

Summary
MHC class I (MHC-I) molecules are the centerpieces of cross-presentation. They are loaded with peptides derived from exogenous sources and displayed on the plasma membrane to communicate with CD8 T cells, relaying a message of tolerance or attack. The study of cross-presentation has been focused on the relative contributions of the vacuolar versus cytosolic pathways of antigen processing and the location where MHC-I molecules are loaded. While vacuolar processing generates peptides loaded onto vacuolar MHC-I molecules, how and where exogenous peptides generated by the proteasome and transported by TAP meet MHC-I molecules for loading has been a matter of debate. The source and trafficking of MHC-I molecules in dendritic cells have largely been ignored under the expectation that these molecules came from the Endoplasmic reticulum (ER) or the plasma membrane. New studies reveal a concentrated pool of MHC-I molecules in the endocytic recycling compartment (ERC). These pools are rapidly mobilized to phagosomes carrying microbial antigens, and in a signal-dependent manner under the control of Toll-like receptors. The phagosome becomes a dynamic hub receiving traffic from multiple sources, the ER-Golgi intermediate compartment for delivering the peptide-loading machinery and the ERC for deploying MHC-I molecules that alert CD8 T cells of infection.
Keywords: Dendritic Cells, Major Histocompatibility Complex, Antigen Presentation/Processing, Toll-like Receptors/Pattern Recognition Receptors, Phagocytosis, Bacterial
Introduction
Classic MHC-I presentation
All nucleated cells express major histocompatibility complex (MHC) class I (MHC-I) molecules, whose presence on the cell surface serves to display an array of peptides, 8–9 amino acids long, derived from various different proteins within the cell. In this manner, cells present a snapshot of their proteins to the immune system in the form of a peptide-MHC-I complex that is engaged by the T cell receptor (TCR) of CD8 T cells (1). When a foreign peptide is displayed, such as a peptide from a virus that has infected the cell, antigen-experienced CD8 T cells with TCR specificity to that peptide kill the infected cell. On professional antigen presenting cells such as dendritic cells (DC), the cognate TCR and peptide-MHC complex interaction only signals the activation of naïve T cells when a signal in the form of a T cell co-stimulatory molecule is also expressed (2, 3). This second signal can activate either CD8 or CD4 T cells and is controlled by pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs) (4–7), which signal detection of microbial components (8).
Formation of the peptide-MHC-I complex in all cells including professional antigen presenting cells occurs constitutively and does not require inductive signals from PRRs. Loading of MHC-I molecules by peptides occurs within the endoplasmic reticulum (ER) (1). Peptides are derived from the activity of the cytosolic proteasome and are translocated into the ER via the activity of the peptide transporter associated with antigen processing (TAP). MHC-I heavy chain that has newly translocated into the ER is chaperoned by Calnexin and the oxidoreductase ERp57, and associates with its partner β2-microglobulin (β2m) followed by interaction with a set of proteins collectively called the peptide loading complex (PLC) (1). TAP is a component of the PLC, which is also comprised of other components including ERp57, Calreticulin, and Tapasin, which collectively mediate translocation of cytosolic proteasome-generated peptides into the ER lumen, peptide trimming, and loading onto heavy chain-β2m complexes. It is the activity of the PLC that underlies the ability of MHC-I molecules to present the wide array of peptides derived from not only cellular proteins but also infecting microorganisms. Because MHC-I are released from the PLC and exported out of the ER only upon binding of peptides, their stable expression at the plasma membrane is inherently linked to successful MHC-I assembly (1).
Cross-presentation and the subcellular pathways behind it
Besides the classic presentation of endogenous peptides, MHC-I molecules can also present peptides from extracellular proteins in a process termed cross-presentation. Like classic MHC-I presentation, cross-presentation is an important component of establishing tolerance to self-tissues, but also in mobilizing CD8 T cell responses against intracellular microorganisms as well as tumors (9, 10). Because of these reasons, the mechanisms and regulation of cross-presentation has instigated intense investigation. A number of pathways and subcellular locations have been described where cross-presentation might occur. One pathway depicts degradation of cargo proteins, internalized either by endocytosis or phagocytosis, by endosomal or phagosomal proteases most notably cathepsin S, and resultant peptides loaded onto MHC-I molecules independently of proteasomal degradation and TAP function. This pathway is known as the vacuolar pathway of cross-presentation (9). A second pathway depicts translocation of internalized proteins from endosomes or phagosomes to the cytoplasm where they undergo degradation by the proteasome. This pathway is called the cytosolic pathway of cross-presentation (9, 11). Peptides generated this way then follow two possible routes: 1) Transport via phagosomal/endosomal TAP back into the phagosomes/endosomes from which they originated where they are then loaded onto MHC-I in those compartments (9), and 2) Transport via ER TAP into the ER lumen for loading onto ER-resident heavy chain-β2m complexes (9). MHC-I loading has been implicated to occur within endosomes based on evidence pointing to peptide loading in early endosomes and not the ER, with cross-presented antigen being transported to the plasma membrane from those endosomes (12). Other evidence supported endosomal/phagosomal loading based on delivery of the MHC-I PLC (including TAP) from the ER-Golgi intermediate compartment (ERGIC) to phagosomes or endosomes (13). This delivery is mediated via vesicular traffic from the ERGIC to phagosomes/endosomes guided by pairing of the ER SNARE Sec22b with the plasma membrane SNARE Syntaxin 4, which is present on phagosomes and presumably also endosomes (13).
Another important distinction in the two pathways of cross-presentation, vacuolar versus cytosolic, is the reliance on TAP. The vacuolar pathway is TAP-independent – no involvement of the proteasome and no need for re-entry into the vacuole to meet MHC-I molecules for loading. The cytosolic pathway is necessarily TAP-dependent – those proteasome-generated peptides derived from an extracellular source of proteins must be brought into close proximity with MHC-I molecules. Notably, the source of MHC-I molecules that can be loaded within phagosomes or endosomes has not been extensively investigated. For endocytosed cargo, evidence points to loading of internalized plasma membrane MHC-I molecules that are recycled back to the plasma membrane for cross-presentation to CD8 T cells (14, 15) (more on this below). Until recently (16), MHC-I molecules were assumed to come from the ER, the ERGIC or endocytic pathways through the plasma membrane.
Regulation of cross-presentation by signals from PRRs – phase by phase during DC maturation
Several reports have demonstrated that TLR engagement increases CD8 T cell activation by cross-presented peptide (17–20), a process called cross-priming (21). While some PRRs transduce signals that impact a multitude of cellular functions including internalization, others serve as endocytic receptors mediating delivery of antigen to different subcellular compartments (22). Thus, depending on the particular PRR, its contribution to cross-presentation is distinct and necessarily reflects different mechanisms. Besides TLR-dependent enhancement of cross-presentation, plenty of evidence also supports TLR-independent enhancement of cross-presentation. In vivo and in vitro studies have shown that the cross-presentation of antigens complexed with IgG depends on the expression of γ-chain containing activating IgG Fc receptor (FcγR) by bone marrow derived DC (BMDC) (23) and CD8α− splenic DC (24). In this case, FcRγ-chain immunoreceptor tyrosine-based activation motif (ITAM) signaling is critical for cross-presentation (25). Upon binding to the model antigen ovalbumin (OVA), the mannose receptor (MR) undergoes polyubiquitination of the single Lysine residue in its cytoplasmic domain, an event that mediates cytosolic transport of endocytosed OVA into the cytoplasm and enables cross-presentation (26, 27). The RIG-I ligand 3pRNA is a robust inducer of CTL cross-priming via its signaling through the adaptor MAVS and induction of a type I interferon response (28). Cross-presentation by human DC is enhanced when antigen is targeted to the C-type lectin receptors Langerin on Langerhans cells (29) and DC-SIGN or DEC-205 on monocyte-derived DC (29, 30). In mice, the signaling PRR that detects necrotic cells, DNGR-1 (CLEC9A), is essential for cross-presentation of dying vaccinia virus-infected cells to CD8 T cells, and its absence is associated with an increased viral load, delayed resolution of primary infection, and loss of protection (31). Recognition of membrane bound heat shock proteins on the surface of dying cells by the lectin-like oxidized LDL receptor 1 promotes cross-presentation of cellular antigen from these dying cells (32). Because PRR signaling can enhance phagocytosis, co-stimulation and inflammatory cytokine production by DC (33), all of which in turn impact T cell activation, the effects of PRRs on cross-presentation can be indirect. Nevertheless, a number of studies focused on the direct effects of TLR signals, have now unequivocally shown control over multiple facets of the process behind the assembly of cross-presented peptide-MHC-I complexes (see below).
Late phase of DC maturation
Mature BMDC that have been treated with lipopolysaccharide (LPS) in vitro for an extended period of time (24–40 hours) are unable to cross-present IgG-complexed antigens (34), likely a reflection of their shutdown in antigen uptake (34, 35). Stimulation of human DC by TLR3 or TLR4 ligands inhibits their subsequent cross-presentation of viral antigens derived from virally infected apoptotic cells (36). Systemic administration of TLR ligands including LPS, CpG, poly I:C also inhibits cross-presentation in vivo (37). In this scenario, DC maturation by inflammatory cytokines may prevent bystander DC from cross-priming T cells (37). It has been proposed that shut down in cross-presentation after DC maturation serves to confine cross-priming to those antigens encountered simultaneously with the PRR signals that initially triggered DC activation (37, 38). Besides decreased antigen uptake, the blockade in cross-presentation by mature DC can also be due to the LPS-dependent upregulation in the levels of the transcription factor EB (TFEB) (39), which is a master regulator of lysosome biogenesis (40, 41). When forced TFEB expression is engineered in immature BMDC, phagosomal acidification and proteolysis are increased and cross-presentation is reduced in vitro and in vivo (39).
Intermediate phase of DC maturation
BMDC that had already been stimulated with LPS, but were not quite fully mature (so-called ‘intermediate DC’ 3–16 hours post-LPS), exhibited increased in vitro and in vivo cross-presentation of antigen-antibody complexes that engage FcγR (34, 42), as well as phagocytic bead-bound endotoxin-free antigen (42). Cross-presentation was evaluated using the T cell hybridoma B3Z as a readout, whose activation is induced by TCR engagement in the absence of co-stimulation (34, 42). ‘Intermediate DC’ exhibited increased endocytosis, expression of proteasome subunits and proteasome activity, as well as TAP activity (34). Despite having lower phagocytic activity than immature DC, the increased cross-presentation of phagocytic antigen by ‘intermediate DC’ is dependent on TLR4-mediated preservation of phagosomal antigen due to a slower rate of phagolysosomal fusion (and thus acidification) in those already stimulated DC (42). This contrasts with TLR-dependent enhancement of phagolysosomal fusion during the first hour of phagocytosis of bacteria by ‘naïve’ macrophages (43). Interestingly, reorganization and clustering of lysosomes in ‘intermediate DC’, a process mediated by the GTPase RAB34 (44, 45), is responsible for preventing their fusion with phagosomes (42). These observations made in vitro have potential implications in vivo where activated DC are proposed to enter a ‘surveillance’ state that allows continuous sampling of the tissue microenvironment and efficient cross-presentation. Murine DC have been reported to retain the capture and cross-presentation of soluble antigens after maturation in vivo (46). Presumably, the TLR-induced ‘surveillance’ state of DC in vivo overlaps with the initial phases of infection when microbes are still present but tissue destruction and resultant availability of self-antigens are absent.
