

Abstract
Importance
Wiskott-Aldrich syndrome (WAS) is a rare primary immunodeficiency associated with severe microthrombocytopenia. Partially HLA-matched allogeneic hematopoietic stem cell (HSC) transplantation is associated with significant co-morbidity.
Objective
To assess the outcome and safety of autologous HSC gene therapy in WAS.
Design
Gene-corrected autologous HSC were infused in 7 consecutive WAS patients (age range: 0.8 to 15.5 years, mean 7 years) following myeloablative conditioning.
Setting
and participants: Patients with severe WAS lacking HLA-matched related or unrelated HSC donors were treated between December 2010 and January 2014. The follow up of patients in this intermediate analysis ranged from 9 to 42 months.
Intervention
A single infusion of gene-modified CD34+ cells with an advanced lentiviral vector.
Main Outcome(s) and Measure(s)
Primary outcomes were improvement at 24 months in eczema, the frequency and severity of infections, bleeding tendency, autoimmunity and reduction in disease-related days of hospitalization. Secondary outcomes were improvement in immunological and haematological parameters, and evidence for safety through vector integration analysis.
Results
Six out of the 7 patients were alive at the time of last follow-up (mean and median follow-up time: 28 and 27 months respectively) and showed sustained clinical benefit. One patient died 7 months after treatment from pre-existing drug- resistant herpes virus infections. Eczema and susceptibility to infections resolved in all 6 patients. Autoimmunity improved in 5/5 patients. No severe bleeding episodes were recorded after treatment, and at last follow up 6/6 patients were free from blood product support and thrombopoietic agonists. Hospitalization days were reduced from 25 days (median) in the 2 years pretreatment to 0 days (median) in the 2 years post treatment. All 6 surviving patients exhibited high-level, stable engraftment of functionally corrected lymphoid cells. The degree of myeloid cell engraftment and of platelet reconstitution correlated with the dose of gene-corrected cells administered. No evidence of vector-related toxicity was observed clinically or by molecular analysis.
Conclusions and Relevance
This study demonstrated the feasibility of the use of gene therapy in patients with Wiskott-Aldrich syndrome. Controlled trials with larger numbers of patients are necessary to assess long-term outcome and safety.
Introduction
Wiskott-Aldrich Syndrome (WAS, OMIM#301000) is a complex, X-linked primary immunodeficiency caused by loss-of-function mutations in the WAS gene. The condition affects the immune and haematopoietic system and has a broad spectrum of severity1. The WAS protein (WASp) is a key regulator of the actin cytoskeleton in all hematopoietic lineages2. WASp deficiency causes characteristic microthrombocytopenia and lymphoid and myeloid cell dysfunction, the severity of which is usually correlated with WASp expression levels. A clinical scoring system is used to stratify disease severity3. Patients with a score from 3 to 5 display a WAS phenotype characterized by a tendency to bleed, persistent eczema, susceptibility to severe opportunistic bacterial and viral infections, autoimmune and inflammatory complications, and an elevated risk of lymphoid malignancies3–5. In the absence of definitive treatment, patients with classical WAS do not survive beyond their second or third decade of life. Although hematopoietic stem cell (HSC) transplantation is usually curative, the use of human leukocyte antigen (HLA) partially-matched HSCs is associated with a high incidence of complications6–10. Gene therapy based on the transplantation of autologous, gene-corrected HSCs may be an effective and potentially safer alternative.
The first gene therapy trial for WAS used a Moloney-leukemia-virus-derived gamma-retroviral vector (MLV). Although this therapy provided significant clinical benefit as characterized by partial or complete resolution of immunodeficiency, autoimmunity, and bleeding diathesis, it was associated with an unacceptably high risk of insertional mutagenesis with activation of several proto-oncogenes leading to leukemia in 7 of the 9 evaluable patients11.
We developed and tested a self-inactivating lentiviral vector for WAS gene correction (referred to below as LV-w1.6 WASp) in which a 1.6 kb fragment of the proximal promoter of the WAS gene is used to express the full-length coding sequence of the human WAS gene in cells of the hematopoietic lineage12–14. In a recently published study, 3 young children with a moderate form of WAS were treated with this vector. They showed stable engraftment of WASp-expressing cells, and improvements in terms of immune function, platelet count and clinical score15. Here, we report the first results of a two-center study designed to assess the feasibility of HSC gene therapy in severe WAS patients.
Methods
Clinical protocol
Seven consecutive patients with confirmed WAS were enrolled at Great Ormond Street Hospital (London, UK) (EUDRACT number: 2007-004308-11) and Necker Children’s Hospital (Paris, France) (EUDRACT number: 2009-011152-22) in an open label study between December 2010 and January 2014. The final dates of follow-up were between May 28th and November 12th 2014.
The study protocol was approved by the UK and French drug regulatory agencies and the appropriate investigational review boards such as the Gene Therapy Advisory Committee (GTAC) in the UK and the Ethical Committee for the Protection of the Persons Submitted to a Clinical Trial (CPP idF2). Informed consent or assent was obtained after the benefits and risks of the trial were explained to the patients or their parents/legal guardians. Hematopoietic stem cells were collected from the patient bone marrow (BM) or mobilized peripheral blood (MPB) and were genetically-modified ex vivo during myeloablative conditioning of patients. All patients were placed in sterile confinement and received a low-intensity conditioning regimen with busulfan (4 mg/kg/day) and fludarabine (40 mg/m2/day) for three days. At the end of the conditioning procedure, transduced cells were infused to the patient without cryopreservation. Anti-CD20 antibody and/or alemtuzumab were added if autoimmune disease was present. Autoimmunity was defined clinically as cutaneous or large vessel vasculitis, arthritis, and cytopenia of one or more hematopoietic lineages in association with the presence of auto-antibodies. All the transduced cell products met the specifications required for product release and infusion. The synopsis of the study is provided in supplemental material.
Outcomes
Primary study objectives were to assess the outcomes following gene therapy judged by improvement in clinical manifestations including frequency and severity of infections, of bleeding episodes, of autoimmune manifestations, and eczema. Secondary objectives were based on biological tests including platelet counts, lymphocyte subset analysis, and lymphocyte function (mitogen and antigen-induced proliferation, serum Ig levels). Secondary objectives of safety were evaluated clinically (including manifestations of clonal proliferation or leukaemia) and by assessment of the frequency of vector integration sites close to relevant proto-oncogenes and their abundance within the engrafted and transduced cell population.
The severity of disease in patients was scored according to the following criteria3,5 : A score of 1 accounts for microthrombocytopenia which is universal to all patients; a score of 2 includes mild eczema, immunodeficiency and occasional mild infections; a score of 3 refers to more severe immunodeficiency associated with recurrent and more protracted infections; a score of 4 is given if eczema or infections are persistent and do not respond easily to conventional treatments; and a score of 5 is assigned to very severe clinical forms that additionally develop autoimmune or malignant complications.
