

Abstract
The KF, sucrose (table sugar) exploited as quenching system in solution phase parallel synthesis. Excess of electrophiles were covalently trapped with hydroxyl functionality of sucrose and due to polar nature of sucrose derivative was solubilize in water. Potassium fluoride used to convert various excess electrophilic reagents such as acid chlorides, sulfonyl chlorides, isocyanates to corresponding fluorides, which are less susceptible for hydrolysis and subsequently sucrose traps these fluorides and dissolves them in water thus removing them from reaction mixture. Various excess electrophilic reagents such as acid chlorides, sulfonyl chlorides, and isocyanates were quenched successfully to give pure products in excellent yields.
Keywords: KF, Sucrose, Acid chlorides, Sulfonyl chlorides, Isocyanates, Parallel synthesis
Background
The generation and use of combinatorial chemical libraries for the identification of novel chemical leads or for optimization of a promising lead candidate has emerged as a potentially powerful tool for acceleration of the drug discovery process (Terrett et al. 1995; Gallop et al. 1994; Gordon et al. 1994; Janda 1994; Pavia et al. 1993). The combinatorial chemistry has already yielded several compounds that are currently undergoing clinical trials. The pharmaceutical industry needs large and diverse molecular libraries. The screening of large number of compounds can be quickly lead to early structure-activity relationships (SARs), and may provide a practical starting point for drug discovery program, where little or no information is known about the target.
The amide and sulfonamide functionalities are the key structural moieties in many pharmaceutically active compounds such as paracetamol (analgesic and antipyretic), nateglinide (for treatment of type 2 diabetes), probenecid (uricosuric drug), and sotalol (for cardiac arrhythmias) (Fig. 1). Similarly, urea functionality act as a non hydrolysable surrogate of amide bonds in many pharmaceutically active molecules (Majer and Randad 1994; Kruijtzer et al. 1997; Decieco et al. 1997). Therefore practical method for rapid synthesis of amide, sulfonamide and urea containing molecules are of great interest in drug discovery and lead optimization.
Fig. 1.
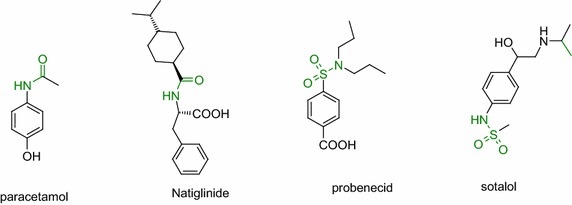
Pharmaceutically active compounds containing amide, sulphone amide groups
Solution-phase parallel synthesis is an excellent way to form libraries of small molecules containing amide, sulfonamide and urea functionalities. However, during the synthesis of library, the chemistry for solution phase parallel synthesis require complete conversion of reactants with little or no formation of by-products or impurities to simplify the tedious purification processes. The solid phase synthesis offers benefit of easy and fast purification to separate excess reagents and side products from the desired compounds attached to the insoluble carrier. However, due to heterogeneous reaction conditions, salvation of bound species, and mass transfer of reagents are the limitations of solid phase synthesis. The range of chemistry applied to the solid-phase synthesis has limitations.
In earlier reports, the solid phase quenchers in the form of reagents on the solid phase or ion exchange resins have been used in quenching reactions to eliminate the reactive entites (Thompson and Ellman 1996). Potassium sarcosinate and fluorous-tethered as quencher were also reported to achieve final compounds in good purity and quantities (Nikam et al. 1998; Lindsley et al. 2002; Curran 2001; Zhang et al. 2003; Zhang et al. 2002). Here, we report sucrose as readily available, environment friendly and cost effective novel quenching agent for the solution phase parallel synthesis.
Results and discussion
In medicinal chemistry program we need rapid synthesis of various acylated and sulfonated derivatives of benzyl amines. During the parallel synthesis, for the complete conversion of the substrate a little excess of an electrophilic reagent was always added, this resulted the impure products and required tedious column chromatographic purifications. Therefore, a new and rapid process for eliminating these excess electrophiles is required. To overcome this issue, KF and sucrose treatment was found to be a novel quenching system in these reactions. As a prototype we chose benzylamine as substrate (Fig. 2). Thus, in a typical reaction of benzyl amine (1.0 equivalent) was treated with an excess of electrophile 1.4 equivalents, e.g. acid chloride, sulfonyl chloride, or isocyanate in presence of triethylamine in DMF. After stirring for 6 h, potassium fluoride (1.5 equivalents) was added to the reaction and the reaction mixture was stirred for an additional 0.5 h. Further sucrose (1.5 equivalents) was added and reaction mixture stirred for additional 0.5 h. Water was then added to the reaction mixture under stirring and the final product was obtained by simple filtration or extraction with EtOAc. The reaction and the purity of the product were monitored by TLC and 1H-NMR, which indicated pure product with no evidence of electrophile used. The products were isolated in excellent yields with high purity. The excess of acid chlorides can also be quenched using aqueous Sodium carbonate, while few aromatic acid chlorides do not get quenched in aqueous base and remain intact; but can be effectively quenched by KF sucrose quenching system (Table 1).
Fig. 2.