Before DC maturation
When immature DC encounter antigen and TLR ligands simultaneously, cross-presentation is also enhanced. Immature BMDC deficient in the expression of TLR signaling adaptors TRIF and MyD88 showed significantly impaired cross-presentation of peptides derived from phagocytosed TLR ligand bearing cargo despite the provision of exogenous co-stimulation to primary antigen-specific naïve CD8 T cells, and with similar results using B3Z (16). Single deficiencies in either MyD88 or TRIF showed that MyD88, and not TRIF, is critical (16). Early work showed that TLRs control cross-presentation by increasing the efficiency of MHC-I peptide loading in human monocyte-derived DC through promotion of NOX2 activity (47). By sustaining the production of low levels of reactive oxygen species, which counteract the activity of the ATP-dependent vacuolar proton pump (v-ATPase) (48), NOX2 favors a relatively more alkaline phagosome luminal pH most conducive for preservation of potential cross-presented peptides (49–51). Concordantly, increased phagosomal acidification and proteolysis induced by TFEB overexpression in immature BMDC reduces cross-presentation as mentioned above (39). These adaptations unique to DC contribute – along with other factors such as limited vacuolar degradation (34, 52, 53), coordinate induction of immunoproteasome subunits, TAP1, Tapasin, and other components of the MHC-I loading machinery (54), as well as translocation of phagosomal/endosomal proteins to the cytosol for proteasomal degradation (11) – to the specialization of DC in cross-presentation (55).
Signaling from TLR4 has been reported to increase delivery of TAP to endosomes in a manner dependent on the TLR signaling adaptor MyD88, thus stimulating cross-presentation of endocytosed cargo (12). Confocal microscopy on BMDC revealed enrichment of MHC-I molecules on phagosomes carrying beads conjugated to TLR ligands, such as LPS and Pam2Csk4, and not plain beads (16) suggesting an influence by compartmentalized TLR signaling specific to phagosomes carrying TLR ligand. MHC-I enrichment peaked at 4.5 hours post phagocytosis and depended on MyD88 (not TRIF) expression in the DC (16). Of note, liquid chromatography-tandem mass spectrometry (LC-MS/MS) conducted on proteins from phagosomes isolated at 1.5 hours post phagocytosis of beads carrying antigen (with no added LPS) also showed MHC-I enrichment specifically in phagosomal proteins from ‘intermediate DC’ stimulated earlier with LPS compared to unstimulated BMDC (42). It remains to be formally tested whether TLR signal dependent MHC-I enrichment is phagosome autonomous, i.e. does a DC that had already been stimulated with LPS indiscriminately enrich MHC-I on all its phagosomes regardless of the TLR ligand of their cargo (self or non-self)? Phagosome autonomous MHC class II (MHC-II) presentation of antigen has been reported (56). Nevertheless, MHC-I enrichment on phagosomes in the presence of TLR signaling could be due to increased trafficking to phagosomes or an inability to exit phagosomes. The latter possibility is unlikely given that the levels of MHC-I molecules on the plasma membrane increase over time after TLR stimulation (16, 57, 58), and more importantly, cross-presentation of specific peptide is enhanced with TLR signaling (16). These observations point to a TLR dependent mechanism that mediates active MHC-I recruitment to phagosomes. How are these MHC-I molecules recruited to phagosomes and where do they come from? Before following up on these findings, a review of subcellular trafficking of MHC-I molecules is necessary.
The cell biology of subcellular trafficking of MHC-I molecules
Beginning in the ER and en route to the plasma membrane
As discussed above, the PLC in the ER mediates loading of MHC-I molecules with proteasome-generated peptides that have been translocated into the ER lumen by TAP. Once loaded, MHC-I molecules associate with transport receptor BAP31, accumulate at ER exit sites and traffic via export vesicles to the ERGIC (59–61). The ERGIC is a stable sub-compartment of the ER into which ER-derived cargo is shuttled from ER-exit sites on its way to the Golgi (62). It functions primarily in the folding and quality control of newly synthesized proteins, and notably harbors components of the MHC-I PLC such as TAP and Calreticulin (13). While loading of MHC-I with peptide occurs primarily in the ER, it can also occur during recycling between the ER and Golgi (63, 64), and the presence of PLC components in the ERGIC ensures both peptide loading and proper MHC-I folding (65). Only then are MHC-I released from this cycle and exported to the plasma membrane. MHC-I accumulate in the ERGIC when misfolded, for instance in the absence of peptides with good affinity of loading due to deficiency for TAP or Calreticulin (63, 66, 67). Using an antibody called AF6-88.5 that detects fully assembled MHC-I H2-Kb (68), these molecules in resting DC fail to co-localize with the lectin ERGIC-53, Calreticulin, Calnexin or TAP2 suggesting that these MHC-I molecules have passed the ERGIC quality control and have successfully been exported to the plasma membrane (16).
Clathrin-independent endocytosis of plasma membrane MHC-I
Two major pathways of endocytosis mediate the internalization of proteins, clathrin-mediated endocytosis (CME), which internalizes proteins such as transferrin receptor (TfR) and low-density lipoprotein receptor with specific signals in their cytoplasmic domains (69), and clathrin-independent endocytosis (CIE), which mediates endocytosis of proteins such as CD59 and β1-integrin (70, 71). Endosomes carrying cargo proteins from either CME or CIE fuse with early sorting endosomes (72). There are then two fates for these cargo proteins, either sorting to lysosomes (presumably for degradation) or sorting to a recycling pathway and returning to the plasma membrane.
Trafficking of MHC-I molecules has extensively been studied in cells other than professional phagocytes (Figure 1). Plasma membrane MHC-I molecules lack the cytoplasmic tails conferring clathrin/AP2 localization (73), and in HeLa cells are continuously internalized via both clathrin and dynamin independent endocytosis regulated by the ADP ribosylation factor 6 (ARF6) (74, 75). Thus, MHC-I molecules are internalized via clathrin independent endocytosis. The cytoplasmic domain of MHC-I contains a tyrosine and two serine residues conserved across 15 widely divergent vertebrate species (15). The tyrosine residue encoded by exon 6 is part of an intracellular targeting motif that is responsible for MHC-I endolysosomal trafficking in both BMDC and spleen derived GM-CSF/TNF-α cultured DC (15). A single point mutation substituting a phenylalanine residue for the conserved tyrosine led to abnormal accumulation of MHC-I molecules in the Golgi, disrupted cross-presentation but not classic antigen presentation in vitro, and strikingly also diminished generation of CTL responses against two immunodominant viral epitopes after infection with vesicular stomatitis virus or Sendai virus (15).
Figure 1. The intracellular traffic of MHC class I molecules.

Clathrin-independent and clathrin dependent endocytosis are depicted. MHC class I molecules undergo clathrin-independent endocytosis mediated by the small GTPase ARF6. After internalization, vesicles carrying MHC-I molecules fuse with sorting endosomes marked by RAB5 and EEA-1. There are two destinations out of sorting endosomes: endolysosomal compartments or the plasma membrane. A fraction of MHC-I molecules is routed to endolysosomal compartments and in resting BMDC appears to be small compared to the total MHC-I pool. MHC-I molecules have also been reported in multivesicular bodies (MVB), specifically MIIC, because of their observed co-localization with MHC class II molecules as well as endosomal and lysosomal markers in human Langerhans cells. Direct trafficking of MHC-I molecules from the ER to endolysosomal compartments has been reported in BMDC mediated by the chaperone CD74, which associates with MHC-I molecules in the ER. Two possible routes take MHC-I molecules back to the plasma membrane, the ‘fast recycling’ route regulated by RAB4 and RAB35, and the ‘slow recycling’ route regulated by RAB11a, whose activity is important for trafficking MHC-I molecules to a transitory perinuclear compartment called the endocytic recycling compartment (ERC). The ERC is comprised of a network of tubular and endosomal structures, some of which appear to be connected by bridges when observed by high-resolution microscopy. MHC-I molecules traffic out of the ERC is regulated by EHD1 and RAB22a through tubular endosomes formed via bending by the EHD1 interacting protein MICAL-L1.
Redirecting traffic of MHC-I molecules to late endosomes and lysosomes
In HeLa cells, a fraction of MHC-I molecules internalized through CIE is diverted to late endosomes and lysosomes converging first with clathrin-dependent cargo such as transferrin receptor within an early sorting EEA1+ endosome (76). In resting BMDC, the fraction of MHC-I molecules that co-localizes with LAMP-1+ late endosomal/lysosomal compartments represents around 10% (16). Presumably, these MHC-I are internalized from the plasma membrane. Within CD34+ precursor-derived human Langerhans cells, MHC-I molecules have been observed to co-localize with HLA-DM and HLA-DR (MHC-II) in late endosomal and lysosomal compartments termed as MIIC (54). Immunoelectron microscopy revealed staining of MHC-I molecules on both intraluminal vesicles and limiting membranes of multivesicular structures (54, 77). These structures also stained for MHC-II (77) as well as the late endosomal markers CD63 and the mannose-6-phosphate receptor (54). Internalized proteins destined for degradation, exocytosis or storage are usually incorporated into intraluminal vesicles of multivesicular bodies (78). The fate of MHC-I molecules that localize to late endosomal/lysosomal compartments in DC is not clear.
Notably, an ER to endolysosomal pathway of MHC-I trafficking chaperoned by the invariant chain (CD74), which is known for its trafficking of MHC-II molecules to the endocytic pathway, has been reported in DC. The possibility for the existence of such a pathway had come from older studies describing co-localization and association of CD74 with MHC-I molecules (77, 79, 80). CD74 deficiency reduced the endolysosomal localization of MHC-I while it had no effect on MHC-I internalization from the plasma membrane (81). The consequences of reduced endolysosomal localization of MHC-I on cross-presentation were profound. Less OVA-peptide loaded MHC-I molecules – detected by immunofluorescence microscopy using the only monoclonal antibody specific to a peptide-MHC-I complex (82) (and which is not sensitive enough to detect cross-presented peptides by flow cytometry (16)) – were observed co-localizing with LAMP-1+ endolysosomal compartments in CD74 deficient splenic DC (81). Whether MHC-I molecules are loaded in these LAMP-1+ endolysosomal compartments and exit from these compartments to the plasma membrane is not defined. CD74 deficiency impaired in vitro cross-presentation by DC and diminished virus-specific CD8 T cell responses to VSV infection (81). MHC-I and CD74 interacted intracellularly forming a complex in a pre-Golgi compartment within BMDC, and deletion of the cytosolic CD74 endosomal trafficking motif abrogated soluble antigen cross-presentation (81). These results suggest that trafficking of the MHC-I/CD74 complex is dictated by the itinerary of CD74. The evidence from these studies discussed above collectively indicates that the fraction of MHC-I molecules present within endolysosomal compartments at steady state can either come from the ER via CD74 or from the plasma membrane.
