

Abstract
Background
The genetic and antigenic characteristics of influenza A viruses (IAV) within and between species change over time due to antigenic shift and drift. Although pigs are known to play a key role in the epidemiology of IAV between species, little is known about the molecular evolution of IAV hemagglutinin (HA) in pigs.
Objectives
The aim of this study was to evaluate the HA drift of an H1N1 IAV after infecting weaned pigs with or without maternally derived passive immunity.
Methods
Three‐ to four‐week‐old piglets born either to vaccinated or unvaccinated sows were contact‐infected upon exposure with an IAV‐infected pig. Nasal swabs were collected daily from each pig and tested for IAV by RRT‐PCR. Full‐length HA sequences were obtained directly from positive nasal swabs and compared between groups.
Results
Synonymous and non‐synonymous mutations were detected in pigs with and without passive immunity. Most of the non‐synonymous mutations occurred within the HA1 region of the HA. Changes within HA1 region were only identified in antigenic site B in pigs without passive immunity and in antigenic sites A, B, and D in pigs with passive immunity. However, there was no association between the immune status of the pig and the amino acid substitutions observed.
Conclusions
Overall, we demonstrated that amino acid substitutions within antigenic sites can happen in weaned pigs with or without passive immunity shortly after infection.
Keywords: Antigenic drift, passive immunity, Swine influenza
Introduction
Influenza A viruses (IAV) have a segmented negative‐sense RNA genome that is able to reassort with other IAV strains (antigenic shift) and/or evolve by the accumulation of mutations throughout the genome (antigenic drift).1 Two proteins are known as major IAV antigens, hemagglutinin (HA) and neuraminidase (NA), and their genotypes are associated with the host species that each virus infects.2 Accumulation of mutations and gene exchange between IAV during infection are expected to influence viral fitness and transmission within and between species.3, 4 Factors responsible for mutations in IAV are not completely understood; however, a viral non‐proofreading polymerase,5 immune selection,6 and intrahost characteristics are known to play key roles.7, 8, 9 Although wild waterfowl are considered the natural reservoirs for IAV, pigs can be intermediate hosts10 with a propensity for generating reassortant viruses11 and sustaining infections that result in new viruses of risk to other species, including humans.12
Herd prevalence estimates indicate that IAV infections are endemic and widespread in pigs.13 In the United States, after the emergence of the 2009 pandemic H1N1 (pH1N1) in swine, the HA gene of H1 subtype clustered in five different phylogenetic groups (α, β, γ, δ1, and δ2), which illustrates the broad diversity of IAV in pigs.14 Active surveillance in US swine has shown that 90% of the herds surveyed throughout a 2‐year period tested virus‐positive for IAV.15 Furthermore, these herds were positive for multiple IAV strains of a variety of subtypes resulting from several reassortment events.16 Such IAV diversity in pigs increases the challenges faced in both understanding IAV evolution and controlling IAV in pigs.
Suckling pigs serve as an important source of IAV as they can be asymptomatically infected and can transport the virus to multiple geographical locations at weaning. Virus shedding occurs even if passive17 or active immunity18 is present, and different genotypes of IAV can co‐circulate in swine populations regardless of their immune status,9 which may result in the emergence of novel reassortant strains. However, little is known about genetic diversity and selective evolution of IAV in suckling piglets with passive immunity. Given the central role that suckling pigs play in the dissemination and emergence of IAV strains in pig farms, the objective of this study was to evaluate the degree of antigenic drift in the HA of a H1N1 virus in pigs with passive immunity under experimental conditions.
Materials and methods
Study design and sample selection
Samples from this study were part of a transmission study described elsewhere.17 Briefly, an IAV‐negative swine herd was selected for the study. The herd was considered negative for IAV based on serology against the nucleoprotein (NP) using an ELISA (FlockChek AI MultiS‐Screen Ab Test Kit, IDEXX Laboratories Inc., Westbrook, ME, USA).19 Sows were either vaccinated with an experimental vaccine (PASSIV‐VAC, n = 3) or left unvaccinated (NAIVE, n = 3). The vaccine was adjuvanted, inactivated, prepared with H1N1 A/Swine/IL/02450/08 virus (α‐cluster, NCBI accession number: CY099052.1), and administered intramuscularly at 5 and 2 weeks prior to farrowing.