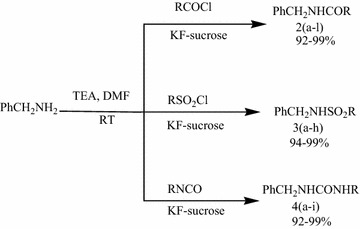
Synthesis of amide 2; sulfonamide 3 and urea derivatives 4, reaction mixture was room temperature for 5–6 h
Table 1.
Isolated yields of amide 2, sulfonamide 3 and urea 4 derivatives
| Amide 2 | R | Yield (%) | Sulfonamide 3 | R | Yield (%) | Urea 4 | R | Yield (%) |
|---|---|---|---|---|---|---|---|---|
| a | Cyclopentyl (Sudrik et al. 2002) | 98 | a | Phenyl (José et al. 2011) | 96 | a | 3-Cyano phenyl | 98 |
| b | t-Butyl (Martínez-Asencio et al. 2011a) | 99 | b | p-Tolyl (Gao et al. 2005) | 99 | b | 1-Naphthyl | 99 |
| c | 4-Fluro-phenyl (Mamat et al. 2011) | 99 | c | 4-Fluro-phenyl (Ramunno et al. 2012) | 97 | c | 4-Fluro phenyl | 97 |
| d | Cyclopropyl (Yang and Shi 2005) | 94 | d | 4-Chlorophenyl (Ramunno et al. 2012) | 98 | d | 2-Trifluromethyl-phenyl | 98 |
| e | Cyclohexyl (Saito et al. 2008) | 98 | e | 4-Trifluromethoxy phenyl (Shi et al. 2009) | 94 | e | 2-Methoxy-phenyl | 97 |
| f | n-Butyl (Dermer and King 1943) | 97 | f | 4-Methoxy phenyl (Martínez-Asencio et al. 2011b) | 99 | f | Phenyl | 99 |
| g | 2-Furyl (Gernigon et al. 2012) | 99 | g | 2,4-Dichloro phenyl | 98 | g | 4-Cyano phenyl | 92 |
| h | 4-Nitro phenyl (Bahrami et al. 2010) | 92 | h | 3,4-Dichloro phenyl | 97 | h | 4-Triflurometh -oxy phenyl | 94 |
| i | 4-Methoxy-phenyl (Kokare et al. 2007) | 99 | i | 2,5-Dimethoxyphenyl | 99 | |||
| j | 2,4-Dichloro-phenyl (Richard Cremlyna et al. 1989) | 95 | ||||||
| k | Ethyl (Lee et al. 1999) | 97 | ||||||
| l | Phenyl (Nowrouzi and Jonaghani 2012) | 99 |
The excess of electrophiles were covalently trapped with hydroxyl functionality of sucrose and dissolve in water. Role of potassium fluoride is to convert various excess electrophilic reagents such as acid chlorides, sulfonyl chlorides, isocyanates to corresponding fluorides (Ishikawa et al. 1981; Kimura and Suzuki 1989; Dang and Olofson 1990), which are less reactive than acid chlorides. Acid fluorides, on the other hand, are known to be more stable to hydrolysis than acid chlorides and hence avoid the formation of by-products. KF is readily soluble in water (92 g/100 ml at 18 °C, 102 g/100 ml at 25 °C) and the reaction in absence of KF yields impure product, which further required column purification. The role of KF is to convert chlorides to corresponding fluorides making them less susceptible to hydrolysis and generation of difficult-to-remove impurities and subsequently sucrose traps these fluorides and dissolves them in water thus removing them from reaction mixture.
After the aqueous quench no sucrose or its derivatives were seen on TLC or in 1H NMR of the products. This methodology worked with a larger excess of electrophiles in the reaction, which needed to be quenched with excess amounts of potassium fluoride and sucrose. This methodology can be easily automated on a synthesizer for synthesizing the desire array of compounds in several gram quantities of acylated products.
Conclusion
In summary, we have developed a simple solution-phase parallel synthesis using a novel quencher for the excess electrophiles. The quencher system KF, sucrose is readily available, environment friendly, cost effective and was found to be very efficient in trapping excess acid chlorides, sulfonyl chlorides and isocyanates to give water soluble by products which could be removed by an aqueous workup.
Methods
Reagents used
Reactions were monitored by thin layer chromatography (TLC), carried out on 0.2 mm silica gel 60 F254 (Merck) plates using UV light (254 and 366 nm) for detection and compounds were purified by column chromatography by using silica gel of 5–20 µm (Merck, 60–120 mesh). Column dimension 39 × 2 cm and elution volume used is about 200–400 ml for each product. Common reagent grade chemicals are either commercially available and were used without further purification or were prepared by standard literature procedures.
Characterization
The 1H spectra were recorded on a Bruker XL 300 spectrometer (300 MHz). Chemical shifts were reported in parts per million using tetramethylsilane as an internal standard and were given in δ units. The solvent for NMR spectra was DMSO-d6 unless otherwise stated. Elemental analyses were performed on a Hosli CH-Analyzer and are within ±0.4 of the theoretical percentage. All reactions were monitored by thin layer chromatography, carried out on 0.2 mm silica gel 60 F254 (Merck) plates using UV light (254 and 366 nm) for detection. Common reagents grade chemicals are either commercially available and were used without further purification or prepared by standard literature procedures.