Trafficking through recycling endosomes – fast and slow recycling
Once a cargo protein is sorted into the recycling pathway, it faces two options. It can go through ‘fast recycling’ where it is returned to the plasma membrane directly from the sorting endosome regulated by RAB4 (83–85). ‘Fast recycling’ of MHC-I molecules in immature human monocyte derived DC has been reported to be dependent on RAB35 (86), which functions in endosomal trafficking (87, 88). Cargo can also go through ‘slow recycling’ by being transported first into a transitory organelle called the endocytic recycling compartment (ERC) (87, 89–92). The ERC is typically perinuclear and localized near the microtubule-organizing center (MTOC) (although not always) (87). Activity of the small GTPase RAB11a regulates traffic from sorting endosomes into the ERC (93, 94), but new evidence suggests that there may be more than one route of entry into ERC depending on the nature of the cargo (95). RAB11a has also been described to transport proteins to the trans-Golgi network (TGN), which is in close proximity to the ERC (96). Expression of a constitutively active (CA) form of Rab11aQ70L in CHO cells was shown to increase transferrin levels in RAB11a+ ERC (93). On the other hand, expression of a dominant negative allele of RAB11a in CHO cells diminished co-localization of transferrin with the ERC (93), and inhibited recycling of β1-integrin (97) and MHC-I molecules in HeLa cells (98). It also inhibited transport of the Shiga toxin B subunit and a marker for TGN, TGN38, from endosomes to the TGN (96).
Once internalized, ≈50% of MHC-I molecules in HeLa cells divert from sorting endosomes and enter into round ellipsoidal tubules that harbor ARF6 and appear devoid of transferrin receptor (74–76). These tubules may be components of tubular recycling endosomes (TRE) that can be as long as 10 μm in length and 200 nm in diameter, and are associated with endocytic regulatory proteins known as C-terminal Eps15 homology domain (EHD) proteins (99). TRE usually serve as conduits for recycling to the plasma membrane, but they may also carry MHC-I molecules out of sorting endosomes and into ERC. In support of this possibility, recent work shows that TRE preferentially facilitate trafficking of CIE cargo, and that some TRE originate from sorting endosomal membranes (95). TRE formation requires generation of phosphatidic acid (100), which recruits Syndapin II and the RAB8 and EHD1 interacting protein called ‘molecules interacting with CasL-like 1’ (MICAL-L1) to the endosomal membrane to mediate its bending and tubulation (101, 102).
By confocal microscopy with a resolution of ≈300 nm, the ERC appears as a compact perinuclear region suggesting a structure enclosed by a single limiting membrane. A side-by-side comparison of the ERC by three dimensional structured illumination microscopy (3D SIM) at ≈110 nm resolution confirmed the closely packed Rab11a region (95). SIM showed that the ERC contains a complex combination of multiple independent structures and endosomal membranes linked by connections up to 500 nm long (95). Surprisingly, SIM showed a high degree of co-localization of RAB11a with transferrin, but limited co-localization with CD59, which is internalized by CIE and is transported via EHD1 and MICAL-L1 containing TRE (103). Direct stochastic optical reconstruction microscopy (dSTORM) imaging at ≈10 nm precision of transferrin and CD59 cargos, which were chased from the sorting endosomes into the ERC, revealed that these two cargos were mostly segregated within the ERC and already arrive segregated from the sorting endosomes (95). Whether MHC-I molecules behave like CD59 was not studied. These data show that while both CME and CIE cargos are internalized into the ERC, they remain within distinct subdomains of the ERC (95). These findings have important implications to the manner in which CME and CIE cargos enter and exit the ERC (more below).
Proteins that enter the long recycling pathway and transit through the ERC must then exit the ERC to return to the plasma membrane. Exit of transferrin receptor from the ERC to the plasma membrane is thought to require GTP hydrolysis of RAB11a, explaining why Rab11aQ70L expression leads to accumulation in ERC rather than increased exit back to the plasma membrane (93). Much evidence supports a role for TRE in mediating outgoing plasma membrane-bound traffic from the ERC (75, 101, 102, 104, 105). EHD1 mediates recycling of transmembrane proteins that have been internalized by CME and CIE (104, 106, 107), and in particular promotes recycling of MHC-I back to the plasma membrane (104). The small GTPase RAB22a has also been associated with these MHC-I+ tubular recycling compartments in HeLa cells and its inactivation is required for final fusion of these tubules with the plasma membrane (98).
In CD34+ precursor-derived human Langerhans cells, LPS induces rapid mobilization of MHC-I molecules from endosomal compartments to the plasma membrane (54). In immature human monocyte-derived DC, ≈56% of the total MHC class I molecules were shown to reside internally, whereas only 23% of the total MHC-I remained internal following stimulation with LPS (58). In human monocyte-derived DC, TLR signaling alone was insufficient for tubulation of the ERC and further ligation of MHC-I and ICAM-1 on DC by the TCR and LFA-1 on CD8 T cells was necessary (108). ERC tubulation in these human DC is mediated by MICAL-L1 (109). Interestingly, disintegration of the tubular ERC in human DC with Nocodazole, an agent that depolymerizes microtubules, reduced the ability of the DC to activate antigen-specific CD8 T cells (108).
The ERC houses large reserves of MHC-I molecules in resting DC
In BMDC and CD8α+ splenic DC, a subset specialized in cross-presentation, a concentrated pool of MHC-I molecules is present within the ERC marked by RAB11a and the vesicle associated membrane proteins, VAMP3/cellubrevin and VAMP8/endobrevin (16). A smaller fraction of MHC-I also colocalized with Rab11a to the ERC-proximal TGN (16). Not surprisingly, some MHC-I molecules also co-localized with transferrin receptor, which undergoes ‘slow recycling’ through the ERC (93, 94). No MHC-I co-localization with EEA1+/Rab5+ early/sorting endosomes could be detected, and as mentioned above this pool of MHC-I was excluded from the ERGIC (16). Interestingly, a pool of MHC-I molecules that did not colocalize with Tapasin or KDEL motif-containing ER proteins had previously been observed in murine BMDC as well as a primary human peripheral blood-derived DC and a DC-like human cell line (57, 58). One study has reported MHC-I co-localization with transferrin in RAB3b/3c+ perinuclear recycling tubular structures within a DC2.4 murine cell line (110). Surprisingly, other RABs such as RAB5, RAB8, RAB10, RAB33a, RAB34 and RAB35 also localized to RAB3b/3 vesicles. siRNA-mediated silencing of these RABs all impaired cross-presentation of phagocytic antigen in a DC2.4/B3Z readout system (110). The relationship of the RAB3b/3c+ recycling structures to sorting endosomes and ERC is not defined.
RAB11a is important for maintaining these MHC-I pools in DC without affecting the levels of plasma membrane MHC-I molecules (16). Expression of a constitutively active allele of Rab11aQ70L led to an accumulation of MHC-I molecules within the ERC of BMDC, while lentiviral mediated silencing of RAB11a led to the disappearance of these stores (16). Based on the recycling paths of CIE cargo reviewed above, the most likely source of MHC-I molecules within the ERC of DC is the plasma membrane. The kinetics of MHC-I movement through the ‘slow recycling’ pathway in DC have not been studied. Interestingly, CD1a, unlike other CD1 isoforms, is also internalized by CIE (111), and like MHC-I has been shown to co-localize with RAB11a in ERC and not sorting endosomes within freshly isolated human epidermal Langerhans cells (112). Like MHC-I, CD1a recycling in HeLa cells is also RAB22a dependent (111).
ERC-Phagosome communication and its role in phagocytosis
With this background and understanding of the subcellular localization and trafficking of MHC-I molecules, we will go back to the question of where MHC-I molecules on TLR ligand bearing phagosomes come from. A vesicular trafficking pathway from the ERGIC to phagosomes is mediated by Sec22b, which delivers the MHC-I PLC (13). An examination of the patterns of Sec22b staining around phagosomes with or without TLR ligands did not reveal any differences, however, while MHC-I co-localized with Sec22b on phagosomes only when phagosomes carried LPS (16). The disparate phagosomal accumulation of Sec22b (TLR-independent) and MHC-I (TLR dependent) hinted at the arrival of MHC-I on phagosomes from a source other than the ERGIC. Plus, mature MHC-I molecules could not be detected in the ERGIC of DC to begin with (16, 57, 58). Despite some co-localization of MHC-I with the TGN marker TGN38 in resting DC, the absence of this marker on phagosomes made it unlikely that the TGN was contributing MHC-I molecules to the phagosome. On the other hand, concomitant accumulation of RAB11a, ERC SNAREs VAMP3 and VAMP8 (113), but not non-ERC SNAREs VAMP2 and VAMP7 betrayed the ERC origin of MHC-I molecules on TLR ligand bearing phagosomes (16).
Macrophages (and DC) can internalize large phagocytic cargo, which consume a significant portion of their plasma membrane and thus necessitate the availability of an additional source of membrane to replenish the plasma membrane and/or contribute to the phagosomal membrane (114). RAB11a has been reported to localize to nascent phagosomes in macrophages, and expression of a dominant negative allele of Rab11a impaired FcγR-mediated phagocytosis (115). Macrophages lack a distinct ERC, thus it was proposed that RAB11a delivery to incoming phagosomes reflected a contribution of endomembrane from an extensive network of recycling endosomes in order to extend membrane around phagosomes and facilitate phagocytosis (115). In contrast to these findings, there are no defects in phagocytosis in the absence of RAB11a in BMDC (16), which could reflect DC to macrophage differences or simply the ability of other endomembranes, perhaps ERGIC-derived (13), to compensate in DC. Such a possibility would be consistent with recruitment of the ER SNARE Sec22b to phagosomes, which occurs irrespective of the TLR ligand content of phagosomes and can thus contribute membranes to all phagosomes regardless of whether they carry microbial cargo or not (16). ER membranes were proposed in a much earlier study to contribute to phagosome formation, albeit in macrophages (116). ER proteins such as Calnexin and Calreticulin were enriched in phagosomes, and reticular structures staining with the ER enzyme Glucose-6-phosphatase were observed connected to phagosomes regardless of their cargo – inert beads, Salmonella typhimurium, Leishmania parasites, or red blood cells – and visible when phagocytosis was slowed down with inhibitors of phosphatidylinositol 3-kinase or phagosome acidification (116). A subsequent study contested these findings showing that ER-phagosome fusion in macrophages is a rare event (117). In retrospect, TAP, Calnexin and Calreticulin are also present in the ERGIC and can be detected along with Sec22b and ERGIC-53 in phagosomal proteins (13, 16). On the other hand, the ER resident protein ERp72 or the cis-Golgi resident ER-Golgi SNARE YKT6 could not be detected in phagosomal proteins attesting first to the purity of the phagosomal preparations (given the ease in which they could carry over membrane from the abundant ER or Golgi), but also to the fact that the presence of ER-associated proteins is selective (16). Only ER proteins that are also in the ERGIC can be detected on phagosomes while proteins from the ER proper are not. Therefore, it would appear that Sec22b-mediated vesicular traffic to phagosomes emanates from the ERGIC and not the ER proper.