Vector production
The LV-w1.6 WASp vector has been described elsewhere14,16. Clinical vector batches were manufactured at Genethon (Evry, France) according to Good Manufacturing Practice and were purified, concentrated and titered for infectious particles (infectious genomes (i.g.)/mL)16. The batches used in the study are described in Supplementary Table S1. Batch 1 was used in London, and batches 2 and 3 were used in Paris.
Biological analyses
Lymphocyte phenotypes, functions and T-cell receptor repertoires (beta, alpha, gamma and delta chains) were analyzed as described elsewhere17–20. Signal joint T-cell receptor excision circles (TREC) were determined using a real-time qPCR21. TREC content was expressed in copies per 1×105 peripheral blood mononuclear cells (PBMCs) (control range value between 150 to 2500 copies/105 PBMCs). The vector copy number (VCN) per cell was measured by qPCR detection of the vector’s HIV Psi sequence with normalization against the copy number of the albumin gene, as described elsewhere22 (for more details, see supplementary methods). Lymphoid, myeloid, naive and memory T cell subpopulations were sorted by flow cytometry, using the corresponding fluorescence-labeled monoclonal antibodies. Natural killer (NK) cells cytotoxicity was evaluated against K562 target cells as described in supplementary methods.
CD34+ cell gene transfer procedure
Patients’ bone marrow cells harvested under general anesthesia, were separated with lymphoprep (Eurobio, Oslo, Norway) and centrifuged in order to collect mononuclear cells (MNC). Patient’s MPB was collected by apheresis. The positive selection of CD34+ cells from MNC or from MPB was performed using immunomagnetic beads and an immunomagnetic enrichment device (CliniMACs, Miltenyi Biotec, Bergish – Gladbach, Germany). Purified CD34+ cells were seeded on cell culture bags pre-coated with clinical grade Retronectin™ (Takara Bio Co) in serum free medium (X-Vivo 20, Biowhittacker/Lonza) and clinical grade stem cell factor (SCF) (300 ng/ml), FLT3-L (300 ng/ml), thrombopoietin (TPO) (100 ng/ml) and IL-3 (20 ng/ml) (all from Peprotech). After 24 hours of prestimulation, cells were transduced twice with LV-w1.6.WASp (1×108ig/ml) each time for 18 hours. At the end of the transduction procedure, washed cells were resuspended in 4% human serum albumin and transferred in a sterile bag for infusion to the patient. Aliquots of cells were further cultured for 14 days to measure stable proviral integration by qPCR and WASp expression by flow cytometry.
Integration site analysis and clonality assays
Ligation-mediated PCR was used to sequence vector integration sites in different cell subpopulations (the cell selection procedure is provided in supplementary methods)23–25. At least 3 independent replicates were analyzed for all samples. Deep sequencing was carried out using both the 454/Roche and Illumina techniques. Data sets analyzed are summarized in the Supplementary Report. Assays of a DNA preparation were judged to be successful if they detected at least 80 different break sites in the human genome associated with unique adaptor ligation positions. All integration site sequence data were deposited at the NCBI SRA accession: SRP050221.
Statistical analysis was carried out using R version 3.1.2. An extensive discussion of statistical methods and results is presented in the Supplementary Report. Briefly, The distributions of integration sites (relative to genomic features) were summarized using the receiver operating characteristic curve method (Supplementary Figure S1 A and B)26. The abundance of cell clones was quantified using the SonicLength method25. Clumping of integration site sequences was analyzed using scan statistics27
Results
Clinical presentation
At enrollment, the age of the patients ranged from 0.8 to 15.5 years (median: 7). All but one of the patients (P6) had a disease score of 5 (Table 1). All patients experienced severe thrombocytopenia, which led to severe bleeding episodes in P2, P3, P4, P5 and P7 (intracerebral hemorrhage in P3, gastro-intestinal hemorrhage in P5). Furthermore, all patients had eczema and associated recurrent skin infections. Three of the 7 patients experienced recurrent, severe infections requiring hospitalization. Six patients had autoimmune disease, with P2 and P3 being most profoundly affected. In P2, a combination of severe lower limb vasculitis and arthritis became refractory to conventional treatment and prevented ambulation. Patient 3 had severe autoimmune cytopenias that led to splenectomy at the age of 3 years. He also experienced a lymphoproliferative disorder with generalized lymphoadenopathy, liver enlargement, and renal infiltration. His primary immunodeficiency was responsible for severe viral infections due to cytomegalovirus (CMV), herpes-simplex viruses (HSV1) and varicella-zoster-virus (VZV) which led to several hospitalizations from the age of 7 up to the time of gene therapy treatment including perioral herpes infections with facial cellulitis, severe respiratory tract infection to Klebsiella pneumoniae aggravated by a cerebral hematoma causing coma and requiring intensive care unit hospitalization. He developed sequelaes following his stroke requiring rehabilitation therapy. The recurrent herpes virus infections were treated with several cycles of acyclovir, ganciclovir and foscarnet requiring hospitalisation (Table 1). The HSV1 genotype was found resistant to acyclovir in the months that preceded gene therapy.
Table 1.