At 3 weeks of age, piglets were weaned, and 30 and 39 piglets were selected from the PASSIV‐VAC and NAIVE sows, respectively, and moved to the University of Minnesota animal isolation units. Nine pigs from the NAIVE group were randomly selected to be used as seeder pigs. Each seeder pig was infected intranasally and intratracheally with 0·5 ml in each location of an inoculum containing 107 TCID50 of influenza A/Swine/IA/00239/04 H1N1 virus (β‐cluster, NCBI accession number: EU139832.1). The remaining pigs (n = 60) were randomly allocated into groups of 10 (three replicates per group). One seeder pig was mixed with pigs in each replicate for a total of six seeder pigs. There were samples from three additional infected pigs that followed the same procedures as the seeder pigs, and their samples were included in the analysis to increase the power of the study. All seeder pigs were considered within the NAÏVE group for analysis.
Sows in the PASSIV‐VAC group were confirmed not to have been exposed to IAV before vaccination by testing serum samples by NP‐ELISA as mentioned above. Piglets in the NAIVE group were also confirmed to not have immunity to IAV via serum sample tests using the same ELISA. Additionally, all piglets in the PASSIV‐VAC and NAIVE groups were confirmed to be IAV negative prior to exposure to the seeder pig by testing nasal swabs by RRT‐PCR.20 The challenge and vaccine viruses were selected based on their limited HA cross‐reactivity and are considered reference strains within their clusters.21
After pigs were exposed to the seeder pigs, nasal swabs were collected daily for 14 days and tested by RRT‐PCR targeting the matrix gene (M).20 The seeder pigs remained with the rest of the pigs throughout the study. For sequencing purposes, one positive sample from each animal was conveniently selected based on the lowest cycle threshold (CT) value obtained on the matrix RRT‐PCR, and all infection days were represented. Full‐length HA gene amplification was performed directly from the selected nasal swabs. Blood samples were collected prior to infection and at the termination of the study. All sera were assayed by hemagglutination inhibition (HI) test22 against the challenge virus, A/Swine/IA/00239/04, and the vaccine virus, A/Swine/IL/02450/08.
Sequencing and sequence analysis
To establish virus HA genetic and protein relatedness between the challenge and vaccine virus, full‐length HA sequences of A/Swine/IA/00239/04 (EU139832.1) and A/Swine/IL/02450/08 (CY099052.1) were first aligned and compared using CLUSTAL W (MegAlign, LaserGene Core 9; DNASTAR, Madison, WI, USA).
From each selected sample, HA gene was amplified using previously described primers,23 and PCR products were confirmed by electrophoresis in 2% agarose gel and stained with ethidium bromide. The expected band for HA (1780 base pairs) was excised from the gel and eluted using QIAquick Gel Extraction Kit (QIAGEN, Valencia, CA, USA) following manufacturers' recommendations and submitted to the Biomedical Genome Center (BMGC) of the University of Minnesota for sequencing using a primer walking scheme.
The trace files obtained from each sample were assembled using the HA sequence of A/swine/New Jersey/11/76(H1N1) as a reference. The template was then removed, and the consensus sequence for each sample was established. All contigs were initially assembled using the default parameters of SeqManPro (LaserGene Core 9; DNASTAR, Madison, WI, USA), and the quality of each nucleotide trace was evaluated visually in each position. Only nucleotide calls with clear peaks were considered for analysis. Each trace file was trimmed when peaks were considered ambiguous to obtain complete sequences with good‐quality reads. Each consensus sequence was annotated using the influenza annotation tool of the NCBI (http://www.ncbi.nlm.nih.gov/genomes/FLU/Database/annotation.cgi) and trimmed accordingly to obtain the HA coding region.
Complete sequences within each group were aligned and compared by CLUSTAL W (MegAlign, LaserGene Core 9; DNASTAR, Madison, WI, USA) using the consensus from all sequences as reference, and nucleotide differences were classified as either transitions (purine↔purine or pyrimidine↔pyrimidine) or transversions (purine↔pyrimidine) according to the base pair interchange identified, and their phylogenetic distance was compared using a median‐joining network algorithm.24 The proportion of synonymous (SM) and non‐synonymous (NSM) mutations was assessed and compared between groups. In some cases, full‐length HA sequences were not obtained, thus short reads for those samples were aligned and polymorphisms were inferred for only the regions sequenced.