The role of Rab11a and the ERC in cross-presentation
An intact ERC is critical for the positive edge that TLR signals impart on cross-presentation. The ERC can be dispersed from its perinuclear region with Nocodazole leading to the scattering of the ERC pool of MHC-I into smaller peripheral pools that no longer co-localize with RAB11a (16). The ERC can also be disrupted in BMDC by lentiviral-mediated silencing of RAB11a, which dissolves the ERC reserves of MHC-I molecules perhaps because they can no longer be shuttled from early sorting endosomes (16). With either Nocodazole treatment or RAB11a silencing, the net effect on cross-presentation is severe impairment. Peptides derived from phagocytic cargo can no longer be cross-presented to antigen-specific CD8 T cells despite the presence of TLR ligands within that phagocytic cargo (16). On the other hand, expression of a constitutively active RAB11a in BMDC increases MHC-I trafficking to the ERC achieving an effect opposite to that of depleting the MHC-I stores. Despite the fact that this happens equally efficiently in both TRIF and MyD88 deficient as well as wild type BMDC, the well-stocked ERC stores of MHC-I fail to restore phagosomal MHC-I accumulation and cross-presentation in the absence of TLR signals (16). Ramping up the numbers of MHC-I molecules in the ERC, however, does lead to earlier detection of MHC-I molecules on TLR ligand carrying phagosomes (16). Therefore, while RAB11a expression is critical for TLR-regulated cross-presentation, its role is primarily to traffic MHC-I molecules into the ERC (94).
RAB11a has been reported to control TLR4 trafficking to Escherichia coli carrying phagosomes, and importantly this conclusion was based on significant inhibition of phagosomal accumulation of TLR4 and its sorting adaptor TRAM that is necessary for TRIF signaling, upon RAB11a suppression (118). But this outcome, similar to that with MHC-I described above (16), likely reflects dissolution of the ERC and with it the source of TLR4 that can traffic to phagosomes. When the ERC is intact, RAB11a accumulation on TLR4 signaling phagosomes is likely a reflection of recruitment and retention of ERC cargo proteins, RAB11a, VAMP3, VAMP8, MHC-I (16), and concordantly TLR4 (118) specifically on those phagosomes carrying TLR4 ligands. While both TLR4 and TLR2 signaling trigger phagosomal MHC-I enrichment on phagosomes carrying their respective ligands (16), TLR4 is the only TLR that has so far been reported to reside in RAB11a+ ERC, and the only TLR whose cell biological basis for signal transduction is understood (119). While TLR2 has been reported to colocalize with RAB11a+ structures in human monocytes (120), further work is necessary to elucidate the relationship between RAB11a and TLR2 signaling. It remains possible that other TLRs and PRRs are also present in ERC. Collectively, the evidence from these studies suggests that vectorial trafficking of TLR4 with MHC-I from ERC to phagosomes further remodels those cross-presenting phagosomes to conduct additional TRIF-mediated signaling functions such as type I IFN-β production (118), which has been shown to stimulate cross-presentation (121). Another important note here is that the impaired cross-presentation resultant from RAB11a silencing is not because of interrupting the enhancing effects of IFN-β on cross-presentation because the addition of exogenous biologically active IFN-β to RAB11a-silenced or TLR signaling deficient BMDC does not restore cross-presentation (16).
Besides the reported presence of TLR4 in the ERC, another protein that has been reported to be localize to the plasma membrane and RAB11a+ ERC – albeit in CHO cells and bone marrow derived macrophages – is the NADPH oxidase flavocytochrome b558, the integral membrane catalytic core of the phagocytic NADPH oxidase (122). Flavocytochrome b558 colocalizes with a GFP-tagged RAB11 protein around nascent phagosomes (122), consistent with the previous report discussed above (115). Flavocytochrome b558 is composed of two subunits p22phox and gp91phox (also known as NOX2) (where phox denotes phagocyte oxidase) (123), and NOX2 plays a significant role in cross-presentation (51, 124). It could thus be argued that the crux of a role for the ERC in cross-presentation is its phagosomal delivery of NOX2 (rather than MHC-I molecules). However, the subcellular localization of NOX2 is different in BMDC where NOX2 resides in inhibitory lysosome related organelles containing lysosomal proteins LAMP-1/LAMP-2 and RAB27a (49). Furthermore, while RAB11a and MHC-I co-localize in BMDC, NOX2 co-localizes with neither, and instead shows almost 100% co-localization with LAMP-1 in resting BMDC and on LAMP-1+ phagosomes in BMDC, and irrespective of the presence of TLR ligands within those phagosomes (16). These findings highlight the differences between macrophages and DC (55) (which incidentally also include higher expression of TFEB mRNA in macrophages than in BMDC (39)), but they also reveal that the trafficking of MHC-I and NOX2 to phagosomes is subject to different rules.
Discovery of the ERC as a major reserve for MHC-I molecules was made in BMDC cultured in GM-CSF, which facilitated study of the cell biological basis of cross-presentation. The subject of recent debate (125–127), GM-CSF cultured DC have been instrumental in revealing fundamental aspects of myeloid cell biology (127). These cultured DC have been proposed to model inflammatory DC (128), such as the DC-SIGN/CD209+ monocyte derived DC, which are recruited to lymph nodes from the blood by LPS and Gram-negative bacteria in a TLR4-dependent manner, and actively cross-present peptides derived from bacteria (129). Enhancing cross-presentation by TLR4 in this context would serve to rapidly activate CD8 T cells for host defense against infection. Consistent with this possibility, human plasmacytoid DC, for example, have been reported to contain a major intracellular pool of MHC-I that co-localizes exclusively with transferrin receptor (130). This pool may underlie their ability to serve as primary mediators of anti-viral responses by allowing prompt cross-presentation of peptides derived from internalized viruses (131). Importantly, the existence of an MHC-I+ ERC may form one mechanistic basis for the superior ability of DC subsets such as conventional splenic and lymph node resident CD8α+ DC to cross-present peptides derived from cellular sources in vivo. Other mechanisms may include the lower levels of TFEB expression in splenic CD8α+ DC compared to other splenic DC and macrophages (39). Like BMDC, the specialized RAB11a+ compartment within CD8α+ splenic DC cells may also provide a critical resource of MHC-I for the cross-presenting phagosome (16).
How are MHC-I molecules selectively recruited to TLR ligand bearing phagosomes?
The discussion so far has left us with MHC-I molecules in the ERC in one hand, and their TLR/MyD88 signal dependent enrichment on TLR ligand bearing phagosomes in the other hand. Because the phagosomal enrichment of ERC derived proteins such as VAMP8, RAB11a, and MHC-I is dependent on TLR signaling and TLR ligands within the phagocytic cargo, it stands to reason that the recruitment of MHC-I molecules from the ERC to phagosomes is subject to regulation. Vesicles that derive from the ERC and fuse with phagosomes likely mediate this recruitment. Vesicular membrane fusion is mediated by SNARE family members, which along with RAB GTPases contribute to the specificity of vesicular transport via their localization to distinct subcellular compartments (132). SNAP23/syntaxin/VAMP complexes constitute the minimal components for membrane fusion (132), and increasing evidence shows that these SNARE complexes can be regulated by phosphorylation (133). Phosphorylation of SNAP23 by the inhibitor of nuclear factor κB kinase subunit 2 (IKK2, also known as IKKβ), a subunit of the IκB kinase (IKK), stabilizes SNAP23/syntaxin 4/VAMP2 or VAMP8 containing SNARE complexes and mediates mast cell degranulation (133, 134). IKK-2 mediated phosphorylation of SNAP23 is also crucial for SNAP23/syntaxin 11/VAMP8 complex formation and controls platelet secretion (135). Phosphorylation of SNAP23 is thought to induce full zippering of the SNARE-pins as often observed in regulated exocytosis, and this could occur via multiple mechanisms (133). For example, phosphorylation of SNAP23 has been shown to inhibit its interaction with syntaxin 4, allowing entry of syntaxin 4 into ternary complexes with VAMP8 (133). Alternatively, phosphorylation of SNAP23 may increase its binding to syntaxin 4 and VAMP8, a possibility supported by observations in stimulated mast cells where most of the phosphorylated SNAP23 was bound to syntaxin 4 and VAMP2 (133).
IKK2 is activated by phosphorylation downstream of TLR as well as many cytokine activated signaling pathways that converge on NF-κB activation (136). An important role for IKK2 in cross-presentation is supported by evidence showing that treatment of BMDC with a specific IKK2 inhibitor abrogates the TLR-regulated phagosomal recruitment of MHC-I and the cross-presentation of microbial antigen (16). When TLR signaling is engaged during phagocytosis of TLR ligand containing cargo, MyD88-dependent phosphorylation of IKK2 activates this kinase enabling its phosphorylation of phagosomal SNAP23 (16). The prediction is that MyD88 signals are compartmentalized impacting only those phagosomes carrying TLR ligands and leading to phosphorylation of SNAP23 specifically on those phagosomes. Evidence in support of this compartmentalization comes from earlier studies showing phagosome autonomous maturation in DC such that only phagosomes carrying TLR ligands undergo invariant chain degradation and peptide loading of MHC-II molecules (56). Phospho-SNAP23 thus serves as a flag on phagosomal membranes by facing the cytosol and serving as the docking site for outgoing traffic from the ERC. This mechanism serves to deliver to phagosomes the critical numbers of MHC-I molecules once TLRs signal infection and the increased need for cross-presentation (Figure 2).
Figure 2. Toll-like receptor signals divert the traffic of MHC-I molecules from the ERC to phagosomes.