Characteristics of patients and the infused patient-autologous gene therapy products(a)
| P1 | P2 | P3 | P4 | P5 | P6 | P7 | |
|---|---|---|---|---|---|---|---|
|
| |||||||
| Age at GT (years or months) | 10 years | 15.5 years | 10 years | 10 months | 3 years | 7 years | 3.5 years |
|
| |||||||
| WAS Gene mutation | 401C/T p.A134V |
c.1453+1 G>C |
c.628delT p.S210HfsX51 |
c.1295delG p.G432EfsX13 |
c.257G>A p.R86H |
100C>T p.R34X |
1271dup(ex10) Leu425Profsx70 |
|
| |||||||
| Revertant cells | − | + | + | − | − | + | − |
|
| |||||||
| Mutant WASp expression | + | − | − | − | − | − | + |
|
| |||||||
| Clinical manifestations, in the 2 years preceding GT(b) | |||||||
| Bleeding(c) | Bruising | Severe Splenectomized | Severe Splenectomized Multiple platelet transfusions | Severe Multiple platelet transfusions | Severe Multiple platelet transfusions; N-plate | Bruising (moderate) | Severe Multiple Platelet transfusions; N-plate |
| Eczema | Severe | Severe | Mild | Mild | Severe | Severe | Severe |
| Infections | Severe | 2 episodes of sepsis, severe VZV | Severe chronic VZV, HSV, CMV, EBV infections | Mild | Mild | Severe | Gastroenteritis (2 episodes) |
| Autoimmunity | Recurrent arthritis Renal Disease | Severe lower limbs vasculitis and arthritis, unable to walk | Pancytopenia | Severe Thrombocytopenia Mild skin vasculitis | Severe Thrombocytopenia | None | Severe Thrombocytopenia Mild cutaneous vasculitis |
| Days in hospital for disease-related complications | <5 | 47days | >6 months | 26 days(b) (in 10 months) | 19 days | <5 | 21days |
|
| |||||||
| Clinical score(d) | 5 | 5 | 5 | 5 | 5 | 3 | 5 |
|
| |||||||
| Date of GT | March/03/2011 | May/27/2011 | Nov/18/2011 | April/20/2012 | June/15/2012 | Nov/16/2012 | January/24/2014 |
|
| |||||||
| HSC origin | BM | MPB | MPB | BM | BM | BM | MPB |
|
| |||||||
| Total CD34+ cell dose ×106/kg | 2 | 11 | 11 | 7.3 | 6.8 | 3.1 | 15 |
|
| |||||||
| VCN | 0.7 | 1.3 | 1.2 | 2.8 | 0.6 | 1.7 | 0.6 |
|
| |||||||
| Date of last Fup | Nov/12/2014 | May/28/2014 | Death July/11/2012 | Oct/04/2014 | June/11/2014 | Nov/12/2014 | Nov/03/2014 |
|
| |||||||
| Fup (months)(d) | 42 | 36 | 7 | 30 | 24 | 24 | 9 |
|
| |||||||
| Clinical status 2 years post GT or at last Fup | A&W | A&W | Died of opportunistic viral infections | A&W | A&W | A&W | A&W |
| Bleeding | Bruising (resolved after splenectomy) | No | NA | Bruising | Bruising | Bruising (mild) | Bruising |
| Eczema | No | Very mild | NA | No | No | No | No |
| Infections | No | Localized zoster (1 episode) | NA | No | Mild gastroenteritis (1 episode) | No | No |
| Autoimmunity | No | Mild vasculitis, able to walk | NA | No | No | No | No |
| Days in hospital (post initial GT period), over following 2 years | 0 | 5 | Continuous hospitalization until death | 0 | 5 | 0 | 0(d) |
| Clinical score at last Fup | <1 | 5 (mild autoimmunity) | NA | <1 | <1 | <1 | 1 |
P1 and P6 were included in London, and P2, P3, P4, P5 and P7 were included in Paris; P: patient; GT: gene therapy; HSC: hematopoietic stem cells; BM: bone marrow; MPB: mobilized peripheral blood; VCN: vector copy number per cell in cultured transduced CD34+ cells; Revertant cells are detected in blood and have recovered some expression of WASp as a result of selected spontaneous somatic mutations; Fup: Follow-up; A&W: alive and well; NA: not applicable; Severe eczema is defined by intense redness, thickness/swelling, and may include diffuse or superinfected lesions; Severe infection indicates infections requiring hospitalization and parenteral treatment ; Severe thrombocytopenia is defined as platelet counts below 10,000/mm3.
P4 had a shorter period of observation since he was treated at the age of 10 months
Thrombocytopenia was initially below 20,000/mm3 in P1, P2, P3 and P6 and below 10,000/mm3 in P4, P5 and P7. P2 and P3 were splenectomized prior to GT. P1 was successfully splenectomized after GT
P3, P4, P5 had more than 20 platelet transfusions, P7 more than 10, P5 and P7 received additional N-plate therapy.
P7 has a shorter follow up of 9 months.
“+” Revertant cells and mutant protein are present and detectable in blood cells
“−” Revertant cells and mutant protein are absent and not detectable in blood cells
Gene transfer
Autologous CD34+ HSCs (from bone marrow in 4 cases and from MPB in 3 cases; Table 1) were transduced ex vivo with the LV-w1.6 WASp lentiviral vector, and were immediately re-infused into conditioned patients. The median dose of CD34+ cells per kg of body weight infused was 7.3×106 (range: 2×106 to 15×106) and the mean vector copy number (VCN) in CD34+ cells was 1.27 ± 0.8 (range: 0.6 to 2.8) (Table 1).
Outcome after gene therapy
Six of the 7 patients treated with gene therapy were evaluable over a period of at least 9 months (mean and median follow-up time: 28 and 27 months respectively) for primary outcomes. Patient 3 died as a consequence of opportunistic herpes viral infections that became drug-resistant post gene therapy including severe peri-oral necrotizing ulcerative lesions caused by HSV-1 and a bilateral CMV retinitis with high blood viral load (ie 4.6 log copies/ml) (Supplementary Table S2). This was associated with an inflammatory pulmonary syndrome characterized by multiple foci of bronchoalveolar parenchymal condensation responsible of oxygen dependence developed in association with diffuse aspergillus- related lesions. Clinical outcome rapidly worsened. A lung biopsy showed an extensive fibrosis. The patient eventually died of septic shock in the intensive care unit seven months post gene therapy. In December 2014, the other 6 treated patients were alive and well and displayed significant clinical improvements (Table 1). In terms of the primary outcomes, eczema and susceptibility to infections have resolved in all cases. Minor, non-recurrent only infections were observed in 2 patients (Table 1). Patient1 has not had any further episodes of arthritis. Patient 2 displayed major improvement in peripheral vasculitis and was able to return to normal physical activity without need for a wheelchair. Patient 7 recovered completely from vasculitis skin lesions. With the exception of occasional bruising, there were no post-treatment recurrences of the severe, recurrent bleeding episodes that had previously affected P2, P4, P5 and P7. From month 7 onwards, none of the 6 patients required regular blood product support or treatment with recombinant stimulators of platelet production (P4 received N-plate until month 13). Days of additional disease-related hospitalization once the initial gene therapy was completed was reduced to 0–5 days over the next 2 years for patients 1,2,4,5 and 6 and over a 9 month period for patient 7. P1 had a 7 day hospitalization for elective splenectomy in year 3 after engraftment. After gene therapy, patients remained on their pre-treatment regimen of immunoglobulin substitution and prophylactic antibiotics, although immunoglobulins have recently been discontinued in P4 and P6 pending evaluation of functional responses to a typical childhood vaccination schedule.
Gene marking and WASp expression in leukocytes
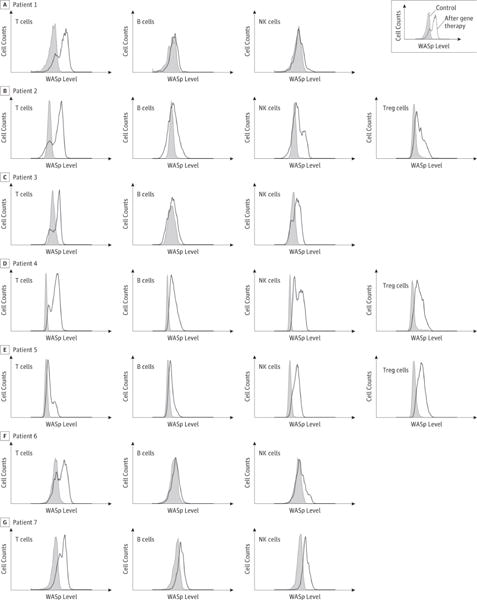
The presence of vector-positive cells in blood was readily detectable one month after treatment. Q-PCR revealed an increase over time in gene marking in peripheral blood mononuclear cells (Figure 1). Gene marking in PBMCs was primarily due to transduced lymphocytes, most prominently T cells, but multiple cell lineages were also transduced. At the last follow-up, the VCN ranged from 0.35 to 1.20 in sorted CD3+ T cells, from 0.1 to 1.34 in sorted CD19+ B cells and from 0.04 to 0.7 in sorted natural killer CD56+ cells. The extent of gene marking in CD14+ or CD15+ myeloid cells was more variable, with 0.4 copies per cell in P4, 0.2 copies in P6, 0.1 in P2 and P7, and between 0.01 and 0.03 in P1 and P5 (Figure 1). The intracellular expression of WASp correlated with the levels of transduction in the different cell subsets. At the last follow-up, the proportion of cells expressing WASp was highest in T cells (34% to 84%) and somewhat lower in NK cells (14% to 85%) and B cells (13% to 55%) (Figure 2A–B and Table 2).