Synthesis of benzylamide derivatives 2(a–l)
The mixture of benzyl amine (0.268 g, 2.5 mmol), acid chloride (3.5 mmol) and triethylamine (0.695 ml, 5 mmol) in DMF (5 ml) was stirred at room temperature for 5–6 h (TLC Check, 40 % ethyl acetate/hexane). To the stirring solution KF (88 mg, 1.5 mmol) was added and the reaction mixture was stirred for 0.5 h. Sucrose (0.514 g, 1.5 mmol) was added and stirring was continued for next half an hour. Water (50 ml) was then added to the reaction mixture under stirring. The products 2a–e and 2g–l were isolated by filtration and product 2f was extracted in EtOAc (25 ml).
Cyclopentanecarboxylic acid benzylamide 2a: white crystalline solid; [M.p. 94 °C, lit. mp 94 °C (Sudrik et al. 2002); yield 498 mg, 98 %] Analysis: IR (KBr): 3354, 1654, 1601 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.26 (bs, 1H, NH), 7.19–7.33 (m, 5H, ArH), 4.24 (d, J = 6.0 Hz, 2H, CH2), 2.52–2.72 (m, 1H, CH), 1.45–1.80 (m, 8H, 4 × CH2).
N-Benzyl-2, 2-dimethyl-propionamide 2b: white crystalline solid; [M.p. 83 °C, lit. Mp 81–84 °C (Martínez-Asencio et al. 2011a), yield 474 mg, 99 %] Analysis: IR (KBr): 3358, 1660, 1610 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.04 (t, J = 5.5 Hz, 1H, NH), 7.17–7.33 (m, 5H, ArH), 4.25 (d, J = 6.0 Hz, 2H, CH2), 1.12 (s, 9H, 3 × CH3).
N-Benzyl-4-fluoro-benzamide 2c: white crystalline solid [M.p. 144 °C, lit. mp 143–144 °C (Mamat et al. 2011) yield 567 mg, 99 %] Analysis: IR (KBr): 3365, 1647, 1611 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 9.07 (t, J = 5.9 Hz, 1H, NH), 7.93–8.01 (m, 2H, ArH), 7.22–7.36 (m, 7H, ArH), 4.48 (d, J = 6.0 Hz, 2H, CH2). 13C NMR (300 MHz, DMSO-d6) δ 43.1, 115.5, 115.8, 127.2, 127.7, 128.7, 130.3, 130.4, 131.2, 131.3, 140.0, 162.7, 165.6.
Cyclopropanecarboxylic acid benzylamide 2d: white crystalline solid [M.p. 140 °C, lit.mp 140–141 °C (Yang and Shi 2005) yield 412 mg, 94 %] Analysis: IR (KBr): 3370, 1665, 1605 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.54 (br. s., 1H, NH), 7.20–7.35 (m, 5H, ArH), 4.27 (d, J = 6.0 Hz, 2H, CH2), 1.55–1.64 (m, 1H, CH), 0.62–0.72 (m, 4H, 2 × CH2).
Cyclohexanecarboxylic acid benzylamide 2e: white crystalline solid [M.p. 108 °C, lit. mp 107–109 °C (Saito et al. 2008) yield 532 mg, 98 %] Analysis: IR (KBr): 3345, 1651, 1612 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.20 (br. s., 1H, NH), 7.18–7.33 (m, 5H, ArH), 4.23 (d, J = 5.7 Hz, 2H, CH2), 2.15 (s, 1H, CH), 1.57–1.75 (m, 5H, 5xCH), 1.10–1.42 (m, 5H, 5 × CH).
Pentanoic acid benzylamide 2f: white crystalline solid [M.p. 42 °C, lit.mp 41.1–41.8 °C (Dermer and King 1943); yield 464 mg, 97 %] Analysis: IR (KBr): 3352, 1654, 1603 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.28 (t, J = 5.5 Hz, 1H, NH), 7.19–7.33 (m, 5H, ArH), 4.24 (d, J = 6.0 Hz, 2H, CH2), 2.13 (t, J = 7.4 Hz, 2H, CH2), 1.41–1.55 (m, 2H, CH2), 1.26 (dq, J = 14.9,7.4 Hz, 2H, CH2), 0.78–0.89 (m, 3H, CH3).
Furan-2-carboxylic acid benzylamide 2g: white crystalline solid [M.p. 113 °C, lit. mp 111–113 °C (Gernigon et al. 2012); yield 498 mg, 99 %] Analysis: IR (KBr): 3370, 1657, 1604 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.92 (t, J = 5.9 Hz, 1H, NH), 7.77–7.85 (m, 1H, ArH), 7.20–7.35 (m, 5H, ArH), 7.13 (d, J = 3.4 Hz, 1H, ArH), 6.62 (dd, J = 3.4, 1.9 Hz, 1H, ArH), 4.42 (d, J = 6.0 Hz, 2H, CH2).
N-Benzyl-4-nitro-benzamide 2h: light yellow crystalline solid [M.p. 140 °C, lit. mp 139 °C (Bahrami et al. 2010); yield 590 mg, 92 %] Analysis: IR (KBr): 3365, 1667, 1617, 1545 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 9.39 (t, J = 6.0 Hz, 1H, NH), 8.31–8.36 (m, 2H, ArH), 8.10–8.15 (m, 2H, ArH), 7.23–7.37 (m, 5H, ArH), 4.52 (d, J = 6.0 Hz, 2H, CH2).