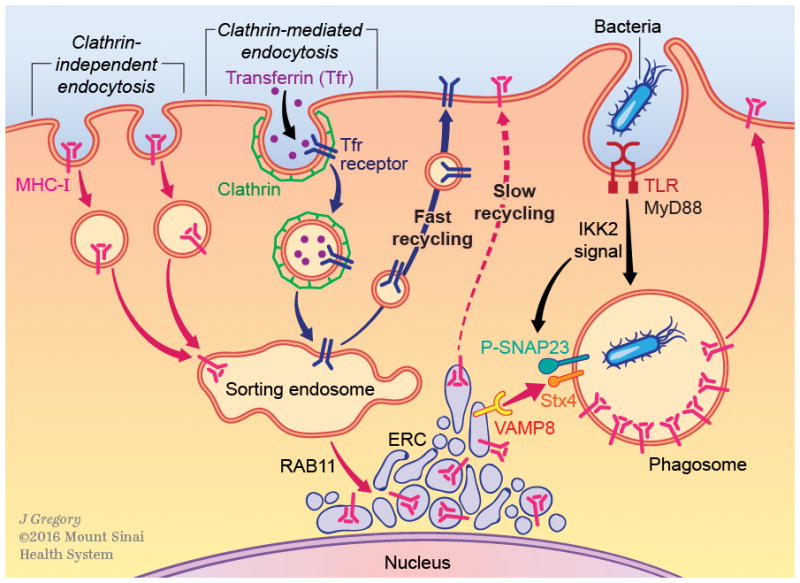
Three types of internalization events are depicted in this schematic. Clathrin-independent endocytosis (CIE) of MHC-I molecules, clathrin-mediated endocytosis (CME) of transferrin and phagocytosis of bacteria. Endosomes carrying the CIE cargo MHC-I and the CME cargo transferrin receptor converge into the sorting endosome, but maintain segregation in distinct subdomains. Although both MHC-I and transferrin receptor can undergo ‘fast recycling’, transferrin receptor is shown here recycling back to the plasma membrane through ‘fast recycling’. MHC-I molecules are shown entering the ERC through ‘slow recycling’. TLR signaling dependent on MyD88 and IKK2 leads to the phosphorylation of the synaptosomal associated protein SNAP23, which serves to stabilize trans-SNARE complexes between phagosomal Q-SNAREs (one candidate is Syntaxin 4) and ERC R-SNARE (VAMP8/cellulbrevin is a candidate). The resultant TLR-induced stable SNARE pins drive fusion between the MHC-I rich ERC and the microbial antigen containing phagosome. The fusion event delivers MHC-I molecules from the ERC to the phagosome. Microbial peptide-loaded MHC-I then travel to the plasma membrane to be displayed for recognition by microbe-specific CD8 T cells.
Expression of a mutant phosphomimetic SNAP23 is sufficient for phagosomal acquisition of VAMP8 (normally recruited with TLR signals) but not VAMP7 (not recruited with TLR signals), supporting the possibility that a SNARE complex comprised of SNAP23, syntaxin 4 and VAMP8 orchestrates ERC-phagosome fusion and subsequent delivery of ERC cargo (MHC-I, TLR4) to phagosomes. Further evidence was provided upon expression of a phosphomimetic mutant form of SNAP23 (SNAP23EE) in TLR signaling deficient DC, which on this background served as a reporter revealing the identity of the molecules that it recruits to the phagosomal membrane. SNAP23EE specifically recruited ERC but not TGN markers along with MHC-I molecules providing additional support for the ERC and not the TGN as the source of phagosomal MHC-I molecules (16). Expression of SNAP23EE overrides TLR signals, and presumably any phagosome carrying self or non-self would consequently be licensed to receive MHC-I molecules. The absence of other TLR-dependent signals should in theory preclude the cross-presentation of a self-antigen in that case.
An additional point to consider is that raising the TLR-dependent flag of phospho-SNAP23 may not divert all outgoing traffic from the ERC specifically to phagosomes carrying TLR ligands. The fraction of traffic that is diverted and whether specific subdomains of the ERC, containing distinct cargo, are trafficked in a TLR-dependent manner remains to be determined. Super-resolution and advanced single-molecule imaging techniques (SIM and dSTORM) have recently revealed that when the ERC acquires cargo from the sorting endosome, it maintains segregation of that cargo (95), which as we discussed above can either be internalized by CME or CIE. Moreover, RAB11a was found to be involved in trafficking CME and not CIE cargo to the plasma membrane, while CIE cargo localized to MICAL-L1 containing TRE (discussed above) (95). These new data demonstrate the complexity of the ERC, and further suggest that multiple routes of trafficking out of the ERC exist, mediated by different molecules and likely subject to different regulation.
The ERGIC and ERC pathways of vesicular traffic contribute distinct components to the cross-presenting phagosome
An updated view of the classic phagosome maturation (137) must now incorporate the recruitment of the ERC and ERGIC as integral parts of the process of phagosome maturation under different cellular cues (Figure 3). The phagosome-bound ERGIC pathway of vesicular traffic is mobilized independently of TLR signals as shown by the abundance of Sec22b, ERGIC-53 and Calnexin on DC phagosomes regardless of their TLR content or TLR signaling (16). The ERGIC pathway delivers components of the MHC-I PLC, most notably TAP, in preparation for cross-presentation (13). Silencing Sec22b in BMDC abrogates cross-presentation (13, 16), because of the blockade in ERGIC-to-phagosome traffic and failure to deliver the MHC-I PLC (13). The MHC-I PLC is recruited to phagosomes in the first few hours following phagocytosis, coinciding with the time when cross-presentation reaches a plateau within 3 hours (138, 139). MHC-I loading by OVA-derived SIINFEKL peptide was shown to occur specifically and autonomously in phagosomes that contained the OVA protein and not in those containing a control protein (138, 139). Transient association of proteasomes with the cytoplasmic side of phagosomes, reported to peak at around 60 minutes after their formation, facilitates the cytosolic proteasome/TAP pathway of generating peptides for cross-presentation (139). These studies along with successful reconstitution of peptide loading in vitro using purified phagosomes (138) argue against loading of MHC-I molecules in the ER and subsequent export to nascent phagosomes. Collectively, the evidence implicates phagosomes as both the source of peptide (derived from phagocytosed cargo) and the site of peptide loading during cross-presentation.
Figure 3. The TLR-dependent ERC pathway and the TLR-independent ERGIC pathway cooperate in the formation of a cross-presenting phagosome.
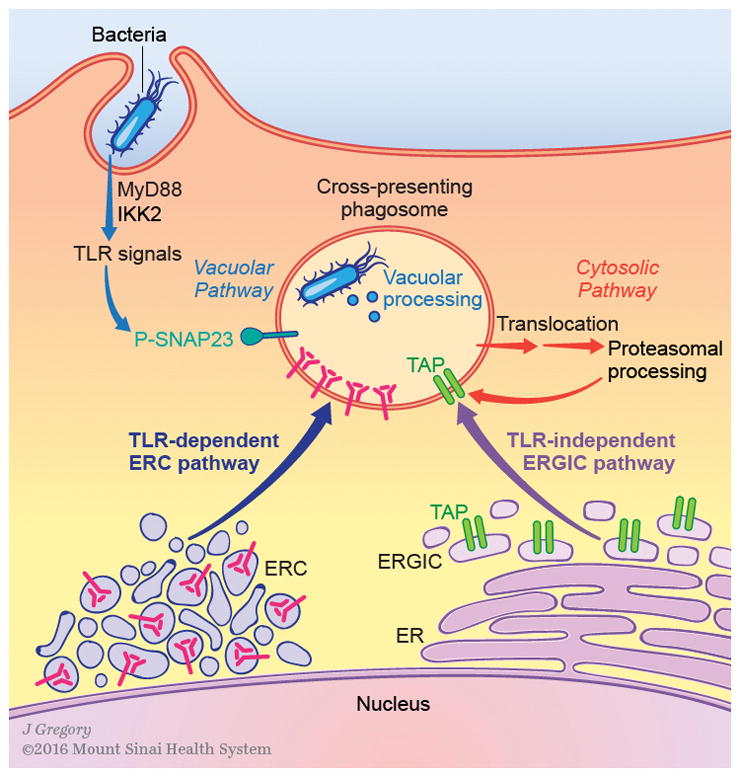
Upon phagocytosis of bacteria, two vesicular pathways are mobilized to greet the bacterium-containing phagosome. The pathway from the ERGIC is dependent on the ER SNARE Sec22b, and serves to deliver the peptide loading machinery including TAP, Tapasin, and Calreticulin. This pathway is mobilized independently of TLR signals, and the precise nature of the signals initiating its mobilization has not been defined. The pathway from the ERC is controlled by MyD88-IKK2 dependent TLR signaling which phosphorylates SNAP23 on phagosomes. Phospho-SNAP23 (pSNAP-23) flags microbial antigen containing phagosomes for receiving MHC-I molecules from the ERC in a TLR-dependent manner driven by a pSNAP-23 stabilized ERC-phagosome SNARE complex. Therefore, while delivery of the peptide loading complex occurs independently of TLR signals, delivery of MHC-I molecules is regulated by TLR signals. In the meantime, bacterial protein antigens derived from bacterial degradation are subjected to proteolysis through the vacuolar pathway which relies on the proteolytic activity of vacuolar proteases, or the cytosolic pathway which relies on cytosolic translocation of antigen, degradation by the proteasome, and transport back into phagosomes. The combined activity of these two pathways likely contributes to the diversity of peptides that can be generated from exogenous cargo and which in turn can be cross-presented by MHC-I molecules. Together with the vacuolar and cytosolic pathways of antigen processing, the ERC and ERGIC pathways of vesicular traffic equip phagosomes for cross-presentation of microbial antigen.
The TLR regulated ERC pathway of vesicular traffic serves a purpose different than the ERGIC pathway. The ERC is the source of MHC-I molecules that are recruited to microbial antigen carrying phagosomes for peptide loading (16). Unlike Sec22b silencing, RAB11a silencing impairs cross-presentation because it destroys the ERC stores of MHC-I (16). The ERC pathway of cross-presentation coexists with the vacuolar and cytosolic pathways of cross-presentation such that ERC-derived MHC-I molecules can either be loaded by peptides generated through the activity of vacuolar proteases such as cathepsin S (the vacuolar pathway) and/or they can be loaded by proteasome-TAP dependent peptides (the cytosolic pathway) as described above. Longer peptides for example, such as those derived from melanoma antigens gp100 and MelanA/MART1, appear to have a preference for vacuolar processing and are independent of the involvement of Sec22b and TAP (140). Nevertheless, by using peptides generated through both the vacuolar and cytosolic pathways, MHC-I molecules can be loaded by a broader repertoire of peptides that contribute to a diverse CD8 T cell response. The ERC pathway adds one more significant dimension to cross-presentation. It is critical for the positive edge that TLR signals impart on cross-presentation. Instead of assembling new correctly folded MHC-I molecules in the ER, the ERC maintains pre-assembled stores of MHC-I molecules, which can readily be recruited to phagosomes containing microbial cargo under the guidance of TLR signals.
Future perspective
The studies reviewed here represent a first glimpse into the impact of intracellular MHC-I traffic on cross-presentation. Studying the heterogeneous subsets of DC residing in lymphoid and non-lymphoid tissues, in mice and in humans, should reveal a tremendous amount of new information regarding the cell biology and regulation of intracellular MHC-I trafficking in these different DC subsets, in different tissues, and under steady state versus inflammatory conditions. The variety of strategies that microbial pathogens use to evade detection by CD8 T cells should shed light on the critical steps in MHC-I trafficking and how they modulate cross-presentation. Combine this with new super resolution imaging techniques, such as structured illumination microscopy (SIM) and three-dimensional direct stochastic optical reconstruction microscopy (dSTORM), and the knowledge we are about to gain is sure to be nothing short of spectacular.
Acknowledgments
J.M.B. is indebted to Priyanka Nair-Gupta for her outstanding contributions. J.M.B. would like to thank her laboratory members for fruitful discussions. J.M.B. and her laboratory are supported by NIH grants DK072201, AI095245 and AI123284, the Burroughs Wellcome Fund, and a Leukemia and Lymphoma Society Scholar Award.