Figure 1. Longitudinal evaluation of gene marking in blood cells after gene therapy.
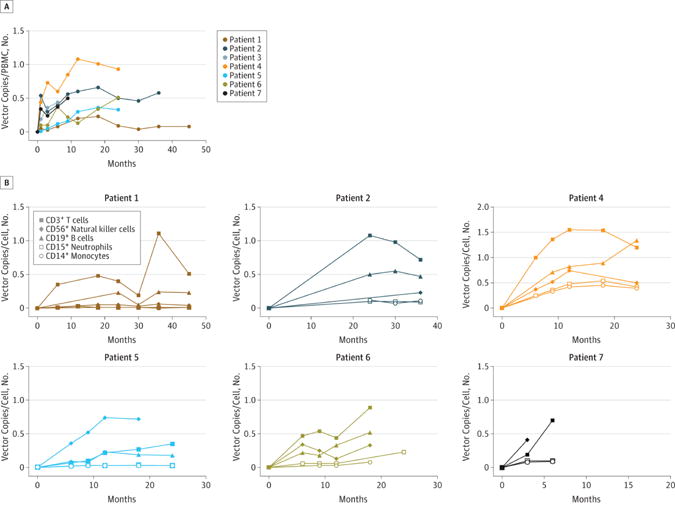
(A). Gene marking in peripheral blood cells over time after gene therapy in patients P1 to P7, as expressed by vector copy numbers per cell (VCN) in PBMC and measured by q-PCR. (B). Gene marking in various blood cell subsets in each patient, expressed as VCN in: CD3+ T cells (filled squares); CD56+ NK cells (filled diamonds); CD19+ B cells (filled triangles); CD15+ neutrophils (open squares) and CD14+ monocytes (open circles).
Figure 2. Lymphoid engraftment and expression of the WASp transgene measured by flow cytometry.
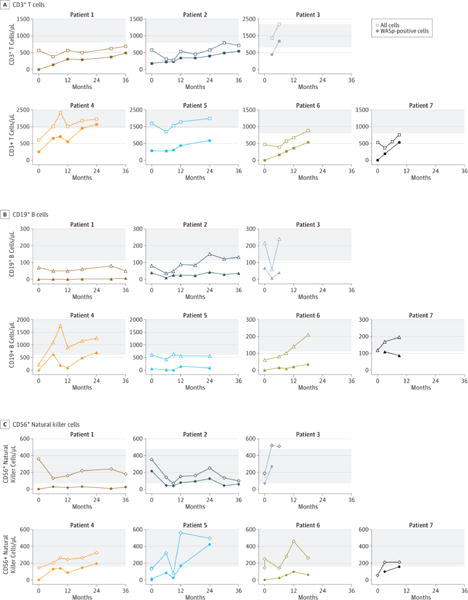
(A) Time course of lymphoid cell subsets recovery in blood after gene therapy: The total (open symbols) or WASp+ (filled symbols) levels of CD3+ T cells (squares), CD19+ B cells (triangles) and CD56+ NK cells (diamonds) were measured in blood over time. Tinted areas indicate values in aged-matched individuals. (B) WASp expression in lymphoid cell subpopulations at different times after gene therapy: Expression of WASp using specific antibodies is indicated by a thick line in CD3+ T lymphocytes, CD19+ B lymphocytes, CD56+ NK cells and CD4+ CD25+ FoxP3+ regulatory T cells (Tregs). Control antibody staining is shown by a gray overlay. WASp marking in T, B and NK cells of P1 to P7 performed respectively at months 30, 30, 6, 24, 24, 12 were shown. WASp marking in Tregs of P2, P4, P5 were performed respectively at months 30, 18, 18.
Table 2.
Most recent immunological analysis of patients
| Patient | P1 | P2 | P3† | P4 | P5 | P6 | P7 | ||
|---|---|---|---|---|---|---|---|---|---|
|
|
|||||||||
| Age at last Fup years) | 14 | 18 | 11 | 3 | 5 | 9 | 4.3 | ||
| Time of analysis (months following gene therapy) | 42 | 36 | 6 | 30 | 24 | 24 | 9 | Control range < 7 years | ≥ 7 years |
|
| |||||||||
| CD3/μl | 840 | 715 | 2106 | 1921 | 1984 | 970 | 756 | 1400–3700 | 800–2200 |
| WASp+ T cells (%) | 71§ | 76 | 54 | 84§ | 34 | 61§ | 70 | 100 | 100 |
| CD4/μl | 560 | 440 | 972 | 1256 | 1271 | 730 | 434 | 700–2200 | 530–1300 |
| CD8/μl | 180 | 187 | 1053 | 471 | 620 | 180 | 196 | 490–1300 | 330–920 |
| Naive CD4+ T cells (%) | 37 | 23 | 1 | 27§ | 32§ | 54 | 22 | 60–72 | 43–55 |
| Naïve CD8+ T cells (%) | 33 | 17 | 0.3 | 38§ | 15§ | 70 | 22 | 52–68 | 52–68 |
| TREC/105 PBMC | 847§ | 612§ | ND | 1118§ | 1444§ | 130§ | ND | >150 | >150 |
| PHA-induced proliferation | + | + | + | + | + | + | + | + | + |
| Anti-CD3 induced proliferation | +/− | + | ND | + | + | + | + | + | + |
| Ag-induced proliferation* | ND | 2* | 0 | 1*§ | 1* | ND | 1* | NA | NA |
| CD56/μl | 190 | 99 | 297 | 324§ | 496 | 240 | 210 | 160–950 | 70–480 |
| WASp+ NK cells (%) | 14 § | 60 | ND | 60§ | 85 | 23 | 74 | 100 | 100 |
| CD19/μl | 70 | 132 | 270 | 1260§ | 558 | 170 | 196 | 610–2600 | 110–570 |
| WASp+ B cells (%) | 13 § | 27 | 17 | 55§ | 15 | 15 § | 44 | 100 | 100 |
| Platelets ×109/L | 217§§ | 87 | 71** | 44 | 6 | 13 | 10 | >100 | >100 |
| MPV (fl) | 9.1§§ | 7.8§ | ND | 9.1§ | ND | 9.1 | ND | 5–12 | 6.5–12 |
| WASp+ platelets | ND | + | ND | + | + | ND | ND | + | + |
Patient 3 died at 7 months from opportunistic infections.