Statistical methods
The nucleotide substitution mean was obtained for each group (sum of the number of nucleotides substituted/total number of nucleotides sequenced in all samples) and compared between groups using a one‐tailed t‐test. Results were considered statistically significant at P < 0·05. Additionally, the odds of non‐synonymous mutations (NSM/SM) were compared between groups using only complete HA sequences. Finally, the frequency of non‐synonymous mutations present more than once was compared between groups (including complete and incomplete sequences). For these last two comparisons, tabular methods were used and considered statistically different when chi‐square P < 0·05. If the count per cell in each comparison was lower than 5, then a Fisher's exact test was used.
HA protein models
To better visualize the location of the amino acid differences identified, the structure of the translated HA1 regions was modeled using A/Swine/Iowa/15/30 (H1N1) as template (Protein Data Bank (PDB) ID:1RUY) with the tools offered in swissmodel.expasy.org.25 The protein structure of the A/Swine/Iowa/15/30 was selected because it is of swine origin, has the HA crystalized, and is available as a reference strain in the protein database. Each amino acid substitution detected was mapped within the predicted model for the reference sequence, and its location was compared with the five antigenic sites previously identified for A/PR/8/1934 (PDB: 1RU7)26, 27 using PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC, New York, NY, USA. The nucleotide numbering for HA followed the numbering for the entire HA gene, while the amino acid numbering was segregated by sig_peptide, HA1, and HA2 portions, such that each region of the molecule was identified by a specific amino acid where they began and/or ended.
Results
The number of days that pigs tested positive and the PCR‐positive samples selected for sequencing and comparison are shown in Table 1. Prior to exposure, all pigs in the NAIVE group tested negative to IAV by ELISA (S/N ratio ≥ 0·673), and all pigs in the PASSIV‐VAC group tested positive (mean: 0·254, min: 0·09, max: 0·495). HI titers for each group before and after contact with the seeder pig are shown in Table 2. HI titers against the challenge strain increased in both groups after infection.
Table 1.
Tables indicate the pigs in the PASSIV‐VAC and NAIVE groups. Numbers in the top indicate the days post‐contact (DPC) for each group. Gray boxes indicate the samples that tested positive by RRT‐PCR for IAV, and the numbers within the gray boxes indicate the CT value of the samples that were used for HA sequencing. The days where all pigs tested negative were excluded for simplicity (R: replicate, S: seeder)
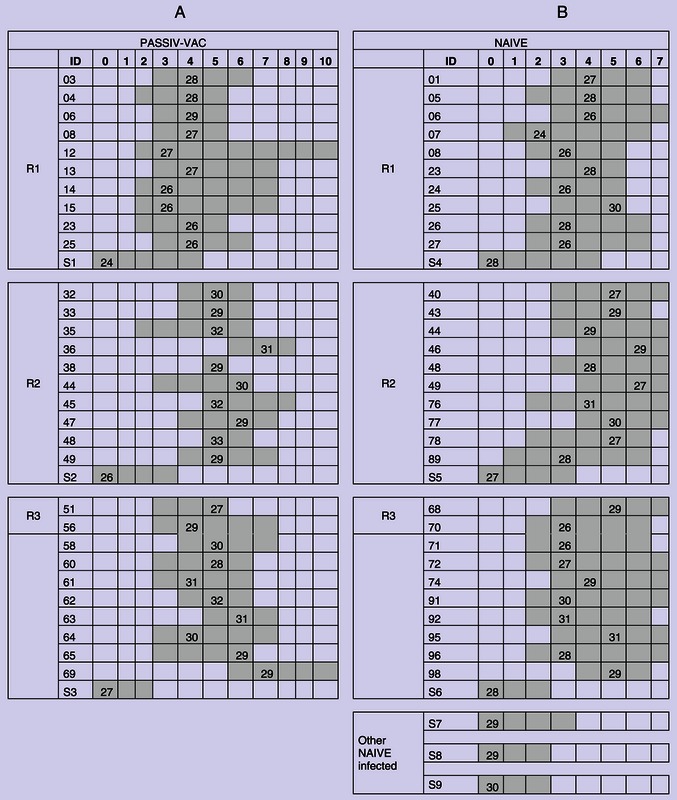
Table 2.