Footnotes
J.M.B. does not have financial or personal relationships that could be viewed as presenting a potential conflict of interest.
References
- 1.Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annual review of immunology. 2013;31:443–473. doi: 10.1146/annurev-immunol-032712-095910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwartz RH. Costimulation of T lymphocytes: the role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell. 1992;71:1065–1068. doi: 10.1016/s0092-8674(05)80055-8. [DOI] [PubMed] [Google Scholar]
- 3.Mueller DL, Jenkins MK, Schwartz RH. Clonal expansion versus functional clonal inactivation: a costimulatory signalling pathway determines the outcome of T cell antigen receptor occupancy. Annual review of immunology. 1989;7:445–480. doi: 10.1146/annurev.iy.07.040189.002305. [DOI] [PubMed] [Google Scholar]
- 4.Liu Y, Janeway CA., Jr Microbial induction of co-stimulatory activity for CD4 T-cell growth. Int Immunol. 1991;3:323–332. doi: 10.1093/intimm/3.4.323. [DOI] [PubMed] [Google Scholar]
- 5.Liu Y, Janeway CA., Jr Cells that present both specific ligand and costimulatory activity are the most efficient inducers of clonal expansion of normal CD4 T cells. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:3845–3849. doi: 10.1073/pnas.89.9.3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 7.Janeway CA., Jr Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54(Pt 1):1–13. doi: 10.1101/sqb.1989.054.01.003. [DOI] [PubMed] [Google Scholar]
- 8.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 9.Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. 2012;12:557–569. doi: 10.1038/nri3254. [DOI] [PubMed] [Google Scholar]
- 10.Kurts C, Robinson BW, Knolle PA. Cross-priming in health and disease. Nat Rev Immunol. 2010;10:403–414. doi: 10.1038/nri2780. [DOI] [PubMed] [Google Scholar]
- 11.Rock KL, Shen L. Cross-presentation: underlying mechanisms and role in immune surveillance. Immunological reviews. 2005;207:166–183. doi: 10.1111/j.0105-2896.2005.00301.x. [DOI] [PubMed] [Google Scholar]
- 12.Burgdorf S, Scholz C, Kautz A, Tampe R, Kurts C. Spatial and mechanistic separation of cross-presentation and endogenous antigen presentation. Nature immunology. 2008;9:558–566. doi: 10.1038/ni.1601. [DOI] [PubMed] [Google Scholar]
- 13.Cebrian I, et al. Sec22b regulates phagosomal maturation and antigen crosspresentation by dendritic cells. Cell. 2011;147:1355–1368. doi: 10.1016/j.cell.2011.11.021. [DOI] [PubMed] [Google Scholar]
- 14.Basha G, Lizee G, Reinicke AT, Seipp RP, Omilusik KD, Jefferies WA. MHC class I endosomal and lysosomal trafficking coincides with exogenous antigen loading in dendritic cells. PloS one. 2008;3:e3247. doi: 10.1371/journal.pone.0003247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lizee G, et al. Control of dendritic cell cross-presentation by the major histocompatibility complex class I cytoplasmic domain. Nature immunology. 2003;4:1065–1073. doi: 10.1038/ni989. [DOI] [PubMed] [Google Scholar]
- 16.Nair-Gupta P, et al. TLR Signals Induce Phagosomal MHC-I Delivery from the Endosomal Recycling Compartment to Allow Cross-Presentation. Cell. 2014;158:506–521. doi: 10.1016/j.cell.2014.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Datta SK, Raz E. Induction of antigen cross-presentation by Toll-like receptors. Springer seminars in immunopathology. 2005;26:247–255. doi: 10.1007/s00281-004-0174-2. [DOI] [PubMed] [Google Scholar]
- 18.Mantegazza AR, Magalhaes JG, Amigorena S, Marks MS. Presentation of phagocytosed antigens by MHC class I and II. Traffic. 2013;14:135–152. doi: 10.1111/tra.12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nair P, Amsen D, Blander JM. Co-ordination of incoming and outgoing traffic in antigen-presenting cells by pattern recognition receptors and T cells. Traffic. 2011;12:1669–1676. doi: 10.1111/j.1600-0854.2011.01251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nair-Gupta P, Blander JM. An Updated View of the Intracellular Mechanisms Regulating Cross-Presentation. Front Immunol. 2013;4:401. doi: 10.3389/fimmu.2013.00401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bevan MJ. Cross-priming. Nature immunology. 2006;7:363–365. doi: 10.1038/ni0406-363. [DOI] [PubMed] [Google Scholar]
- 22.Moretti J, Blander JM. Insights into phagocytosis-coupled activation of pattern recognition receptors and inflammasomes. Current opinion in immunology. 2014;26:100–110. doi: 10.1016/j.coi.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Regnault A, et al. Fcgamma receptor-mediated induction of dendritic cell maturation and major histocompatibility complex class I-restricted antigen presentation after immune complex internalization. The Journal of experimental medicine. 1999;189:371–380. doi: 10.1084/jem.189.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.den Haan JM, Bevan MJ. Constitutive versus activation-dependent cross-presentation of immune complexes by CD8(+) and CD8(−) dendritic cells in vivo. The Journal of experimental medicine. 2002;196:817–827. doi: 10.1084/jem.20020295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boross P, et al. FcRgamma-chain ITAM signaling is critically required for cross-presentation of soluble antibody-antigen complexes by dendritic cells. Journal of immunology. 2014;193:5506–5514. doi: 10.4049/jimmunol.1302012. [DOI] [PubMed] [Google Scholar]
- 26.Zehner M, et al. Mannose receptor polyubiquitination regulates endosomal recruitment of p97 and cytosolic antigen translocation for cross-presentation. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:9933–9938. doi: 10.1073/pnas.1102397108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zehner M, Burgdorf S. Regulation of antigen transport into the cytosol for cross-presentation by ubiquitination of the mannose receptor. Molecular immunology. 2013;55:146–148. doi: 10.1016/j.molimm.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 28.Hochheiser K, et al. Cutting Edge: The RIG-I Ligand 3pRNA Potently Improves CTL Cross-Priming and Facilitates Antiviral Vaccination. Journal of immunology. 2016 doi: 10.4049/jimmunol.1501958. [DOI] [PubMed] [Google Scholar]
- 29.Fehres CM, et al. Cross-presentation through langerin and DC-SIGN targeting requires different formulations of glycan-modified antigens. Journal of controlled release: official journal of the Controlled Release Society. 2015;203:67–76. doi: 10.1016/j.jconrel.2015.01.040. [DOI] [PubMed] [Google Scholar]
- 30.Saluja SS, et al. Targeting human dendritic cells via DEC-205 using PLGA nanoparticles leads to enhanced cross-presentation of a melanoma-associated antigen. International journal of nanomedicine. 2014;9:5231–5246. doi: 10.2147/IJN.S66639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iborra S, Izquierdo HM, Martinez-Lopez M, Blanco-Menendez N, Reis e Sousa C, Sancho D. The DC receptor DNGR-1 mediates cross-priming of CTLs during vaccinia virus infection in mice. The Journal of clinical investigation. 2012;122:1628–1643. doi: 10.1172/JCI60660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu H, et al. Membrane-bound heat shock proteins facilitate the uptake of dying cells and cross-presentation of cellular antigen. Apoptosis: an international journal on programmed cell death. 2015 Oct 19; doi: 10.1007/s10495-015-1187-0. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 33.Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327:291–295. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gil-Torregrosa BC, et al. Control of cross-presentation during dendritic cell maturation. European journal of immunology. 2004;34:398–407. doi: 10.1002/eji.200324508. [DOI] [PubMed] [Google Scholar]
- 35.Garrett WS, et al. Developmental control of endocytosis in dendritic cells by Cdc42. Cell. 2000;102:325–334. doi: 10.1016/s0092-8674(00)00038-6. [DOI] [PubMed] [Google Scholar]
- 36.Weck MM, Grunebach F, Werth D, Sinzger C, Bringmann A, Brossart P. TLR ligands differentially affect uptake and presentation of cellular antigens. Blood. 2007;109:3890–3894. doi: 10.1182/blood-2006-04-015719. [DOI] [PubMed] [Google Scholar]
- 37.Wilson NS, et al. Systemic activation of dendritic cells by Toll-like receptor ligands or malaria infection impairs cross-presentation and antiviral immunity. Nature immunology. 2006;7:165–172. doi: 10.1038/ni1300. [DOI] [PubMed] [Google Scholar]
- 38.Villadangos JA, Schnorrer P, Wilson NS. Control of MHC class II antigen presentation in dendritic cells: a balance between creative and destructive forces. Immunological reviews. 2005;207:191–205. doi: 10.1111/j.0105-2896.2005.00317.x. [DOI] [PubMed] [Google Scholar]
- 39.Samie M, Cresswell P. The transcription factor TFEB acts as a molecular switch that regulates exogenous antigen-presentation pathways. Nature immunology. 2015;16:729–736. doi: 10.1038/ni.3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nature reviews Molecular cell biology. 2013;14:283–296. doi: 10.1038/nrm3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sardiello M, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 42.Alloatti A, et al. Toll-like Receptor 4 Engagement on Dendritic Cells Restrains Phago-Lysosome Fusion and Promotes Cross-Presentation of Antigens. Immunity. 2015;43:1087–1100. doi: 10.1016/j.immuni.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 43.Blander JM, Medzhitov R. Regulation of phagosome maturation by signals from toll-like receptors. Science. 2004;304:1014–1018. doi: 10.1126/science.1096158. [DOI] [PubMed] [Google Scholar]
- 44.Wang T, Hong W. Interorganellar regulation of lysosome positioning by the Golgi apparatus through Rab34 interaction with Rab-interacting lysosomal protein. Molecular biology of the cell. 2002;13:4317–4332. doi: 10.1091/mbc.E02-05-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kasmapour B, Gronow A, Bleck CK, Hong W, Gutierrez MG. Size-dependent mechanism of cargo sorting during lysosome-phagosome fusion is controlled by Rab34. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:20485–20490. doi: 10.1073/pnas.1206811109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Drutman SB, Trombetta ES. Dendritic cells continue to capture and present antigens after maturation in vivo. Journal of immunology. 2010;185:2140–2146. doi: 10.4049/jimmunol.1000642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vulcano M, et al. Toll receptor-mediated regulation of NADPH oxidase in human dendritic cells. Journal of immunology. 2004;173:5749–5756. doi: 10.4049/jimmunol.173.9.5749. [DOI] [PubMed] [Google Scholar]
- 48.Trombetta ES, Ebersold M, Garrett W, Pypaert M, Mellman I. Activation of lysosomal function during dendritic cell maturation. Science. 2003;299:1400–1403. doi: 10.1126/science.1080106. [DOI] [PubMed] [Google Scholar]