TREC: T-cell receptor excision circle; PHA: phytohemagglutinin; Ag: antigen; (*) positive T cell proliferation with any of the following antigens tested (tetanus toxoid, varicella zoster virus, candidin and purified protein derivative), numbers “2” and “1”, indicate the number of antigen to which the patient’s cell proliferate; (+): comparable to the control; MPV: mean platelet volume; ND: not determined; NA: not applicable.
value measured at the previous time point. (**)during platelet transfusion.
Measured at month 42, after splenectomy. The control values for lymphocytes are from W.T. Shearer et al study (Shearer W.T. et al, J. Aller.Immunol, 2003). The standards for the other parameters have been established by our local hospital diagnostic laboratories.
+/− : proliferation below the normal value.
Immune reconstitution
Recovery of normal absolute T cell counts was achieved in 4 of the 6 evaluable patients (P1, P4, P5, and P6) while values remained just below the normal range in P2 and P7 (Table 2 and Figure 2A). Normal counts of CD4+ T cells were demonstrated in the same 4 patients. Low absolute CD8+ T cell count was observed in 4 out of 6 patients (Table 2 and Supplementary Figure S2). In all patients, ongoing thymopoiesis was evidenced by recovery of circulating naïve T-cells and detection of TRECs. One patient has normal absolute naive CD4+ T-cell counts, although for 5 of the 6 evaluable patients this remained below the normal range (Supplementary Figure S2, and Table 2). For P3, the follow-up period was too short to evaluate immune reconstitution, although T cell numbers recovered rapidly after treatment and were transgene-positive prior to his death 7 months after gene therapy. The presence of anti- CMV specific T cells was detected by pp65 CMV-specific tetramers as well as IFNγ+ immuno-staining (Supplementary Table S2). This is an evidence of partial, albeit functionally incomplete specific T cell response.
Expression of functional WASp is known to provide T cells with a significant growth and survival advantage; this is especially true for the memory subset and effector populations that normally express higher levels of WASp than naïve T cells28–30. Accordingly, VCN were repeatedly found to be higher in memory T cells than in naïve T cells in P4 (12 months after gene therapy, we found: 2.1 versus 1.1 vector copies/cell respectively). Although WASp is not essential for natural regulatory T cell (nTreg) generation, it is required for nTreg function31,32. After gene therapy, CD4+/CD25+/FoxP3+ Treg cells expressed the WAS transgene in all 3 patients evaluated (P2, P4 and P5) (Figure 2B).
At last follow-up absolute NK cell counts were within the normal range in all patients and the function was normal in the sample available for testing (P7) (Figure 2A, Table 2 and Supplementary Figure S3). At last follow-up, B cell counts were within the normal range in 3 out of the 6 patients (P2, P4 and P6) (Figure 2A, Table 2).
Immunoscope analysis revealed that the transduced T cells displayed a polyclonal TCR repertoire (Supplementary Figure S4). As a consequence of impaired T-cell receptor (TCR)-mediated signaling, T cells from WAS-patients typically show defective proliferation after stimulation with anti-CD3 monoclonal antibodies33–35. After gene therapy, all patients displayed normal proliferative responses to phytohemagglutinin (PHA) and anti-CD3 antibody -mediated proliferation was also observed in all patients except P1 (Table 2). A positive response to one or more microbial antigens (tetanus toxoid, varicella-zoster virus, Candida albicans and purified protein derivative) was detected in P2, P4, P5 and P7 (Table 2).
WASp-deficient dendritic cells (DCs) and monocytes fail to form the actin-rich adhesion structures known as podosomes36,37. After gene therapy, monocyte-derived DCs from P2, P4 and P5 were assayed for their ability to assemble podosomes upon adhesion to fibronectin. The fraction of podosome-positive DCs in P4, P2 and P5 was 50%, 20% and <5%, respectively, demonstrating that WASp expression (which was correlated with myeloid cell gene marking) restored the DCs’ ability to regulate cytoskeletal rearrangements.
As of December 2014, B-cell function could not be fully evaluated because 4 out of 6 patients were still receiving immunoglobulin (Ig) replacement therapy but 2 patients (P6 and P4) had recently stopped Ig treatment. In P4, the analysis of the B-cell phenotype 24 months after gene therapy showed B cell subsets within normal ranges compared with healthy age-matched reference values most probably due to WASp expression restoration in B cells38 (Supplementary Figure S5). Moreover, serum IgE levels which are generally elevated in WAS patients, have dropped significantly in P2 and become very low in P7 (Supplementary Table S3).
Platelet reconstitution
Platelets counts and platelet volumes were variably ameliorated by gene therapy in the 6 evaluable patients (Table 2 and Figure 3A). Although all patients remained thrombocytopenic, the detected platelets were predominantly WASp-positive (Figure 3B). The patients’ mean platelet volume (range: 8.1 to 9.3 fl) was in the normal range for healthy controls in our centers (7.5 to 10.1 fl). Patients receiving the highest doses of transduced CD34+ cells appeared to exhibit the most pronounced increases in platelet counts but findings were not statistically tested (Table 1 and 2, Figures 3A and 3C). P1 was splenectomised for quality of life reasons 3 years after gene therapy and recovered normal platelet counts.
Figure 3. Platelet reconstitution after gene therapy.
(A) The change over time in platelet counts in each individual patient. The lower normal value is indicated by a horizontal dotted bar. Filled triangles indicate platelet transfusions and open triangles indicate administration of romiplostim. (B) WASp expression in platelets, as measured by flow cytometry and represented as in Fig. 2B. (C) The correlation between platelet reconstitution and the number of transduced cells infused. The platelet count at last follow-up (Table 2) was plotted against the number of transduced CD34+ cells infused per kg of body weight, which was calculated by taking account of VCN values below 1.0 in the infused product (Table 1).
Analysis of the vector integration site distribution
More than 5 million genomic integration site sequence reads were collected from sorted cell samples from the 6 patients, yielding over 90,000 unique integration sites. As expected for a lentiviral vector, correlation of the integration site distribution with genomic features revealed preferential integration within transcription units and association with epigenetic markers of transcription, in the infused gene therapy products (pre-transplant) and in circulating blood cells at different time points after gene therapy (post-transplant)23,39 (Supplementary Figure S1 A–B). Analysis of genomic regions with high frequencies of integration revealed clusters of sites27 in both the pre-transplant and post-transplant samples (also referred to as “clumps” in the Supplementary Report). Clusters were spread over large genomic distances (>0.5 Mb) and did not overlap with the 5′ ends of the genes implicated in the occurrence of adverse events in previous gene therapy trials (i.e. LMO2, CCND2, and MECOM/EVI1) (Supplementary Figure S1 C–D). Gene-rich regions showed more clusters, occasionally overlapping with cancer-associated genes in some patients at statistically non-significant frequency. Clones associated with genes of concern were a far smaller proportion of sites in the WAS-lentiviral trial than the WAS-gammaretroviral trial (~0.06% versus ~2% respectively; p<10e-6), and none showed persistent expansion over time (Supplementary Figure S6) (for further analysis, see the Supplementary methods and the Supplementary Report). Thus, in contrast to what has been observed in patients treated with gammaretroviral vectors11,40,41, we did not detect an association between recurrent integration near specific cancer-associated genes and cell amplification or persistence.