N-Benzyl-4-methoxy-benzamide 2i: white crystalline solid [M.p. 129 °C, lit. mp125 °C (Kokare et al. 2007), yield 597 mg, 99 %] Analysis: IR (KBr): 3345, 1649, 1601 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.89 (t, J = 6.0 Hz, 1H, NH), 7.84–7.91 (m, 2H, ArH), 7.20–7.34 (m, 5H, ArH), 6.97–7.04 (m, 2H, ArH), 4.46 (d, J = 6.0 Hz, 2H, CH2), 3.8 (s, 3H, OCH3).
N-Benzyl-2, 4-Dichloro-benzamide 2j: white crystalline solid [M.p. 125 °C, lit. mp 125 °C (Richard Cremlyna et al. 1989); yield 666 mg, 95 %] Analysis: IR (KBr): 3355, 1654, 1619 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 9.02 (s, 1H, NH), 7.69 (s, 1H, ArH), 7.49 (s, 2H, ArH), 7.22–7.37 (m, 5H, ArH), 4.44 (d, J = 6.0 Hz, 2H, CH2).
N-Benzyl-propionamide 2 k: white crystalline solid [M.p. 51 °C, lit. mp 49–51 °C (Lee et al. 1999); yield 396 mg, 97 %] Analysis: IR (KBr): 3349, 1657, 1608 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.25 (br. s., 1H, NH), 7.19–7.34 (m, 5H, ArH), 4.24 (d, J = 6.0 Hz, 2H, CH2), 2.13 (q, J = 7.6 Hz, 2H, CH2), 0.97–1.15 (m, 3H, CH3).
N-Benzyl-benzamide 2l: white crystalline solid [M.p. 104 °C, lit. mp 104–106 °C (Nowrouzi and Jonaghani 2012); yield 523 mg, 99 %] Analysis: IR (KBr): 3358, 1656, 1610 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 9.04 (t, J = 5.9 Hz, 1H, NH), 7.87–7.92 (m, 2H, ArH), 7.43–7.56 (m, 3H, ArH), 7.20–7.35 (m, 5H, ArH), 4.48 (d, J = 6.0 Hz, 2H, CH2). Calculated for C14H13NO (211.27): C, 79.59; H, 6.20; N, 6.63. Found: C, 79.57; H, 6.21; N, 6.62.
Synthesis of sulfonamides derivatives 3(a–h)
The mixture of benzylamine (0.268 g, 2.5 mmol), sulfonyl chloride (3.5 mmol) and triethylamine (0.695 ml, 5 mmol) in DMF (5 ml) was stirred at room temperature for 5–6 h (TLC Check, 40 % ethyl acetate/hexane). To the stirring solution KF (88 mg, 1.5 mmol) was added and the reaction mixture was stirred for 0.5 h. Sucrose (0.514 g, 1.5 mmol) was added and stirring was continued for next half an hour. Water (50 ml) was then added to the reaction mixture under stirring. The product was isolated by filtration and washed with 10 ml water.
N-Benzyl-benzenesulfonamide 3a: white crystalline solid [M.p. 90 °C, lit. mp 89–90 °C (José et al. 2011); yield 594 mg, 96 %] Analysis: IR (KBr): 3365, 1601, 1340 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 9.04 (bs. s 1H, NH), 7.87–7.92 (m, 2H, ArH), 7.43–7.56 (m, 3H, ArH), 7.20–7.35 (m, 5H, ArH), 4.48 (d, J = 6.0 Hz, 2H, CH2).
N-Benzyl-4-methyl-benzenesulfonamide 3b: white crystalline solid [M.p. 111 °C, lit. mp 111 °C (Gao et al. 2005); yield 647 mg, 99 %] Analysis: IR (KBr): 3361, 1615, 1338 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.22 (bs, 1H, NH), 7.69–7.94 (m, 2H, ArH), 7.33–7.52 (m, 2H, ArH), 7.19–7.32 (m, 5H, ArH), 4.00 (s, 2H, CH2), 1.90 (s, 3H, CH3).
N-Benzyl-4-fluoro-benzenesulfonamide 3c: white crystalline solid [M.p. 99 °C, lit. mp 96–98 °C (Ramunno et al. 2012), yield 644 mg, 97 %] Analysis: IR (KBr): 3355, 1611, 1338 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.22 (bs, 1H, NH), 7.69–7.94(m, 2H, ArH), 7.33–7.52 (m, 2H, ArH), 7.19–7.32 (m, 5H, ArH), 4.00 (s, 2H, CH2). 13C NMR (300 MHz, DMSO-d6) δ: 46.6, 116.5, 116.8, 127.6, 128.1, 128.7, 129.9, 130.0, 137.6, 137.7, 137.9, 162.8, 166.1.
N-Benzyl-4-chloro-benzenesulfonamide 3d: white crystalline solid [M.p. 105 °C, lit. mp 104–106 °C (Ramunno et al. 2012) yield 690 mg, 98 %] Analysis: IR (KBr): 3365, 1614, 1335 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.25 (bs, 1H, NH), 7.75–7.98 (m, 2H, ArH), 7.38–7.54 (m, 2H, ArH), 7.19–7.32 (m, 5H, ArH), 4.02 (s, 2H, CH2).