- 49.Jancic C, et al. Rab27a regulates phagosomal pH and NADPH oxidase recruitment to dendritic cell phagosomes. Nature cell biology. 2007;9:367–378. doi: 10.1038/ncb1552. [DOI] [PubMed] [Google Scholar]
- 50.Mantegazza AR, et al. NADPH oxidase controls phagosomal pH and antigen cross-presentation in human dendritic cells. Blood. 2008;112:4712–4722. doi: 10.1182/blood-2008-01-134791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Savina A, et al. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell. 2006;126:205–218. doi: 10.1016/j.cell.2006.05.035. [DOI] [PubMed] [Google Scholar]
- 52.Inaba K, et al. The formation of immunogenic major histocompatibility complex class II-peptide ligands in lysosomal compartments of dendritic cells is regulated by inflammatory stimuli. The Journal of experimental medicine. 2000;191:927–936. doi: 10.1084/jem.191.6.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lennon-Dumenil AM, et al. Analysis of protease activity in live antigen-presenting cells shows regulation of the phagosomal proteolytic contents during dendritic cell activation. The Journal of experimental medicine. 2002;196:529–540. doi: 10.1084/jem.20020327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.MacAry PA, Lindsay M, Scott MA, Craig JI, Luzio JP, Lehner PJ. Mobilization of MHC class I molecules from late endosomes to the cell surface following activation of CD34-derived human Langerhans cells. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:3982–3987. doi: 10.1073/pnas.071477498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Savina A, Amigorena S. Phagocytosis and antigen presentation in dendritic cells. Immunological reviews. 2007;219:143–156. doi: 10.1111/j.1600-065X.2007.00552.x. [DOI] [PubMed] [Google Scholar]
- 56.Blander JM, Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature. 2006;440:808–812. doi: 10.1038/nature04596. [DOI] [PubMed] [Google Scholar]
- 57.Delamarre L, Holcombe H, Mellman I. Presentation of exogenous antigens on major histocompatibility complex (MHC) class I and MHC class II molecules is differentially regulated during dendritic cell maturation. The Journal of experimental medicine. 2003;198:111–122. doi: 10.1084/jem.20021542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ackerman AL, Cresswell P. Regulation of MHC class I transport in human dendritic cells and the dendritic-like cell line KG-1. Journal of immunology. 2003;170:4178–4188. doi: 10.4049/jimmunol.170.8.4178. [DOI] [PubMed] [Google Scholar]
- 59.Spiliotis ET, Manley H, Osorio M, Zuniga MC, Edidin M. Selective export of MHC class I molecules from the ER after their dissociation from TAP. Immunity. 2000;13:841–851. doi: 10.1016/s1074-7613(00)00081-9. [DOI] [PubMed] [Google Scholar]
- 60.Paquet ME, Cohen-Doyle M, Shore GC, Williams DB. Bap29/31 influences the intracellular traffic of MHC class I molecules. Journal of immunology. 2004;172:7548–7555. doi: 10.4049/jimmunol.172.12.7548. [DOI] [PubMed] [Google Scholar]
- 61.Abe F, Van Prooyen N, Ladasky JJ, Edidin M. Interaction of Bap31 and MHC class I molecules and their traffic out of the endoplasmic reticulum. Journal of immunology. 2009;182:4776–4783. doi: 10.4049/jimmunol.0800242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Appenzeller-Herzog C, Hauri HP. The ER-Golgi intermediate compartment (ERGIC): in search of its identity and function. Journal of cell science. 2006;119:2173–2183. doi: 10.1242/jcs.03019. [DOI] [PubMed] [Google Scholar]
- 63.Donaldson JG, Williams DB. Intracellular assembly and trafficking of MHC class I molecules. Traffic. 2009;10:1745–1752. doi: 10.1111/j.1600-0854.2009.00979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ghanem E, et al. The transporter associated with antigen processing (TAP) is active in a post-ER compartment. J Cell Sci. 2010;123:4271–4279. doi: 10.1242/jcs.060632. [DOI] [PubMed] [Google Scholar]
- 65.Wearsch PA, Cresswell P. The quality control of MHC class I peptide loading. Current opinion in cell biology. 2008;20:624–631. doi: 10.1016/j.ceb.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Raposo G, van Santen HM, Leijendekker R, Geuze HJ, Ploegh HL. Misfolded major histocompatibility complex class I molecules accumulate in an expanded ER-Golgi intermediate compartment. J Cell Biol. 1995;131:1403–1419. doi: 10.1083/jcb.131.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Van Hateren A, James E, Bailey A, Phillips A, Dalchau N, Elliott T. The cell biology of major histocompatibility complex class I assembly: towards a molecular understanding. Tissue Antigens. 2010;76:259–275. doi: 10.1111/j.1399-0039.2010.01550.x. [DOI] [PubMed] [Google Scholar]
- 68.Kuhns ST, Pease LR. A region of conformational variability outside the peptide-binding site of a class I MHC molecule. Journal of immunology. 1998;161:6745–6750. [PubMed] [Google Scholar]
- 69.Kirchhausen T, Owen D, Harrison SC. Molecular structure, function, and dynamics of clathrin-mediated membrane traffic. Cold Spring Harbor perspectives in biology. 2014;6:a016725. doi: 10.1101/cshperspect.a016725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sandvig K, Torgersen ML, Raa HA, van Deurs B. Clathrin-independent endocytosis: from nonexisting to an extreme degree of complexity. Histochemistry and cell biology. 2008;129:267–276. doi: 10.1007/s00418-007-0376-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mayor S, Parton RG, Donaldson JG. Clathrin-independent pathways of endocytosis. Cold Spring Harbor perspectives in biology. 2014;6 doi: 10.1101/cshperspect.a016758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jovic M, Sharma M, Rahajeng J, Caplan S. The early endosome: a busy sorting station for proteins at the crossroads. Histology and histopathology. 2010;25:99–112. doi: 10.14670/hh-25.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Neefjes JJ, Stollorz V, Peters PJ, Geuze HJ, Ploegh HL. The biosynthetic pathway of MHC class II but not class I molecules intersects the endocytic route. Cell. 1990;61:171–183. doi: 10.1016/0092-8674(90)90224-3. [DOI] [PubMed] [Google Scholar]
- 74.Naslavsky N, Weigert R, Donaldson JG. Characterization of a nonclathrin endocytic pathway: membrane cargo and lipid requirements. Molecular biology of the cell. 2004;15:3542–3552. doi: 10.1091/mbc.E04-02-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Radhakrishna H, Donaldson JG. ADP-ribosylation factor 6 regulates a novel plasma membrane recycling pathway. J Cell Biol. 1997;139:49–61. doi: 10.1083/jcb.139.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Naslavsky N, Weigert R, Donaldson JG. Convergence of non-clathrin- and clathrin-derived endosomes involves Arf6 inactivation and changes in phosphoinositides. Molecular biology of the cell. 2003;14:417–431. doi: 10.1091/mbc.02-04-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kleijmeer MJ, et al. Antigen loading of MHC class I molecules in the endocytic tract. Traffic. 2001;2:124–137. doi: 10.1034/j.1600-0854.2001.020207.x. [DOI] [PubMed] [Google Scholar]
- 78.Hanson PI, Cashikar A. Multivesicular body morphogenesis. Annual review of cell and developmental biology. 2012;28:337–362. doi: 10.1146/annurev-cellbio-092910-154152. [DOI] [PubMed] [Google Scholar]
- 79.Sugita M, Brenner MB. Association of the invariant chain with major histocompatibility complex class I molecules directs trafficking to endocytic compartments. The Journal of biological chemistry. 1995;270:1443–1448. doi: 10.1074/jbc.270.3.1443. [DOI] [PubMed] [Google Scholar]
- 80.Vigna JL, Smith KD, Lutz CT. Invariant chain association with MHC class I: preference for HLA class I/beta 2-microglobulin heterodimers, specificity, and influence of the MHC peptide-binding groove. Journal of immunology. 1996;157:4503–4510. [PubMed] [Google Scholar]
- 81.Basha G, et al. A CD74-dependent MHC class I endolysosomal cross-presentation pathway. Nature immunology. 2012;13:237–245. doi: 10.1038/ni.2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Porgador A, Yewdell JW, Deng Y, Bennink JR, Germain RN. Localization, quantitation, and in situ detection of specific peptide-MHC class I complexes using a monoclonal antibody. Immunity. 1997;6:715–726. doi: 10.1016/s1074-7613(00)80447-1. [DOI] [PubMed] [Google Scholar]
- 83.Choudhury A, Sharma DK, Marks DL, Pagano RE. Elevated endosomal cholesterol levels in Niemann-Pick cells inhibit rab4 and perturb membrane recycling. Molecular biology of the cell. 2004;15:4500–4511. doi: 10.1091/mbc.E04-05-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.van der Sluijs P, Hull M, Webster P, Male P, Goud B, Mellman I. The small GTP-binding protein rab4 controls an early sorting event on the endocytic pathway. Cell. 1992;70:729–740. doi: 10.1016/0092-8674(92)90307-x. [DOI] [PubMed] [Google Scholar]
- 85.Daro E, van der Sluijs P, Galli T, Mellman I. Rab4 and cellubrevin define different early endosome populations on the pathway of transferrin receptor recycling. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:9559–9564. doi: 10.1073/pnas.93.18.9559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Allaire PD, Marat AL, Dall’Armi C, Di Paolo G, McPherson PS, Ritter B. The Connecdenn DENN domain: a GEF for Rab35 mediating cargo-specific exit from early endosomes. Molecular cell. 2010;37:370–382. doi: 10.1016/j.molcel.2009.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nature reviews Molecular cell biology. 2009;10:597–608. doi: 10.1038/nrm2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kouranti I, Sachse M, Arouche N, Goud B, Echard A. Rab35 regulates an endocytic recycling pathway essential for the terminal steps of cytokinesis. Current biology: CB. 2006;16:1719–1725. doi: 10.1016/j.cub.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 89.Maxfield FR, McGraw TE. Endocytic recycling. Nature reviews Molecular cell biology. 2004;5:121–132. doi: 10.1038/nrm1315. [DOI] [PubMed] [Google Scholar]
- 90.Gruenberg J. The endocytic pathway: a mosaic of domains. Nature reviews Molecular cell biology. 2001;2:721–730. doi: 10.1038/35096054. [DOI] [PubMed] [Google Scholar]
- 91.Hopkins CR. Intracellular routing of transferrin and transferrin receptors in epidermoid carcinoma A431 cells. Cell. 1983;35:321–330. doi: 10.1016/0092-8674(83)90235-0. [DOI] [PubMed] [Google Scholar]
- 92.Yamashiro DJ, Tycko B, Fluss SR, Maxfield FR. Segregation of transferrin to a mildly acidic (pH 6. 5) para-Golgi compartment in the recycling pathway. Cell. 1984;37:789–800. doi: 10.1016/0092-8674(84)90414-8. [DOI] [PubMed] [Google Scholar]