Reciprocal geometric mean HI titers against A/Swine/IA/00239/04 H1N1 (challenge virus) and A/Swine/IL/02450/08 (vaccine strain)
| Group | A/Swine/IA/00239/04 H1N1 | A/Swine/IL/02450/08 | ||
|---|---|---|---|---|
| Pre‐contact | Post‐contact | Pre‐contact | Post‐contact | |
| PASSIV‐VAC | 17 | 70 | 143 | 111 |
| NAIVE | 10 | 357 | 23 | 113 |
Hemagglutinin gene sequence of A/Swine/IA/00239/04 and A/Swine/IL/02450/08 aligned within the β‐ and α‐H1 clusters of swine IAV, respectively, and both followed the same reading frame described for the human pandemic influenza virus A/California/04/2009(H1N1) (GenBank: FJ966082.1). The overall nucleotide and amino acid identity for the HA gene and hypothetical proteins and the identity for each expected antigenic site are summarized in Table 3.
Table 3.
Hemagglutinin nucleotide and amino acid identity between A/Swine/IA/00239/04 (challenge virus) and A/Swine/IL/02450/08 (vaccine strain)
| Sequence length | % Identity | |
|---|---|---|
| HA (nt) | 1701 | 87·9 |
| HA (a.a) | 566 | 88·5 |
| HA1 (nt) | 980 | 87·8 |
| HA1 (a.a) | 327 | 87·5 |
| Antigenic site Aa | 24 | 79·2 |
| Antigenic site B | 22 | 72·7 |
| Antigenic site C | 33 | 75·8 |
| Antigenic site D | 48 | 87·5 |
| Antigenic site E | 34 | 82·4 |
| No antigenic sites | 166 | 94·0 |
Nt, nucleotide sequence; a.a, amino acid sequence.
Note that antigenic sites are not linear. Each position for each antigenic site previously described was compared between sequences used in this study.
Both SM and NSM were observed in both groups of pigs (Table 4). Differences were observed within signal peptide and HA1 region for the NAIVE group (16 complete sequences), and within HA1 and HA2 for the PASSIV‐VAC group (nine complete sequences). Additionally, 12 and 16 partial sequences were obtained in the PASSIV‐VAC and NAIVE groups, respectively, and their polymorphisms are summarized in Table 5. The total number of polymorphisms identified in both groups led to eight different HA alleles (complete sequences different one from each other), and their phylogenetic relatedness is illustrated in Figure 1.
Table 4.
Summary of nucleotide substitutions found in the full‐length HA sequences by group
| Group | Type of substitutiona | Effectb | Nucleotide | Regionf | Frequencyg | ||
|---|---|---|---|---|---|---|---|
| Ref.c | Posd | Vare | |||||
| NAIVE (n = 16)h | Transition | SM | C | 282 | T | HA1 | 1 |
| Transition | NSM | A | 15 | G | Sig_pep | 1 | |
| Transition | NSM | G | 515 | A | HA1 | 5 | |
| Transversion | NSM | A | 617 | C | HA1 | 1 | |
| PASSIV‐VAC (n = 9) | Transition | SM | T | 1194 | C | HA2 | 1 |
| Transition | NSM | G | 515 | A | HA1 | 6 | |
| Transition | NSM | A | 563 | G | HA1 | 1 | |
| Transversion | NSM | G | 454 | C | HA1 | 1 | |
| Transversion | NSM | A | 1382 | C | HA2 | 1 | |
Type of substitution compared with the reference sequence.
Base pair substitution effect on the hypothetical translated protein (SM, synonymous mutation, NSM, non‐synonymous mutation).
Reference nucleotide according to reference sequence.
Position‐based HA numbering.
Variant nucleotide.
HA region where the substitution was found.
Frequency: number of times the specific difference was observed.
Number of full‐length HA sequences.
Table 5.
Nucleotide reads in partial sequences at positions where polymorphisms in the HA full‐length sequences were identified
| Group | Ref.a | Posb | Readc | Freq. d |
|---|---|---|---|---|
| NAIVE (n = 12) e | C | 282 |
C T Nf |
10 1 1 |
| A | 15 | A | 12 | |
| G | 515 |
G A N |
7 3 2 |
|
| A | 617 |
A N |
11 1 |
|
| PASSIV‐VAC (n = 16) | T | 1194 | T | 16 |
| G | 515 |
G A – |
11 4 1 |
|
| A | 563 |
A – |
14 2 |
|
| G | 454 |
G – |
15 1 |
|
| A | 1382 |
A C N |
12 1 3 |
Reference nucleotide according to reference sequence.
Position based on the full‐length HA sequence.
Nucleotide read at the indicated position.
Frequency: number of times the specified read was found.
Number of partial HA sequences obtained.