N-Benzyl-4-trifluromethoxy-benzenesulfonamide 3e: white crystalline solid [M.p. 114 °C, lit. mp 114 °C (Shi et al. 2009); yield 779 mg, 94 %] Analysis: IR (KBr): 3348, 1603, 1341 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.44 (bs, 1H, NH), 7.76–8.01 (m, 4H, ArH), 7.11–7.28 (m, 5H, ArH), 4.04 (s, 2H, CH2).
N-Benzyl-4-methoxy-benzenesulfonamide 3f: white crystalline solid [M.p. 113 °C, lit. mp 112–113 °C (Martínez-Asencio et al. 2011b); yield 686 mg, 99 %] Analysis: IR (KBr): 3357, 1607, 1344 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.42 (bs, 1H, NH), 7.84–7.91 (m, 2H, ArH), 7.20–7.34 (m, 5H, ArH), 6.97–7.04 (m, 2H, ArH), 4.46 (d, J = 6.0 Hz, 2H, CH2), 3.95 (s, 3H, OCH3).
N-Benzyl-2, 4-dichloro-benzenesulfonamide 3g: white crystalline solid [M.p. 115 °C, yield 775 mg, 98 %] Analysis: Calculated for C13H11Cl2NO2S (316.21): C, 49.38; H, 3.51; N, 4.43. Found: C, 49.37; H, 3.52; N, 4.42. IR (KBr): 3358, 1605, 1342 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.56 (s, 1H, NH), 7.86 (d, J = 8.7 Hz, 1H, ArH), 7.67–7.76 (m, 1H, ArH), 7.44–7.60 (m, 1H, ArH), 7.12–7.26 (m, 5H, ArH), 4.09 (s, 2H, CH2).
N-Benzyl-3, 4-dichloro-benzenesulfonamide 3h: white crystalline solid [M.p. 112 °C; yield 767 mg, 97 %] Analysis: Calculated for C13H11Cl2NO2S (316.21): C, 49.38; H, 3.51; N, 4.43. Found: 49.40; H, 3.52; N, 4.42. IR (KBr): 3354, 1605, 1343 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.40 (bs, 1H, NH), 7.86 (d, J = 1.9 Hz, 1H, ArH), 7.80 (d, J = 8.7 Hz, 1H, ArH), 7.65–7.75 (m, 1H, ArH), 7.13–7.29 (m, 5H, ArH), 4.05 (s, 2H, CH2). 13C NMR (300 MHz, DMSO-d6) δ: 46.7, 127.1, 127.7, 128.2, 128.6, 128.8, 131.9, 132.4, 135.7, 137.5, 141.7.
Synthesis of urea derivatives 4(a–i)
The mixture of benzylamine (0.268 g, 2.5 mmol), isocyanate (3.5 mmol) and triethylamine (0.695 ml, 5 mmol) in DMF (5 ml) was stirred at room temperature for 5–6 h (TLC Check, 50 % ethyl acetate/hexane). To the above stirring solution, KF (88 mg, 1.5 mmol) was added and the reaction mixture was stirred for 0.5 h. Sucrose (0.514 g, 1.5 mmol) was added and stirring was continued for next half an hour. Water (50 ml) was then added to the reaction mixture under stirring. The product was isolated by filtration and washed with 10 ml water.
1-Benzyl -3-(3-cyano-phenyl)-urea 4a: white crystalline solid [M.p. 176 °C; yield 616 mg, 98 %] Analysis: IR (KBr): 3454, 2202, 1640, 1602 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.94 (s, 1H, NH), 7.95 (t, J = 1.7 Hz,1H, NH), 7.60 (ddd, J = 8.1, 2.3, 1.3 Hz, 1H, ArH), 7.43 (t, J = 7.9 Hz, 1H, ArH), 7.19–7.39 (m, 6H, ArH), 6.81 (t, J = 5.9 Hz, 1H, ArH), 4.31 (d, J = 6.0 Hz, 2H, CH2). 13C NMR (300 MHz, DMSO-d6) δ: 43.2, 111.9, 119.4, 120.6, 122.7, 125.0, 127.2, 127.6, 128.8, 130.5, 140.5, 141.8, 155.4.
1-Benzyl-3-naphthalen-1-yl-urea 4b: white crystalline solid [M.p. 203 °C; yield 684 mg, 99 %] Analysis: Calculated for C18H16N2O (276.34): C, 78.24; H, 5.84; N, 10.14. Found: C, 78.22; H, 5.85; N, 10.13. IR (KBr): 3374, 1638, 1605 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.64 (s, 1H, NH), 8.11 (d, J = 7.6 Hz, 1H, ArH), 8.05 (dd, J = 7.7, 0.9 Hz, 1H, ArH), 7.85–7.98 (m, 1H, ArH), 7.66 (s, 1H, ArH), 7.24–7.59 (m, 10H, ArH), 7.06 (t, J = 5.9 Hz, 1H, ArH), 4.39 (d, J = 5.7 Hz, 2H, CH2). 13C NMR (300 MHz, DMSO-d6) δ: 46.7, 127.1, 127.7, 128.2, 128.6, 128.8, 131.9, 132.4, 135.7, 137.5, 141.7.