- 93.Ren M, Xu G, Zeng J, De Lemos-Chiarandini C, Adesnik M, Sabatini DD. Hydrolysis of GTP on rab11 is required for the direct delivery of transferrin from the pericentriolar recycling compartment to the cell surface but not from sorting endosomes. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:6187–6192. doi: 10.1073/pnas.95.11.6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ullrich O, Reinsch S, Urbe S, Zerial M, Parton RG. Rab11 regulates recycling through the pericentriolar recycling endosome. J Cell Biol. 1996;135:913–924. doi: 10.1083/jcb.135.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Xie S, Bahl K, Reinecke JB, Hammond GR, Naslavsky N, Caplan S. The endocytic recycling compartment maintains cargo segregation acquired upon exit from the sorting endosome. Molecular biology of the cell. 2016;27:108–126. doi: 10.1091/mbc.E15-07-0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wilcke M, Johannes L, Galli T, Mayau V, Goud B, Salamero J. Rab11 regulates the compartmentalization of early endosomes required for efficient transport from early endosomes to the trans-golgi network. J Cell Biol. 2000;151:1207–1220. doi: 10.1083/jcb.151.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Powelka AM, et al. Stimulation-dependent recycling of integrin beta1 regulated by ARF6 and Rab11. Traffic. 2004;5:20–36. doi: 10.1111/j.1600-0854.2004.00150.x. [DOI] [PubMed] [Google Scholar]
- 98.Weigert R, Yeung AC, Li J, Donaldson JG. Rab22a regulates the recycling of membrane proteins internalized independently of clathrin. Molecular biology of the cell. 2004;15:3758–3770. doi: 10.1091/mbc.E04-04-0342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Naslavsky N, Caplan S. EHD proteins: key conductors of endocytic transport. Trends in cell biology. 2011;21:122–131. doi: 10.1016/j.tcb.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Xie S, Naslavsky N, Caplan S. Diacylglycerol kinase alpha regulates tubular recycling endosome biogenesis and major histocompatibility complex class I recycling. The Journal of biological chemistry. 2014;289:31914–31926. doi: 10.1074/jbc.M114.594291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Giridharan SS, Cai B, Vitale N, Naslavsky N, Caplan S. Cooperation of MICAL-L1, syndapin2, and phosphatidic acid in tubular recycling endosome biogenesis. Molecular biology of the cell. 2013;24:1776–1790. S1771–1715. doi: 10.1091/mbc.E13-01-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sharma M, Giridharan SS, Rahajeng J, Naslavsky N, Caplan S. MICAL-L1 links EHD1 to tubular recycling endosomes and regulates receptor recycling. Molecular biology of the cell. 2009;20:5181–5194. doi: 10.1091/mbc.E09-06-0535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cai B, Katafiasz D, Horejsi V, Naslavsky N. Pre-sorting endosomal transport of the GPI-anchored protein, CD59, is regulated by EHD1. Traffic. 2011;12:102–120. doi: 10.1111/j.1600-0854.2010.01135.x. [DOI] [PubMed] [Google Scholar]
- 104.Caplan S, et al. A tubular EHD1-containing compartment involved in the recycling of major histocompatibility complex class I molecules to the plasma membrane. The EMBO journal. 2002;21:2557–2567. doi: 10.1093/emboj/21.11.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Maldonado-Baez L, Williamson C, Donaldson JG. Clathrin-independent endocytosis: a cargo-centric view. Experimental cell research. 2013;319:2759–2769. doi: 10.1016/j.yexcr.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Grant BD, Caplan S. Mechanisms of EHD/RME-1 protein function in endocytic transport. Traffic. 2008;9:2043–2052. doi: 10.1111/j.1600-0854.2008.00834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lin SX, Grant B, Hirsh D, Maxfield FR. Rme-1 regulates the distribution and function of the endocytic recycling compartment in mammalian cells. Nature cell biology. 2001;3:567–572. doi: 10.1038/35078543. [DOI] [PubMed] [Google Scholar]
- 108.Compeer EB, Flinsenberg TW, Boon L, Hoekstra ME, Boes M. Tubulation of endosomal structures in human dendritic cells by Toll-like receptor ligation and lymphocyte contact accompanies antigen cross-presentation. The Journal of biological chemistry. 2014;289:520–528. doi: 10.1074/jbc.M113.511147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Compeer EB, Boes M. MICAL-L1-related and unrelated mechanisms underlying elongated tubular endosomal network (ETEN) in human dendritic cells. Communicative & integrative biology. 2014;7:e994969. doi: 10.4161/19420889.2014.994969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zou L, et al. The GTPase Rab3b/3c-positive recycling vesicles are involved in cross-presentation in dendritic cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:15801–15806. doi: 10.1073/pnas.0905684106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Barral DC, et al. CD1a and MHC class I follow a similar endocytic recycling pathway. Traffic. 2008;9:1446–1457. doi: 10.1111/j.1600-0854.2008.00781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Salamero J, et al. CD1a molecules traffic through the early recycling endosomal pathway in human Langerhans cells. The Journal of investigative dermatology. 2001;116:401–408. doi: 10.1046/j.1523-1747.2001.01264.x. [DOI] [PubMed] [Google Scholar]
- 113.Stow JL, Manderson AP, Murray RZ. SNAREing immunity: the role of SNAREs in the immune system. Nat Rev Immunol. 2006;6:919–929. doi: 10.1038/nri1980. [DOI] [PubMed] [Google Scholar]
- 114.Aderem A. How to eat something bigger than your head. Cell. 2002;110:5–8. doi: 10.1016/s0092-8674(02)00819-x. [DOI] [PubMed] [Google Scholar]
- 115.Cox D, Lee DJ, Dale BM, Calafat J, Greenberg S. A Rab11-containing rapidly recycling compartment in macrophages that promotes phagocytosis. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:680–685. doi: 10.1073/pnas.97.2.680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gagnon E, et al. Endoplasmic reticulum-mediated phagocytosis is a mechanism of entry into macrophages. Cell. 2002;110:119–131. doi: 10.1016/s0092-8674(02)00797-3. [DOI] [PubMed] [Google Scholar]
- 117.Touret N, et al. Quantitative and dynamic assessment of the contribution of the ER to phagosome formation. Cell. 2005;123:157–170. doi: 10.1016/j.cell.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 118.Husebye H, et al. The Rab11a GTPase controls Toll-like receptor 4-induced activation of interferon regulatory factor-3 on phagosomes. Immunity. 2010;33:583–596. doi: 10.1016/j.immuni.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annual review of immunology. 2015;33:257–290. doi: 10.1146/annurev-immunol-032414-112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nilsen NJ, et al. Cellular trafficking of lipoteichoic acid and Toll-like receptor 2 in relation to signaling: role of CD14 and CD36. Journal of leukocyte biology. 2008;84:280–291. doi: 10.1189/jlb.0907656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Le Bon A, et al. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nature immunology. 2003;4:1009–1015. doi: 10.1038/ni978. [DOI] [PubMed] [Google Scholar]
- 122.Casbon AJ, Allen LA, Dunn KW, Dinauer MC. Macrophage NADPH oxidase flavocytochrome B localizes to the plasma membrane and Rab11-positive recycling endosomes. Journal of immunology. 2009;182:2325–2339. doi: 10.4049/jimmunol.0803476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wallach TM, Segal AW. Stoichiometry of the subunits of flavocytochrome b558 of the NADPH oxidase of phagocytes. The Biochemical journal. 1996;320(Pt 1):33–38. doi: 10.1042/bj3200033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Savina A, et al. The small GTPase Rac2 controls phagosomal alkalinization and antigen crosspresentation selectively in CD8(+) dendritic cells. Immunity. 2009;30:544–555. doi: 10.1016/j.immuni.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 125.Lutz MB, Inaba K, Schuler G, Romani N. Still Alive and Kicking: In-Vitro-Generated GM-CSF Dendritic Cells! Immunity. 2016;44:1–2. doi: 10.1016/j.immuni.2015.12.013. [DOI] [PubMed] [Google Scholar]
- 126.Helft J, et al. Alive but Confused: Heterogeneity of CD11c(+) MHC Class II(+) Cells in GM-CSF Mouse Bone Marrow Cultures. Immunity. 2016;44:3–4. doi: 10.1016/j.immuni.2015.12.014. [DOI] [PubMed] [Google Scholar]
- 127.Guilliams M, Malissen B. A Matter of Perspective: Moving from a Pre-omic to a Systems-Biology Vantage of Monocyte-Derived Cell Function and Nomenclature. Immunity. 2016;44:5–6. doi: 10.1016/j.immuni.2015.12.020. [DOI] [PubMed] [Google Scholar]
- 128.Shortman K, Naik SH. Steady-state and inflammatory dendritic-cell development. Nature reviews Immunology. 2007;7:19–30. doi: 10.1038/nri1996. [DOI] [PubMed] [Google Scholar]
- 129.Cheong C, et al. Microbial stimulation fully differentiates monocytes to DC-SIGN/CD209(+) dendritic cells for immune T cell areas. Cell. 2010;143:416–429. doi: 10.1016/j.cell.2010.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Di Pucchio T, et al. Direct proteasome-independent cross-presentation of viral antigen by plasmacytoid dendritic cells on major histocompatibility complex class I. Nature immunology. 2008;9:551–557. doi: 10.1038/ni.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Di Pucchio T, et al. Direct proteasome-independent cross-presentation of viral antigen by plasmacytoid dendritic cells on major histocompatibility complex class I. Nat Immunol. 2008;9:551–557. doi: 10.1038/ni.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wickner W, Schekman R. Membrane fusion. Nat Struct Mol Biol. 2008;15:658–664. doi: 10.1038/nsmb.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Snyder DA, Kelly ML, Woodbury DJ. SNARE complex regulation by phosphorylation. Cell Biochem Biophys. 2006;45:111–123. doi: 10.1385/CBB:45:1:111. [DOI] [PubMed] [Google Scholar]
- 134.Suzuki K, Verma IM. Phosphorylation of SNAP-23 by IkappaB kinase 2 regulates mast cell degranulation. Cell. 2008;134:485–495. doi: 10.1016/j.cell.2008.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Karim ZA, et al. IkappaB kinase phosphorylation of SNAP-23 controls platelet secretion. Blood. 2013;121:4567–4574. doi: 10.1182/blood-2012-11-470468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hinz M, Scheidereit C. The IkappaB kinase complex in NF-kappaB regulation and beyond. EMBO reports. 2014;15:46–61. doi: 10.1002/embr.201337983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Fairn GD, Grinstein S. How nascent phagosomes mature to become phagolysosomes. Trends in immunology. 2012;33:397–405. doi: 10.1016/j.it.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 138.Guermonprez P, Saveanu L, Kleijmeer M, Davoust J, Van Endert P, Amigorena S. ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature. 2003;425:397–402. doi: 10.1038/nature01911. [DOI] [PubMed] [Google Scholar]
- 139.Houde M, et al. Phagosomes are competent organelles for antigen cross-presentation. Nature. 2003;425:402–406. doi: 10.1038/nature01912. [DOI] [PubMed] [Google Scholar]
- 140.Ma W, et al. Long-Peptide Cross-Presentation by Human Dendritic Cells Occurs in Vacuoles by Peptide Exchange on Nascent MHC Class I Molecules. Journal of immunology. 2016;196:1711–1720. doi: 10.4049/jimmunol.1501574. [DOI] [PubMed] [Google Scholar]