N: Ambiguous nucleotide.
Figure 1.
Network analysis of the different alleles identified among all samples. Each circle represents a different allele, and its size is proportional to the number of sequences by allele identified (total number of sequences included in the analysis = 25). Yellow and blue indicate the sequences from the NAIVE and PASSIV‐VAC groups, respectively. The numbers indicate the position were nucleotide differences were identified.
The nucleotide substitution mean for the NAIVE group (2·94 × 10−4) was lower (P = 0·042) when compared with the mean in the PASSIV‐VAC group (6·4 × 10−4). The odds of NSM in the PASSIV‐VAC group was 1·29 times higher compared with the NAIVE group (Table 6); however, this difference was not statistically significant (P > 0·05). Overall, the most common non‐synonymous mutation in both groups was at position 515, and the odds of this mutation (G515A) in the PASSIV‐VAC group was 1·6 times higher compared with the NAIVE group (Table 7); however, this was not statistically different (P = 0·38). Table 8 summarizes the hypothetical amino acid substitutions found in all groups, and Figure 2 shows their location within the HA1 protein model. All changes in the NAIVE group were mapped within antigenic site B, while changes in the PASSIV‐VAC group were located within antigenic sites A, B, and D. The substitution identified at position 155 found in both groups indicate a change from a polar to a charged amino acid, and all the other substitutions found in the PASSIV‐VAC group did not change the amino chemical nature, but the substitution found in the NAIVE at position 189 did (polar to hydrophobic).
Table 6.
Non‐synonymous mutation (NSM) versus synonymous mutation (SM) in full‐length HA sequences by group
| Non‐Synonymous (NSM) | Synonymous (SM) | Total | |
|---|---|---|---|
| PASSIV‐VAC | 9 | 1 | 10 |
| NAIVE | 7 | 1 | 8 |
| Total | 16 | 2 | 18 |
Odds of NSM in the PASSIV‐VAC = 9/1 = 9; Odds of NSM in the NAIVE = 7/1 = 7; OR = 1·29 (Fisher's exact test P > 0·05).
Table 7.
Nucleotide read at position 515 by group
| Variant = A | Reference = G | Total | |
|---|---|---|---|
| PASSIV‐VAC | 10 | 14 | 24 |
| NAIVE | 8 | 18 | 26 |
| Total | 18 | 32 | 50 |
Odds of NSM at position 515 in the PASSIV‐VAC = 10/14 = 0·714; Odds of NSM at position 515 in the NAIVE = 8/18 = 0·444; OR = 1·61 (P = 0·38).
Table 8.
Summary of hypothetical amino acid substitutions found in HA by group
| Group | Region | Amino acid positiona | Amino Acid | Sited | Frequencye | Chemical naturef | |
|---|---|---|---|---|---|---|---|
| Ref.b | Var.c | ||||||
| NAIVE | H1 | NAg | NA | NA | – | 1 | NA |
| Sig_pep | 5 | Isoleucine | Methionine | – | 1 | Hydrophobic to hydrophobic | |
| H1 | 155 | Glycine | Glutamate | B | 5 | Polar to charged | |
| H1 | 189 | Glutamine | Proline | B | 1 | Polar to hydrophobic | |
| PASSIV‐VAC | H2 | NA | NA | NA | – | 1 | NA |
| H1 | 155 | Glycine | Glutamate | B | 6 | Polar to charged | |
| H1 | 171 | Lysine | Arginine | D | 1 | Charged to charged | |
| H1 | 135 | Alanine | Proline | A | 1 | Hydrophobic to hydrophobic | |
| H2 | 204 | Asparagine | Threonine | – | 1 | Polar to polar | |
Numbering for each hypothetical protein.
Ref: Amino acid predicted in the reference sequence.
Var: Amino acid predicted in the variant sequence.
Site: Antigenic site where the substitution was mapped.
Frequency: number of times the specific difference was observed.
Chemical nature of the amino acid substitution.
NA: No applicable (synonymous mutation).
Figure 2.
Protein model for one HA1 monomer of A/Swine/IA/00239/04 (challenge virus). (A) Mesh model highlighting the spatial distribution within the protein of the amino acids that were found different. Close ups B, C, and D represent the predicted surface of the protein and the area exposed for each amino acid replaced. NAIVE: Q189P (magenta) within antigenic site B; PASSIV‐VAC: K171R (red) within antigenic site D; and A135P (yellow) within antigenic site A. The green spheres represent G155E that was seen in both groups NAIVE and PASSIV‐VAC within the antigenic site D.