1-Benzyl-3-(4-fluoro-phenyl)-urea 4c: white crystalline solid [M.p. 179 °C; yield 592 mg, 97 %] Analysis: Calculated for C14H13FN2O (244.27): C, 68.84; H, 5.36; N, 11.47. Found: C, 68.83; H, 5.35; N, 11.46. IR (KBr): 3354, 1635, 1602 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.87 (s, 1H, NH), 8.2 (t, J = 5.9 Hz, 1H, NH), 7.93–8.01 (m, 2H, ArH), 7.15–7.25 (m, 5H, ArH), 7.25–7.36 (m, 2H, ArH), 4.48 (d, J = 6.0 Hz, 2H, CH2).
1-Benzyl-3-(2-trifluoromethyl-phenyl)-urea 4d: white crystalline solid [M.p. 168 °C; yield 721 mg, 98 %] Analysis: Calculated for C15H13F3N2O (294.28): C, 61.22; H, 4.45; N, 9.52. Found: C, 61.20; H, 4.44; N, 9.53. IR (KBr): 3370, 1645, 1601 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.72 (s, 1H, NH), 7.60–7.97 (m, 4H, ArH), 7.45 (t, J = 5.7 Hz, 1H, NH), 7.25–7.35 (m, 5H, ArH), 4.31 (d, J = 5.7 Hz, 2H).
1-Benzyl-3-(2-methoxy- phenyl)-urea 4e: white crystalline solid [M.p. 177 °C; yield 622 mg, 97 %] Analysis: Calculated for C15H16N2O2 (256.31): C, 70.29; H, 6.29; N, 10.93. Found: C, 70.31; H, 6.28; N, 10.94. IR (KBr): 3365, 1636, 1607 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.72 (s, 1H, NH),7.08–7.36 (m, 7H, ArH), 6.85–6.95 (m, 1H, ArH), 6.69 (bs, 1H, NH), 6.44–6.58 (m,1H, ArH), 4.29 (bs, 2H, CH2), 3.5 (s, 3H, OCH3).
1-Benzyl-3-phenyl-urea 4f: white crystalline solid [M.p. 194 °C; yield 560 mg, 99 %] Analysis: Calculated for C14H14N2O (226.28): C, 74.31; H, 6.24; N, 12.38. Found: C, 74.33; H, 6.25; N, 12.39. IR (KBr): 3364, 1643, 1602 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.60 (s, 1H, NH), 7.08–7.40 (m, 8H, ArH), 6.879 (ddd, J = 8.0, 2.0, 0.9 Hz, 1H, ArH), 6.63 (t, J = 6.0 Hz, 1H, NH), 6.47 (ddd, J = 8.3, 2.6, 0.9 Hz, 1H, ArH), 4.33(d, J = 6.0 Hz, 2H, CH2).
1-Benzyl-3-(4-cyano-phenyl)-urea 4g: white crystalline solid [M.p. 198 °C; yield 578 mg, 92 %] Analysis: Calculated for C15H13N3O (251.29): C, 71.70; H, 5.21; N, 16.72. Found: C, 71.73; H, 5.22; N, 16.71. IR (KBr): 3374, 2201, 1643, 1608 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.80 (s, 1H, NH), 7.39–7.62(m, 1H, ArH), 7.19–7.36 (m, 5H, ArH), 7.32 (bs, 1H, NH), 6.96–7.11 (m, 1H, ArH), 6.62–6.78 (m, 2H), 4.29 (d, J = 6.0 Hz, 2H, CH2).
1-Benzyl-3-(4-trifluoromethoxy-phenyl) urea 4h: white crystalline solid [M.p. 192 °C; yield 730 mg, 94 %] Analysis: Calculated for C15H13F3N2O2 (310.28): C, 58.07; H, 4.22; N, 18.37. Found: C, 58.06; H, 4.21; N, 18.36. IR (KBr): 3374, 1644, 1610 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.80 (s, 1H, NH), 7.5 (d, J = 7.6 Hz, 2H, ArH), 7.18–7.22 (m, 2H, ArH), 7.22–7.36 (m, 5H, ArH), 6.69 (t, J = 6.0 Hz, 1H, NH), 4.30 (d, J = 6.0 Hz, 2H, CH2).
1-Benzyl-3-(2,5-Dimethoxy-phenyl) urea 4i: white crystalline solid [M.p. 179 °C; yield 709 mg, 99 %] Analysis: Calculated for C16H18N2O3 (286.33): C, 67.12; H, 6.34; N, 9.78. Found: C, 67.11; H, 6.33; N, 9.77. IR (KBr): 3374, 1638, 1601 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.03 (s, 1H, NH), 7.8–7.84 (m, 1H, ArH), 7.21–7.38 (m, 6H, ArH & NH), 6.83–6.91 (m, 1H, ArH), 6.38–6.50 (m, 1H, ArH), 4.28 (d, J = 5.7 Hz, 2H), 3.75 (s, 3H, OCH3), 3.65 (s, 3H, OCH3).
Authors’ contributions
RBT: research guide give the idea of KF sugar as new quenching reagent and write this paper. SNC: research students who actual work for the synthesis of compounds 2 and 3 and characterized by spectral and analytical methods, also done literature search. RAW: research students, synthesis compounds 4 and characterized by spectral and analytical methods, communicate this manuscript. All authors read and approved the final manuscript.
Acknowledgements
Authors thank to BCUD, University of Pune for financial support and Department of Chemistry University of Pune for spectroscopic data and the Principal KTHM College, Nashik for facility.
