz, G., Deleuze, C., Jullien, F., Manrique, C., Couraud, F., Crest, M., Giraud, P.: Molecular determinants of emerging excitability in rat embryonic motoneurons. J. Physiol. 541(Pt 1), 25–39 (2002) [DOI] [PMC free article] [PubMed]
- 18.Goldin AL. Diversity of mammalian voltage-gated sodium channels. Ann. N. Y. Acad. Sci. 1999;868:38–50. doi: 10.1111/j.1749-6632.1999.tb11272.x. [DOI] [PubMed] [Google Scholar]
- 19.Beckh S, Noda M, Lubbert H, Numa S. Differential regulation of three sodium channel messenger RNAs in the rat central nervous system during development. EMBO. J. 1989;8(12):3611–3616. doi: 10.1002/j.1460-2075.1989.tb08534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holland KD, Kearney JA, Glauser TA, Buck G, Keddache M, Blankston JR, Glaaser IW, Kass RS, Meisler MH. Mutation of sodium channel SCN3A in a patient with cryptogenic pediatric partial epilepsy. Neurosci. Lett. 2008;433(1):65–70. doi: 10.1016/j.neulet.2007.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lampert A, Hains BC, Waxman SG. Upregulation of persistent and ramp sodium current in dorsal horn neurons after spinal cord injury. Exp. Brain. Res. 2006;174(4):660–666. doi: 10.1007/s00221-006-0511-x. [DOI] [PubMed] [Google Scholar]
- 22.Hayward L, Rodriquez J, Jang G, Tiwari A, Goto J, Cabelli D, Selverstone V, Brown RJ. Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial ALS. J. Biol. Chem. 2002;277:15923–15931. doi: 10.1074/jbc.M112087200. [DOI] [PubMed] [Google Scholar]
- 23.Rotunno MS, Auclair JR, Maniatis S, Shaffer SA, Agar J, Bosco DA. Identification of a misfolded region in superoxide dismutase 1 that is exposed in amyotrophic lateral sclerosis. J. Biol. Chem. 2014;289(41):28527–28538. doi: 10.1074/jbc.M114.581801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O'Connell D, Mruk K, Rocheleau JM, Kobertz WR. Xenopus laevis oocytes infected with multi-drug-resistant bacteria: implications for electrical recordings. J. Gen. Physiol. 2011;138(2):271–277. doi: 10.1085/jgp.201110661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carnevale NT, Hines ML. The NEURON Book. Cambridge: Cambridge University Press; 2006. [Google Scholar]
- 26.Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 1952;117(4):500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nelson ME. Electrophysiological models. In: Koslow S, Subramaniam S, editors. Databasing the Brain: from Data to Knowledge. New York: Wiley; 2005. pp. 285–301. [Google Scholar]
- 28.Zona C, Pieri M, Carunchio I. Voltage-dependent sodium channels in spinal cord motor neurons display rapid recovery from fast inactivation in a mouse model of amyotrophic lateral sclerosis. J. Neurophysiol. 2006;96(6):3314–3322. doi: 10.1152/jn.00566.2006. [DOI] [PubMed] [Google Scholar]
- 29.van Zundert, B., Izaurieta, P., Fritz, E., Alvarez, F.J.: Early pathogenesis in the adult onset neurodegenerative disease amyotrophic lateral sclerosis. J. Cell. Biochem. 113(11), 3301–3312 (2012) [DOI] [PMC free article] [PubMed]
- 30.Bories C, Amendola J, Lamotte d’Incamps B, Durand J. Early electrophysiological abnormalities in lumbar motoneurons in a transgenic mouse model of amyotrophic lateral sclerosis. Eur. J. Neurosci. 2007;25(2):451–459. doi: 10.1111/j.1460-9568.2007.05306.x. [DOI] [PubMed] [Google Scholar]
- 31.Elbasiouny SM, Amendola J, Durand J, Heckman CJ. Evidence from computer simulations in the membrane biophysical properties and dendritic processing of synaptic inputs in mutant superoxide dismutase-1 motoneurons. J Neurosci. 2010;30(16):5544–5558. doi: 10.1523/JNEUROSCI.0434-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quinlan KA, Schuster JE, Fu R, Siddique T, Heckman CJ. Altered post maturation of electrical properties in spinal motoneurons in a mouse model of amyotrophic lateral sclerosis. J Physiol. 2011;589(Pt 9):2245–2260. doi: 10.1113/jphysiol.2010.200659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rojas F, Cortes N, Abarzua S, Dyrda A, van Zundert B. Astrocytes expressing mutant SOD1 and TDP43 trigger motoneuron death that is mediated via sodium channels and nitro-oxidative stress. Front Cell Neurosci. 2014;8(24):1–15. doi: 10.3389/fncel.2014.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vucic S, Kiernan MC. Cortical excitability testing distinguishes Kennedy’s disease from amyotrophic lateral sclerosis. Clin Neurophysiol. 2008;119(5):1088–1096. doi: 10.1016/j.clinph.