Discussion
Influenza in pigs represents a constant risk to other species including humans as pigs serve as a reservoir for IAV that can result in zoonotic infections of pandemic proportions.28 Although it is known that immunity can play a role by inducing IAV change in other animal models, little is known about the degree of virus diversity found in pigs with passive immunity. In this study, we evaluated genetic and antigenic changes in the HA of an H1N1 IAV in pigs with and without passive immunity under experimental conditions with emphasis on changes observed in young pigs early in the infection process. Although nucleotide substitution in swine H1 IAV is believed to be lower compared with human viruses,29 this study demonstrates that genetic changes can occur in young pigs early during infection, and those changes can induce amino acid changes located within antigenic sites. Furthermore, both SM and NSM were observed in the HA protein of pigs with and without passive immunity, and sequences from both groups were genetically related regardless of immune status. Even though the nucleotide substitution mean was higher in the PASSIV‐VAC group compared with the NAIVE group, the odds of NSM in the PASSIV‐VAC group were not significantly higher compared with the NAIVE group. Changes in the chemical nature, especially of G155E identified in 11 pigs (five and six in the NAIVE and PASSIV‐VAC, respectively), indicate that there might be specific changes that occur in early infection to improve viral fitness in a new host. However, the biological significance of this type of change as it relates to immune pressure needs to be further investigated. The dynamic distribution of IAV subtypes in pigs, and the ability of the virus to change early in infection, has been described and suggests that allele fixation can occur rapidly.9
Immune pressure and vaccination have been associated with virus change in other animal models.30 In swine, the use of influenza vaccines is common, and usually, vaccines are administered to breeding females prior to farrowing. According to USDA National Animal Health Monitoring System (NAHMS) data, 70% of large breeding herds (>500 sows) in the United States are vaccinated against IAV (http://www.aphis.usda.gov/animal_health/nahms/swine/downloads/swine2006/Swine2006_is_vacc.pdf). Pre‐farrowing vaccination is practised to enable transfer of passive immunity and minimize clinical disease to progeny. However, passive immunity is generally insufficient to prevent transmission of IAV in pigs.17 It is logical that immune pressure created by vaccination of pigs would drive virus change. However, in our study, we did not see significant genetic differences between immune and naïve pigs, and passive immunity did not affect transmission patterns between groups (results not shown).The similarity of virus change between groups in this study could also be a result of the phenotypic differences between the virus used to prepare the vaccine and the virus used to infect the seeder pig. However, these results are in agreement with a recent study where it was shown that vaccination did not appear to have a major effect on the genetic structure of intrahost viral populations through immune selection.9
We selected samples based on the amount of genetic material present in the sample and ensured that there was representation of infection through all days of the study. Most of the mutations were observed one time post‐infection. Only one mutation was seen in multiple animals. This mutation was at position 155 of HA1, and it was observed in naïve and immune pigs. This mutation resulted in glycine to glutamate substitution, which changes the predicted conformation of the HA structure. Substitution at this same site was also described by Hensley et al.30, but only in immune mice after serial passages. Furthermore, the mutation, E156K of A/Puerto Rico/8/34(H1N1), which aligns with 155 of A/Swine/IA/00239/04 in this study, was mapped within HA antigenic site B and was associated with immune escape. Additionally, a mutation at position 155 was also described by Kuroda et al.31, as a likely rare event in a human sample from a 33‐year‐old male diagnosed with pandemic influenza virus [A/Nagano/RC1/2009(H1N1)] (accession number AB538389) who died from respiratory failure and multiple organ dysfunction syndrome. In the present study, the mutation at position 155 seemed to be common and was not associated with immune status or increase in clinical signs (results not shown). It is possible that this mutation was present in the virus inoculum in low frequency, but that it was replicated more efficiently taking over the most predominant allele identified in the challenge virus. Alternatively, the mutation may have happened in one or several seeder pigs and then transmitted to the study pigs. This mutation may have provided the virus some advantage when transmitting from the experimentally infected seeder pig to the new hosts, but this was outside the scope of this study. Furthermore, none of the other mutations identified were common to both groups.