Competing interests
In organic synthesis particularly while preparing derivatives using electrophiles e.g. acid chloride, sulfonyl chloride, isocyanate etc. many times we have to use slight excess of these reagents to avoid the risk of starting material in the product. But then the work-up procedure became tedious as no. of impurities form due to the use of excess of the reagent. Here, we have developed very simple method using KF and table sugar to avoid all above problems. The product can be isolated just by aqueous work-up. Other workers have also developed new reagents for this purpose, but advantage of our new reagent is inexpensive, easily available and water soluble yield pure product, which does not require further purification.
Contributor Information
Sunil Chavan, Email: sunilchavan1@hotmail.com.
Rahul Watpade, Email: rahulwatpade@gmail.com.
Raghunath Toche, Email: raghunath_toche@rediffmail.com.
References
- Bahrami K, Khodaei MM, Tajik M. Trimethylsilyl chloride promoted selective desulfurization of thiocarbonyls to carbonyls with hydrogenperoxide. Synthesis. 2010;24:4282. doi: 10.1055/s-0030-1258283. [DOI] [Google Scholar]
- Curran DP. Fluorous reverse phase silica gel. A new tool for preparative separations in synthetic organic and organofluorine chemistry. Synlett. 2001;252:1488. doi: 10.1055/s-2001-16800. [DOI] [Google Scholar]
- Dang VA, Olofson RA. Advantages of fluoroformates as carboalkoxylating reagents for polar reactants. J Org Chem. 1990;55:1851. doi: 10.1021/jo00293a033. [DOI] [Google Scholar]
- Decieco CS, Seng JL, Kennedy KE, Covington MB, Welch PK, Arner EC, Magolda RL, Nelson DJ. Amide surrogates of matrix metalloproteinase inhibitors: urea and sulfonamide mimics. Bioorg Med Chem Lett. 1997;7:233. doi: 10.1016/S0960-894X(96)00605-1. [DOI] [Google Scholar]
- Dermer OC, King J. n-benzylamides as derivatives for identifying the acyl group in esters. J Org Chem. 1943;8:168. doi: 10.1021/jo01190a008. [DOI] [Google Scholar]
- Gallop MA, Barrett RW, Dower WJ, Fodor SPA, Gordon EM. Applications of combinatorial technologies to drug discovery. 1. Background and peptide combinatorial libraries. J Med Chem. 1994;37:1233. doi: 10.1021/jm00035a001. [DOI] [PubMed] [Google Scholar]
- Gao F, Deng M, Qian C. The effect of coordination on the reaction of N-tosyl imines with diethylzinc. Tetrahedron. 2005;61(52):12238. doi: 10.1016/j.tet.2005.09.111. [DOI] [Google Scholar]
- Gernigon N, Raed M, Al-Zoubi Hall DG. Direct amidation of carboxylic acids catalyzed by ortho-iodo arylboronic acids: catalyst optimization, scope, and preliminary mechanistic study supporting a peculiar halogen acceleration effect. J Org Chem. 2012;77:8386. doi: 10.1021/jo3013258. [DOI] [PubMed] [Google Scholar]
- Gordon EM, Barrett RW, Dower WJ, Fodor SPA, Gallop MA. Applications of comb inatorial technologies to drug discovery. 2. Combinatorial organic synthesis, library screening strategies, and future directions. J Med Chem. 1994;37:1385. doi: 10.1021/jm00036a001. [DOI] [PubMed] [Google Scholar]
- Ishikawa N, Kitazume T, Yamazaki T, Mochida Y, Tatsund T. Enhanced effect of spray-dried potassium fluoride on fluorination. Chem Lett. 1981;6:761. doi: 10.1246/cl.1981.761. [DOI] [Google Scholar]
- Janda KD. Tagged versus untagged libraries: methods for the generation and screening of combinatorial chemical libraries. Proc Natl Acad Sci USA. 1994;91:10779. doi: 10.1073/pnas.91.23.10779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- José L, Ruano G, Parra A, Marzo L, Yuste F, Mastranzo VM. One-pot synthesis of sulfonamides from methyl sulfinates using ultrasound. Tetrahedron. 2011;67(16):2905. doi: 10.1016/j.tet.2011.02.060. [DOI] [Google Scholar]
- Kimura Y, Suzuki H. Freeze-dried potassium fluoride: Synthetic utility as a fluorinating agent. Tetrahedron Lett. 1989;30:127. doi: 10.1016/S0040-4039(01)80342-7. [DOI] [Google Scholar]
- Kokare ND, Nagawade RR, Rane VP, Shinde DB. Organophosphorus esters of 1-hydroxy-2-phenylbenzimidazole: synthesis and utilization as novel peptide coupling reagents. Synthesis. 2007;05:766. doi: 10.2174/092986607780090847. [DOI] [PubMed] [Google Scholar]
- Kruijtzer JAW, Lefeber DJ, Liskamp RMJ. Approaches to the synthesis of ureapeptoid peptidomimetics. Tetrahedron Lett. 1997;38:5335. doi: 10.1016/S0040-4039(97)01166-0. [DOI] [Google Scholar]