Sequencing of pre-transplant integration sites from a sample of the infused cells revealed up to 2.9×104 unique sites. Replicate analyses of pre-transplant samples from each patient showed relatively little overlap in the integration patterns, which is consistent with very large population sizes. Change in clonality of PBMCs, and also in sorted peripheral blood lymphoid (CD3+ T cells, CD19+, B cells and CD56+ NK cells) and myeloid cells (CD15+ neutrophils and CD14+ monocytes) were monitored over time in the 6 evaluable patients (Supplementary Figure S6). Population sizes in well-characterized samples could be modeled by comparison with the overlap in sites detected in replicate analyses (using the Chao 1 estimator with Jackknife correction, as applied to counts of linker positions)25. The estimated population sizes in PBMCs corresponded to hundreds or thousands of clones. In all analyzed cell types and in all patients, no one clone accounted for more than 10% of the population detected at a given time point. All patients displayed a stable, high diversity Shannon index (Supplementary Figure S1E). These data indicate that reconstitution with gene-corrected cells was highly polyclonal, with intermittent progenitor activity and no lasting clonal expansion.
Common integration sites in myeloid and lymphoid lineages are indicative of the transduction of multipotent progenitors. We focused our analysis on highly pure (>97%) neutrophils, T and B lymphocytes sorted from P4 twelve months after transplantation (Supplementary Figure S7). These neutrophils shared 12% and 14% of their integration sites with T and B lymphocytes, respectively, suggesting that common progenitors were successfully transduced and engrafted in this patient.
Discussion
WAS is a multifaceted disease with a broad spectrum of severity1. There is still a need for novel, effective, well-tolerated treatments – particularly in patients with advanced disease and/or who lack an HLA-matched allogeneic donor10,42. Here, we report on the outcome of HSC gene therapy in 7 severely affected WAS patients, using a lentiviral vector to transfer a WAS expression cassette in repopulating HSC13,14. The protocol incorporated a near-myeloablative and immunosuppressive conditioning regimen, in order to enhance the engraftment of transduced cells. Compared to a recently-reported monocentric study of three WAS patients aged 1 to 6 (and with a clinical score of 3 or 4)15, the children in the present two-center study were older and had more severe disease (with a clinical score of 5 in 6 out of 7 cases, which is a risk factor for allogeneic HSC transplantation). Among 6 of the 7 patients, there was clinical improvement after gene therapy, which was well tolerated. However, one patient died of pre-existing, treatment-refractory, infectious disease. In the 6 surviving patients the infectious complications resolved after gene therapy, and prophylactic antibiotic therapy was successfully discontinued in 3 cases. Severe eczema resolved in all affected patients as did signs and symptoms of autoimmunity. No patient developed hemorrhagic complications after withdrawal of supportive treatment where implemented. T-cell-related function was corrected in all evaluable patients, regardless of their age at the time of treatment or the dose of transduced cells received. A longer-term follow-up will be required to assess the functional reconstitution of humoral immunity, although the evidence of an accumulation of WASp-expressing B cells is encouraging.
Part of our study’s objectives was to evaluate the haematological and immunological outcomes after engraftment of gene-corrected cells in various lineages. In all patients, the degree of gene marking in lymphoid cells was greater than that achieved in myeloid cells. This is consistent with a strong proliferative and/or survival advantage conferred on the lymphoid compartment by WASp expression, as predicted from earlier observations in mice and in cells from patients28,29. This hierarchy in gene marking has also been observed in patients included in two other gene therapy trials11,15 and is in keeping with the results of previous studies of WASp status in patients after allogeneic HSC transplantation.
One of the major, invariable signs of WAS is a tendency to bleed as a result of intrinsic microthrombocytopenia – particularly when compounded by the development of anti-platelet antibodies. Platelet counts and the mean platelet volume increased in 3 of the 6 evaluable patients (P2, P4 and P5) but remained below normal values although no patient experienced any major bleeding episodes after gene therapy. Persistent thrombocytopenia also occurs after allogeneic HSC transplantation when associated with low myeloid chimerism10. The platelet counts measured in our patients and in other individuals treated with lentiviral gene therapy15 are at the low end of the range observed in patients with mixed chimerism after allogeneic HSC transplantation. Recovery of platelet counts may be related to the dose of transduced cells received. These findings suggest that robust engraftment of HSCs is required to fully correct the disease phenotype. Nevertheless, lower-level engraftment enables lymphoid reconstitution as a consequence of the profound selective advantage conferred on these lineages29. Of note, a rapid normalization of platelet counts and platelet volume was obtained in patient P1 following splenectomy 45 months after gene therapy.
The present study is the first to our knowledge to demonstrate clinical improvement after autologous gene therapy using a lentiviral vector in severely affected children and young adult patients in whom more pronounced procedure-related complications would be expected. When considered alongside with another study in younger and less severely affected patients15, this lentiviral vector may represent a safer alternative to a MLV-derived vector used in a recently reported trial in which 7 of the 9 patients developed acute leukemia as a result of insertional oncogenesis11. Interpretation of the results of this type of study is constrained by the small number of patients and the difficulty in performing randomized trials in severe orphan diseases. We therefore cannot draw conclusions on long–term outcome and safety. Further follow-up of these patients, and those reported in a similar study last year15 together with additional clinical trials of this therapy are therefore necessary.
Conclusion
This study demonstrated the feasibility of the use of gene therapy in patients with Wiskott-Aldrich syndrome. Controlled trials with larger numbers of patients are necessary to assess long-term outcome and safety.