It is important to highlight in this study that despite the reduced sample size, changes in antigenic sites were not restricted to pigs with immunity indicating that antigenic drift in pigs can happen early during the infection–transmission chain and may be driven independently from immune selection. This must be further investigated given the risk of swine IAV to human health and the impact of IAV on pig health. Although most of the amino acid changes were observed in antigenic sites of the HA protein considered to be under higher selection pressure, there were changes also observed in the two other regions of HA that stabilize the conformation of the HA protein, the signal peptide and HA2. The biological implications of these substitutions are not known, and further studies are needed to characterize their role in pathogenicity and viral fitness. However, this suggests that amino acid changes outside the antigenic sites may also be important for the virus to adapt and transmit to new hosts.
In summary, this study shows that antigenic drift of IAV can happen in young pigs shortly after infection. Overall, pigs with passive immunity had more nucleotide substitutions compared with naïve pigs. However, in this study, these changes did not always result in phenotypic changes in the HA protein that resulted in new antigenic variants. Nevertheless, this study is important to highlight the role of the suckling pig as a potential source of IAV genetic diversity in pigs. This is especially important given that extensive movement of suckling pigs takes place after weaning in the swine industry. In addition, nucleotide substitutions that induce amino acid changes were detected in naïve animals as well as in pigs with passive immunity. Some of these changes were able to persist throughout the infection period. Furthermore, changes were also identified in non‐antigenic sites indicating that viral adaptation during transmission in pigs is not only dependent on its antigenic characteristics. Overall, this study indicates the complexity of genetic diversity in pigs, and further studies are needed to understand viral evolution and epidemiology of IAV in swine populations and the risk they represent to people.
Acknowledgements
This study was supported in whole or in part with federal funds from the NIH, National Institute of Allergy and Infectious Diseases and Department of Health and Human Services under the contract No. HHSN266200700007C, and the USDA Discretionary GAR Funds AES0060014 as part of the Signature Program Funding at the College of Veterinary Medicine, University of Minnesota. The authors would also like to acknowledge the Minnesota Super Computing Institute at the University of Minnesota for their support during this study.
Diaz et al (2013) Antigenic drift of H1N1 influenza A virus in pigs with and without passive immunity. Influenza and Other Respiratory Viruses 7 (Suppl. 4), 52–60.
References
- 1. Taubenberger JK, Kash JC. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe 2010; 7:440–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ferguson NM, Galvani AP, Bush RM. Ecological and immunological determinants of influenza evolution. Nature 2003; 422:428–433. [DOI] [PubMed] [Google Scholar]
- 3. Rambaut A, Pybus OG, Nelson MI et al The genomic and epidemiological dynamics of human influenza A virus. Nature 2008; 453:615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fanning TG, Reid AH, Taubenberger JK. Influenza A virus neuraminidase: regions of the protein potentially involved in virus‐host interactions. Virology 2000; 276:417–423. [DOI] [PubMed] [Google Scholar]
- 5. Boivin S, Cusack S, Ruigrok RWH, Hart DJ. Influenza A virus polymerase: structural insights into replication and host adaptation mechanisms. J Biol Chem 2010; 285:28411–28417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Long JX, Bushnell RV, Tobin JK et al Evolution of H3N2 influenza virus in a guinea pig model. PLoS One 2011; 6 (7):e20130. doi: 10.1371/journal.pone.0020130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hoelzer K, Murcia PR, Baillie GJ et al Intrahost evolutionary dynamics of canine influenza virus in naive and partially immune dogs. J Virol 2010; 84:5329–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Murcia PR, Baillie GJ, Daly J et al Intra‐ and interhost evolutionary dynamics of equine influenza virus. J Virol 2010; 84:6943–6954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Murcia PR, Hughes J, Battista P et al Evolution of an Eurasian avian‐like influenza virus in naive and vaccinated pigs. PLoS Pathog 2012; 8 (5): e1002730. doi: 10.1371/journal.ppat.1002730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hass J, Matuszewski S, Cieslik D, Haase M. The role of swine as “mixing vessel” for interspecies transmission of the influenza A subtype H1N1: a simultaneous Bayesian inference of phylogeny and ancestral hosts. Infect Genet Evol 2011; 11:437–441. [DOI] [PubMed] [Google Scholar]