- Lee SY, Lee CW, Oh DY. Dephosphonylation of β-carbonyl phosphonates. J Org Chem. 1999;64(19):7017. doi: 10.1021/jo990221r. [DOI] [Google Scholar]
- Lindsley CW, Zhao Z, Leister WH. Fluorous-tethered quenching reagents for solution phase parallel synthesis. Tetrahedron Lett. 2002;43:4225. doi: 10.1016/S0040-4039(02)00748-7. [DOI] [Google Scholar]
- Majer P, Randad RS. A safe and efficient method for preparation of N, N′-unsymmetrically disubstituted ureas utilizing triphosgene. J Org Chem. 1994;59:1937. doi: 10.1021/jo00086a061. [DOI] [Google Scholar]
- Mamat C, Flemming A, Kockerling M, Steinbach J, Wuest FR. Synthesis, structure determination, and (radio-)fluorination of novel functionalized phosphanes suitable for the traceless Staudinger ligation. Synthesis. 2011;19:3311. [Google Scholar]
- Martínez-Asencio A, Yus M, Ramón DJ. Palladium(II) acetate as catalyst for the N-alkylation of aromatic amines, sulfonamides, and related nitrogenated compounds with alcohols by a hydrogen autotransfer process. Synthesis. 2011;22:3730. [Google Scholar]
- Martínez-Asencio A, Ramón DJ, Yus M. N-alkylation of poor nucleophilic amines and derivatives with alcohols by a hydrogen autotransfer process catalyzed by copper(II) acetate: scope and mechanistic considerations. Tetrahedron. 2011;67(17):3140. doi: 10.1016/j.tet.2011.02.075. [DOI] [Google Scholar]
- Nikam SS, Kornberg BE, Ault-Justus SE, Rafferty MF. Novel quenchers for solution phase parallel synthesis. Tetrahedron Lett. 1998;39:1121. doi: 10.1016/S0040-4039(97)10781-X. [DOI] [Google Scholar]
- Nowrouzi N, Jonaghani MH. Highly selective mono-N-benzylation and amidation of amines with alcohols or carboxylic acids using the Ph2PCl/I2/imidazole reagent system. Can J Chem. 2012;90(6):498. doi: 10.1139/v2012-021. [DOI] [Google Scholar]
- Pavia MR, Sawyer TK, Moos WH. The generation of molecular diversity. Bioorg Med Chem Lett. 1993;3:387. doi: 10.1016/S0960-894X(01)80220-1. [DOI] [Google Scholar]
- Ramunno A, Cosconati S, Sartini S, Maglio V, Angiuoli S, La Pietra VL, Maro SD, Giustiniano M, Motta CL, Settimo FD, Marinelli L, Novellino EE. Progresses in the pursuit of aldose reductase inhibitors: the structure based lead optimization step. Eur J Med Chem. 2012;51:216. doi: 10.1016/j.ejmech.2012.02.045. [DOI] [PubMed] [Google Scholar]
- Cremlyna R, Ellisa L, Pinneya A (1989) Phosphorus sulfur silicon and the related elements. doi:10.1080/10426508908040606
- Saito Y, Ouchi H, Takahata H. Carboxamidation of carboxylic acids with 1-tert-butoxy-2-tert-butoxycarbonyl-1,2-dihydroisoquinoline (BBDI) without bases. Tetrahedron. 2008;64:11129. doi: 10.1016/j.tet.2008.09.094. [DOI] [Google Scholar]
- Shi F, Tse MK, Zhou S, Pohl MM, Radnik J, Hubner S, Jähnisch K, Brückner A, Beller M. Green and efficient synthesis of sulfonamides catalyzed by nano-Ru/Fe3O4. J Am Chem Soc. 2009;131(5):1775. doi: 10.1021/ja807681v. [DOI] [PubMed] [Google Scholar]
- Sudrik SG, Chavan SP, Chandrakumar KRS, Pal S, Date SK, Sonawane HR. Microwave specific Wolff rearrangement of α-diazoketones and its relevance to the nonthermal and thermal effect. J Org Chem. 2002;67:1574. doi: 10.1021/jo010951a. [DOI] [PubMed] [Google Scholar]
- Terrett NK, Gardner M, Gordon DW, Kobylecki RJ, Steele Combinatorial synthesis—the design of compound libraries and their application to drug discover. Tetrahedron. 1995;51:8135. doi: 10.1016/0040-4020(95)00467-M. [DOI] [Google Scholar]
- Thompson LA, Ellman JA. Synthesis and applications of small molecule libraries. Chem Rev. 1996;96:555. doi: 10.1021/cr9402081. [DOI] [PubMed] [Google Scholar]
- Yang YH, Shi M. Note Prev. Article next article table of contents ring-expanding reaction of cyclopropyl amides with triphenylphosphine and carbon tetrahalide. J Org Chem. 2005;70(21):8645. doi: 10.1021/jo0516988. [DOI] [PubMed] [Google Scholar]
- Zhang W, Curran DP, Chen CHT. Use of fluorous silica gel to separate fluorous thiol quenching derivatives in solution-phase parallel synthesis. Tetrahedron. 2002;58:3871. doi: 10.1016/S0040-4020(02)00209-0. [DOI] [Google Scholar]
- Zhang W, Chen CHT, Nagashima T. Fluorous electrophilic scavengers for solution-phase parallel synthesis. Tetrahedron Lett. 2003;44:2065. doi: 10.1016/S0040-4039(03)00178-3. [DOI] [Google Scholar]