Supplementary Material
Acknowledgments
The study is sponsored by Genethon, Evry France. We are grateful for expert help from Didier Caizergues (D. Pharm.), Malika Souquières (M.S.), Estelle de Barbeyrac (Ph.D.), Marie-Laurence Gourlay-Chu (M.D.), Hafedh Haddad (M.D.) who are employees of Genethon and helped for preparation and filing of regulatory approvals and for clinical study management. We acknowledge Shabi Soheili (Ph.D.) and Alessandra Magnani (Ph.D.) (employed at Necker Children’s Hospital as post-doctoral fellows) for their expert assistance in the characterization of the NK and B cell population. We thank Valérie Jolaine (M.S.) and Elodie Henry (M.S.) employed by “Assistance Publique/ Hôpitaux de Paris at the URC-CIC Paris Centre for the implementation, monitoring and data management of the study. Thanks to Guillaume Corre (Ph.D.) employed as a post-doctoral fellow at Genethon for useful discussions on data analysis. We are also grateful to the Genethon bioproduction and control and quality-assurance teams for providing the clinical-grade vector for the studies. We thank patients’ families for their continuous support of the study, as well as the medical and nursing staff of the Immunology and Pediatric Hematology Department, Hôpital Necker, Paris, and the Immunology and Bone Marrow Transplantation staff at Great Ormond Street Hospital NHS Trust, London. We also acknowledge the gift of expert scientific and medical input from Dr. William Vainchenker (M.D. Ph.D.).
MC and AJT who are principal investigators of the study on each site acknowledge that they had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
The sponsor, as represented by the co-authors A. Galy, S. Charrier, F. Mavilio, G. Honnet, has been involved in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, approval of the manuscript; and decision to submit the manuscript for publication.
Sources of support:
The study is sponsored by Genethon, Evry France with funds from AFM/Telethon. It is also supported by funds from the European Commission (ERC PIDIMMUN #249816 to AF; ERC Regenerative Therapy #269037 to MC; EC FP7 CELL PID #261387 to MC and AJT). AJT is supported by the Wellcome Trust. The study is also supported by the National Institute of Health Research Biomedical Research Centre at Great Ormond Street Hospital NHS Foundation Trust, Great Ormond Street Hospital Children’s Charity, and the United States National Institutes of Health (grant AI 082020-03 to FDB).
Footnotes
Supplementary material includes: 1 clinical trial synopsis, 3 supplemental Tables, 8 supplemental Figures and 1 report.
Trial Registration: Clinicaltrials.gov identifiers: NCT01347346 and NCT01347242.
References
- 1.Albert MH, Notarangelo LD, Ochs HD. Clinical spectrum, pathophysiology and treatment of the Wiskott-Aldrich syndrome. Current opinion in hematology. 2011 Jan;18(1):42–48. doi: 10.1097/MOH.0b013e32834114bc. [DOI] [PubMed] [Google Scholar]
- 2.Thrasher AJ, Burns SO. WASP: a key immunological multitasker. Nat Rev Immunol. 2010 Mar;10(3):182–192. doi: 10.1038/nri2724. [DOI] [PubMed] [Google Scholar]
- 3.Zhu Q, Zhang M, Blaese RM, et al. The Wiskott-Aldrich syndrome and X-linked congenital thrombocytopenia are caused by mutations of the same gene. Blood. 1995 Nov 15;86(10):3797–3804. [PubMed] [Google Scholar]
- 4.Imai K, Morio T, Zhu Y, et al. Clinical course of patients with WASP gene mutations. Blood. 2004 Jan 15;103(2):456–464. doi: 10.1182/blood-2003-05-1480. [DOI] [PubMed] [Google Scholar]
- 5.Mahlaoui N, Pellier I, Mignot C, et al. Characteristics and outcome of early-onset, severe forms of Wiskott-Aldrich syndrome. Blood. 2013 Feb 28;121(9):1510–1516. doi: 10.1182/blood-2012-08-448118. [DOI] [PubMed] [Google Scholar]
- 6.Pai SY, Notarangelo LD. Hematopoietic cell transplantation for Wiskott-Aldrich syndrome: advances in biology and future directions for treatment. Immunol Allergy Clin North Am. 2010 May;30(2):179–194. doi: 10.1016/j.iac.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobayashi R, Ariga T, Nonoyama S, et al. Outcome in patients with Wiskott-Aldrich syndrome following stem cell transplantation: an analysis of 57 patients in Japan. Br J Haematol. 2006 Nov;135(3):362–366. doi: 10.1111/j.1365-2141.2006.06297.x. [DOI] [PubMed] [Google Scholar]
- 8.Friedrich W, Schutz C, Schulz A, Benninghoff U, Honig M. Results and long-term outcome in 39 patients with Wiskott-Aldrich syndrome transplanted from HLA-matched and -mismatched donors. Immunol Res. 2009;44(1–3):18–24. doi: 10.1007/s12026-008-8063-8. [DOI] [PubMed] [Google Scholar]
- 9.Gennery AR, Slatter MA, Grandin L, et al. Transplantation of hematopoietic stem cells and long-term survival for primary immunodeficiencies in Europe: entering a new century, do we do better? J Allergy Clin Immunol. 2010 Sep;126(3):602–610. e601–611. doi: 10.1016/j.jaci.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 10.Moratto D, Giliani S, Bonfim C, et al. Long-term outcome and lineage-specific chimerism in 194 patients with Wiskott-Aldrich syndrome treated by hematopoietic cell transplantation in the period 1980–2009: an international collaborative study. Blood. 2011 Aug 11;118(6):1675–1684. doi: 10.1182/blood-2010-11-319376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Braun CJ, Boztug K, Paruzynski A, et al. Gene therapy for Wiskott-Aldrich syndrome–long-term efficacy and genotoxicity. Sci Transl Med. 2014 Mar 12;6(227):227ra233. doi: 10.1126/scitranslmed.3007280. [DOI] [PubMed] [Google Scholar]
- 12.Charrier S, Dupre L, Scaramuzza S, et al. Lentiviral vectors targeting WASp expression to hematopoietic cells, efficiently transduce and correct cells from WAS patients. Gene Ther. 2007 Mar;14(5):415–428. doi: 10.1038/sj.gt.3302863. [DOI] [PubMed] [Google Scholar]
- 13.Marangoni F, Bosticardo M, Charrier S, et al. Evidence for long-term efficacy and safety of gene therapy for Wiskott-Aldrich syndrome in preclinical models. Mol Ther. 2009 Jun;17(6):1073–1082. doi: 10.1038/mt.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zanta-Boussif MA, Charrier S, Brice-Ouzet A, et al. Validation of a mutated PRE sequence allowing high and sustained transgene expression while abrogating WHV-X protein synthesis: application to the gene therapy of WAS. Gene Ther. 2009 May;16(5):605–619. doi: 10.1038/gt.2009.3. [DOI] [PubMed] [Google Scholar]
- 15.Aiuti A, Biasco L, Scaramuzza S, et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013 Aug 23;341(6148):1233151. doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Merten OW, Charrier S, Laroudie N, et al. Large-scale manufacture and characterization of a lentiviral vector produced for clinical ex vivo gene therapy application. Hum Gene Ther. 2011 Mar;22(3):343–356. doi: 10.1089/hum.2010.060. [DOI] [PubMed] [Google Scholar]