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 35.Pieri M, Caioli S, Canu N, Mercuri NB, Guatteo E, Zona C. Over-expression of N-type calcium channels in cortical neurons from a mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2013;247:349–358. doi: 10.1016/j.expneurol.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 36.Hadzipasic M, Tahvildari B, Nagy M, Bian M, Horwich AL, McCormick DA. Selective degeneration of a physiological subtype of spinal motor neuron in mice with SOD1-linked ALS. Proc Natl Acad Sci U S A. 2014;111(47):16883–16888. doi: 10.1073/pnas.1419497111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang M, Schuster JE, Fu R, Siddique T, Heckman CJ. Progressive changes in synaptic inputs to motoneurons in adult sacral spinal cord of a mouse model of amyotrophic lateral sclerosis. J Neurosci. 2009;29(48):15031–15038. doi: 10.1523/JNEUROSCI.0574-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin E, Cazenave W, Cattaert D, Branchereau P. Embryonic alteration of motoneuronal morphology induces hyperexcitability in the mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2013;54:116–126. doi: 10.1016/j.nbd.2013.02.011. [DOI] [PubMed] [Google Scholar]
- 39.Pieri M, Albo F, Gaetti C, Spalloni A, Bengtson CP, Longone P, Cavalcanti S, Zona C. Altered excitability of motor neurons in a transgenic mouse model of familial amyotrophic lateral sclerosis. Neurosci Lett. 2003;351(3):153–156. doi: 10.1016/j.neulet.2003.07.010. [DOI] [PubMed] [Google Scholar]
- 40.Schuster JE, Fu R, Siddique T, Heckman CJ. Effect of prolonged riluzole exposure on cultured motoneurons in a mouse model of ALS. J Neurophysiol. 2012;107(1):484–492. doi: 10.1152/jn.00714.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Damme P, Bogaert E, Dewil M, Hersmus N, Kiraly D, Scheveneels W, Bockx I, Braeken D, Verpoorten N, Verhoeven K, Timmerman V, Herijgers P, Callewaert G, Carmeliet P, Van Den Bosch L, Robberecht W. Astrocytes regulate GluR2 expression in motor neurons and their vulnerability to excitotoxicity. Proc Natl Acad Sci U S A. 2007;104(37):14825–14830. doi: 10.1073/pnas.0705046104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bellingham MC. Pre- and postsynaptic mechanisms underlying inhibition of hypoglossal motor neuron excitability by riluzole. J Neurophysiol. 2013;110(5):1047–1061. doi: 10.1152/jn.00587.2012. [DOI] [PubMed] [Google Scholar]
- 43.McGown A, McDearmid JR, Panagiotaki N, Tong H, Al Mashhadi S, Redhead N, Lyon AN, Beattie CE, Shaw PJ, Ramesh TM. Early interneuron dysfunction in ALS: insights from a mutant sod1 zebrafish model. Ann Neurol. 2013;73(2):246–258. doi: 10.1002/ana.23780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Numann R, Catterall WA, Scheuer T. Functional modulation of brain sodium channels by protein kinase C phosphorylation. Science. 1991;254(5028):115–118. doi: 10.1126/science.1656525. [DOI] [PubMed] [Google Scholar]
- 45.Israelson A, Arbel N, Da Cruz S, Ilieva H, Yamanaka K, Shoshan-Barmatz V, Cleveland DW. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron. 2010;67(4):575–587. doi: 10.1016/j.neuron.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev. 2005;57(4):411–425. doi: 10.1124/pr.57.4.5. [DOI] [PubMed] [Google Scholar]
- 47.Fleidervish IA, Libman L. How cesium dialysis affects the passive properties of pyramidal neurons: implications for voltage clamp studies of persistent sodium current. New J Phys. 2008;10:35001. doi: 10.1088/1367-2630/10/3/035001. [DOI] [Google Scholar]
- 48.Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330(9):585–591. doi: 10.1056/NEJM199403033300901. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Exogenous hSOD1 and hNav1.3 are expressed in Xenopus oocytes. a hSOD1 protein was identified in extracts from injected oocytes by Western-blot analysis. The first three lanes represent 5 μg total protein from 23 oocyte lysates whereas the following two lanes represent 5 μg total protein from a single oocyte lysate. There is a non-specific band at ~15 kD that is present in both injected and uninjected oocytes. b Representative traces from oocytes that were uninjected or injected with Nav1.3 mRNA. Proper assembly and trafficking of Nav1.3 channels was confirmed by the presence of Na+ current elicited from a single pulse to 0 mV from a holding potential of –100 mV. The dotted line indicates zero current; scale bar is 1 μA and 10 ms. c hSOD1 protein was identified in oocyte extracts immediately after injection and up to 24 h post-injection. Each lane contains 5 μg total protein from a single oocyte lysate (GIF 55 kb)