- 11. Nelson MI, Lemey P, Tan Y et al Spatial dynamics of human‐origin H1 influenza A virus in North American swine. PLoS Pathog 2011; 7 (6): e1002077. doi: 10.1371/journal.ppat.1002077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Van Reeth K. Avian and swine influenza viruses: our current understanding of the zoonotic risk. Vet Res 2007; 38:243–260. [DOI] [PubMed] [Google Scholar]
- 13. Van Reeth K, Brown IH, Durrwald R et al Seroprevalence of H1N1, H3N2 and H1N2 influenza viruses in pigs in seven European countries in 2002‐2003. Influenza Other Respir Viruses 2008; 2:99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lorusso A, Vincent AL, Harland ML et al Genetic and antigenic characterization of H1 influenza viruses from United States swine from 2008. J Gen Virol 2011; 92:919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Corzo CA, Culhane M, Juleen K et al Active surveillance for influenza A virus among swine, midwestern United States, 2009‐2011. Emerg Infect Dis 2013; 19:954–960. Epub 2013/06/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ducatez MF, Hause B, Stigger‐Rosser E et al Multiple reassortment between pandemic (H1N1) 2009 and endemic influenza viruses in pigs, United States. Emerg Infect Dis 2011; 17:1624–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alllerson M, Deen J, Detmer SE et al The impact of maternally derived immunity on influenza A virus transmission in neonatal pig populations. Vaccine 2013; 31:500–505. Epub 2012/11/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Romagosa A, Allerson M, Gramer M et al Vaccination of influenza a virus decreases transmission rates in pigs. Vet Res 2011; 42:120. doi: 10.1186/1297-9716-42-120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ciacci‐Zanella JR, Vincent AL, Prickett JR, Zimmerman SM, Zimmerman JJ. Detection of anti‐influenza A nucleoprotein antibodies in pigs using a commercial influenza epitope‐blocking enzyme‐linked immunosorbent assay developed for avian species. J Vet Diagn Invest 2010; 22:3–9. [DOI] [PubMed] [Google Scholar]
- 20. Spackman E, Suarez DL. Type A influenza virus detection and quantitation by real‐time RT‐PCR. Methods Mol Biol 2008; 436:19–26. Epub 2008/03/29. [DOI] [PubMed] [Google Scholar]
- 21. Vincent AL, Lager KM, Ma WJ et al Evaluation of hemagglutinin subtype 1 swine influenza viruses from the United States. Vet Microbiol 2006; 118:212–222. [DOI] [PubMed] [Google Scholar]
- 22. Direksin K, Joo H, Goyal SM. An immunoperoxidase monolayer assay for the detection of antibodies against swine influenza virus. J Vet Diagn Invest 2002; 14:169–171. [DOI] [PubMed] [Google Scholar]
- 23. Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR. Universal primer set for the full‐length amplification of all influenza A viruses. Arch Virol 2001; 146:2275–2289. [DOI] [PubMed] [Google Scholar]
- 24. Bandelt HJ, Forster P, Rohl A. Median‐joining networks for inferring intraspecific phylogenies. Mol Biol Evol 1999; 16:37–48. [DOI] [PubMed] [Google Scholar]
- 25. Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS‐MODEL workspace: a web‐based environment for protein structure homology modelling. Bioinformatics 2006; 22:195–201. [DOI] [PubMed] [Google Scholar]
- 26. Deem MW, Pan KY. The epitope regions of H1‐subtype influenza A, with application to vaccine efficacy. Protein Eng Des Sel 2009; 22:543–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Caton AJ, Brownlee GG, Yewdell JW, Gerhard W. The antigenic structure of influenza virus A/PR/8/34 hemagglutinin (H‐1 subtype). Cell 1982; 31:417–427. [DOI] [PubMed] [Google Scholar]
- 28. Rambaut A, Holmes E. The early molecular epidemiology of the swine‐origin A/H1N1 human influenza pandemic. PLoS Curr 2009; 1:RRN1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Furuse Y, Shimabukuro K, Odagiri T et al Comparison of selection pressures on the HA gene of pandemic (2009) and seasonal human and swine influenza A H1 subtype viruses. Virology 2010; 405:314–321. [DOI] [PubMed] [Google Scholar]
- 30. Hensley SE, Das SR, Bailey AL et al Hemagglutinin receptor binding avidity drives influenza A virus antigenic drift. Science 2009; 326:734–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kuroda M, Katano H, Nakajima N et al Characterization of quasispecies of pandemic 2009 influenza A virus (A/H1N1/2009) by de novo sequencing using a next‐generation DNA sequencer. PLoS One 2010; e10256. doi: 10.1371/journal.pone.0010256 [DOI] [PMC free article] [PubMed] [Google Scholar]