- 17.Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000 Apr 28;288(5466):669–672. doi: 10.1126/science.288.5466.669. [DOI] [PubMed] [Google Scholar]
- 18.Hacein-Bey-Abina S, Garrigue A, Wang GP, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008 Sep;118(9):3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lim A, Baron V, Ferradini L, Bonneville M, Kourilsky P, Pannetier C. Combination of MHC-peptide multimer-based T cell sorting with the Immunoscope permits sensitive ex vivo quantitation and follow-up of human CD8+ T cell immune responses. J Immunol Methods. 2002 Mar 1;261(1–2):177–194. doi: 10.1016/s0022-1759(02)00004-2. [DOI] [PubMed] [Google Scholar]
- 20.Hacein-Bey-Abina S, Le Deist F, Carlier F, et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. The New England journal of medicine. 2002 Apr 18;346(16):1185–1193. doi: 10.1056/NEJMoa012616. [DOI] [PubMed] [Google Scholar]
- 21.Poulin JF, Sylvestre M, Champagne P, et al. Evidence for adequate thymic function but impaired naive T-cell survival following allogeneic hematopoietic stem cell transplantation in the absence of chronic graft-versus-host disease. Blood. 2003 Dec 15;102(13):4600–4607. doi: 10.1182/blood-2003-05-1428. [DOI] [PubMed] [Google Scholar]
- 22.Charrier S, Ferrand M, Zerbato M, et al. Quantification of lentiviral vector copy numbers in individual hematopoietic colony-forming cells shows vector dose-dependent effects on the frequency and level of transduction. Gene Ther. 2011 May;18(5):479–487. doi: 10.1038/gt.2010.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang GP, Ciuffi A, Leipzig J, Berry CC, Bushman FD. HIV integration site selection: analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res. 2007 Aug;17(8):1186–1194. doi: 10.1101/gr.6286907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang GP, Garrigue A, Ciuffi A, et al. DNA bar coding and pyrosequencing to analyze adverse events in therapeutic gene transfer. Nucleic Acids Res. 2008 May;36(9):e49. doi: 10.1093/nar/gkn125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berry CC, Gillet NA, Melamed A, Gormley N, Bangham CR, Bushman FD. Estimating abundances of retroviral insertion sites from DNA fragment length data. Bioinformatics. 2012 Mar 15;28(6):755–762. doi: 10.1093/bioinformatics/bts004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berry C, Hannenhalli S, Leipzig J, Bushman FD. Selection of target sites for mobile DNA integration in the human genome. PLoS Comput Biol. 2006 Nov 24;2(11):e157. doi: 10.1371/journal.pcbi.0020157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berry CC, Ocwieja KE, Malani N, Bushman FD. Comparing DNA integration site clusters with scan statistics. Bioinformatics. 2014 Jun 1;30(11):1493–1500. doi: 10.1093/bioinformatics/btu035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rengan R, Ochs HD, Sweet LI, et al. Actin cytoskeletal function is spared, but apoptosis is increased, in WAS patient hematopoietic cells. Blood. 2000 Feb 15;95(4):1283–1292. [PubMed] [Google Scholar]
- 29.Westerberg LS, de la Fuente MA, Wermeling F, et al. WASP confers selective advantage for specific hematopoietic cell populations and serves a unique role in marginal zone B-cell homeostasis and function. Blood. 2008 Nov 15;112(10):4139–4147. doi: 10.1182/blood-2008-02-140715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamaguchi K, Ariga T, Yamada M, et al. Mixed chimera status of 12 patients with Wiskott-Aldrich syndrome (WAS) after hematopoietic stem cell transplantation: evaluation by flow cytometric analysis of intracellular WAS protein expression. Blood. 2002 Aug 15;100(4):1208–1214. doi: 10.1182/blood-2002-01-0211. [DOI] [PubMed] [Google Scholar]
- 31.Maillard MH, Cotta-de-Almeida V, Takeshima F, et al. The Wiskott-Aldrich syndrome protein is required for the function of CD4(+)CD25(+)Foxp3(+) regulatory T cells. J Exp Med. 2007 Feb 19;204(2):381–391. doi: 10.1084/jem.20061338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marangoni F, Trifari S, Scaramuzza S, et al. WASP regulates suppressor activity of human and murine CD4(+)CD25(+)FOXP3(+) natural regulatory T cells. J Exp Med. 2007 Feb 19;204(2):369–380. doi: 10.1084/jem.20061334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Molina IJ, Sancho J, Terhorst C, Rosen FS, Remold-O’Donnell E. T cells of patients with the Wiskott-Aldrich syndrome have a restricted defect in proliferative responses. J Immunol. 1993 Oct 15;151(8):4383–4390. [PubMed] [Google Scholar]
- 34.Snapper SB, Rosen FS, Mizoguchi E, et al. Wiskott-Aldrich syndrome protein-deficient mice reveal a role for WASP in T but not B cell activation. Immunity Jul. 1998;9(1):81–91. doi: 10.1016/s1074-7613(00)80590-7. [DOI] [PubMed] [Google Scholar]
- 35.Zhang J, Shehabeldin A, da Cruz LA, et al. Antigen receptor-induced activation and cytoskeletal rearrangement are impaired in Wiskott-Aldrich syndrome protein-deficient lymphocytes. J Exp Med. 1999 Nov 1;190(9):1329–1342. doi: 10.1084/jem.190.9.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burns S, Thrasher AJ, Blundell MP, Machesky L, Jones GE. Configuration of human dendritic cell cytoskeleton by Rho GTPases, the WAS protein, and differentiation. Blood. 2001 Aug 15;98(4):1142–1149. doi: 10.1182/blood.v98.4.1142. [DOI] [PubMed] [Google Scholar]
- 37.Calle Y, Chou HC, Thrasher AJ, Jones GE. Wiskott-Aldrich syndrome protein and the cytoskeletal dynamics of dendritic cells. J Pathol. 2004 Nov;204(4):460–469. doi: 10.1002/path.1651. [DOI] [PubMed] [Google Scholar]
- 38.Duchamp M, Sterlin D, Diabate A, et al. B-cell subpopulations in children: National reference values. Immunity, Inflammation and Disease. 2014;2(3):131–140. doi: 10.1002/iid3.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schroder AR, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002 Aug 23;110(4):521–529. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- 40.Hacein-Bey-Abina S, Hauer J, Lim A, et al. Efficacy of gene therapy for X-linked severe combined immunodeficiency. The New England journal of medicine. 2010 Jul 22;363(4):355–364. doi: 10.1056/NEJMoa1000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hacein-Bey-Abina S, Pai SY, Gaspar HB, et al. A modified gamma-retrovirus vector for X-linked severe combined immunodeficiency. The New England journal of medicine. 2014 Oct 9;371(15):1407–1417. doi: 10.1056/NEJMoa1404588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ozsahin H, Cavazzana-Calvo M, Notarangelo LD, et al. Long-term outcome following hematopoietic stem-cell transplantation in Wiskott-Aldrich syndrome: collaborative study of the European Society for Immunodeficiencies and European Group for Blood and Marrow Transplantation. Blood. 2008 Jan 1;111(1):439–445. doi: 10.1182/blood-2007-03-076679. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.