

Abstract
Populus (poplar) and Salix (willow) are sister genera in the Salicaceae family. In both lineages extant species are predominantly diploid. Genome analysis previously revealed that the two lineages originated from a common tetraploid ancestor. In this study, we conducted a syntenic comparison of the corresponding 19 chromosome members of the poplar and willow genomes. Our observations revealed that almost every chromosomal segment had a parallel paralogous segment elsewhere in the genomes, and the two lineages shared a similar syntenic pinwheel pattern for most of the chromosomes, which indicated that the two lineages diverged after the genome reorganization in the common progenitor. The pinwheel patterns showed distinct differences for two chromosome pairs in each lineage. Further analysis detected two major interchromosomal rearrangements that distinguished the karyotypes of willow and poplar. Chromosome I of willow was a conjunction of poplar chromosome XVI and the lower portion of poplar chromosome I, whereas willow chromosome XVI corresponded to the upper portion of poplar chromosome I. Scientists have suggested that Populus is evolutionarily more primitive than Salix. Therefore, we propose that, after the “salicoid” duplication event, fission and fusion of the ancestral chromosomes first give rise to the diploid progenitor of extant Populus species. During the evolutionary process, fission and fusion of poplar chromosomes I and XVI subsequently give rise to the progenitor of extant Salix species. This study contributes to an improved understanding of genome divergence after ancient genome duplication in closely related lineages of higher plants.
Keywords: chromosomal rearrangement, genome duplication, genome divergence, Populus, Salix
Introduction
Angiosperms, comprising approximately 420,000 extant species (Govaerts 2001), are the largest and most diverse group of terrestrial plants, and exhibit remarkable diversity in morphology, adaptations, genome size, and chromosome number. Modern angiosperms are derived from a common ancestor during the early Cretaceous period in about 150–300 million years ago (Ma) based on fossil records (Friis et al. 2006). Genomic analysis has revealed that the angiosperm genome has experienced one or more episodes of polyploidy in their evolutionary history (De Bodt et al. 2005; Tate et al. 2005; Soltis et al. 2008). For instance, genomes of modern eudicots share a common paleohexaploidization event (Jaillon et al. 2007), followed by additional events of lineage-specific paleotetraploidizations in some taxa, such as one or more rounds in different legume lineages (Cannon et al. 2006, 2014) and two rounds in Arabidopsis (Simillion et al. 2002; Blanc et al. 2003). Subsequent to the paleopolyploidizations events, substantial chromosomal reshuffling during genome stabilization has played a critical role in speciation in closely related lineages (Town et al. 2006; Tuskan et al. 2006; Velasco et al. 2010; Cheng et al. 2013). For example, genome comparison of Brassica oleracea and its sister species B. rapahas has revealed that 19 major and numerous fine-scale chromosome rearrangements contribute to the divergence of the two species (Liu et al. 2014). In addition, fusion of two chromosomes into a single chromosome is known to have occurred in many closely related plant taxa. Particularly striking examples have been observed in grasses. Five, three, and two dysploid reductions (i.e., reduction in chromosome number) are reported to have taken place by a process during which an entire chromosome is inserted by its telomeres into the centromeric region of another chromosome in finger millet (Srinivasachary et al. 2007), sorghum, and Triticeae (Luo et al. 2009), respectively. This process is suggested to be the dominant evolutionary mechanism in grass family (Luo et al. 2009).
Populus (poplar) and Salix (willow) are sister genera in the Salicaceae, which is widely distributed in the Northern Hemisphere (Heywood 1993). The combination of specific characters, such as rapid growth rate, ease of vegetative propagation, predisposition to hybridize, and utility of the wood, have long made them popular for human utilization (Dickmann and Kuzovkina 2008). Poplar and willow each possess a relatively small genome, which together with increased research attention and the rapidly growing availability of genomic resources, leading to the emergence of Salicaceae species as model systems for genetic research on woody plants. The genomes of willow and poplar species have been sequenced and are publically available (Tuskan et al. 2006; Dai et al. 2014). Genomic analysis has revealed that the two lineages share a common whole-genome duplication event, termed the “salicoid” duplication, which occurred around 58 Ma (Tuskan et al. 2006; Dai et al. 2014). Salix and Populus are indicated to have diverged from a common paleotetraploid ancestor approximately 6 Ma after the “salicoid” duplication event (Tuskan et al. 2006; Dai et al. 2014). Cytogenetic studies show that both genera predominantly comprise diploids and typically have a basic haploid chromosome number of n = 19 (Blackburn and Harrison 1924). Thus, after the “salicoid” duplication event, genome diploidization is presumed to have occurred during the evolutionary process, accompanied by intensive chromosomal reshuffling. Sequencing of the genome of Populus trichocarpa has confirmed the intensive chromosomal rearrangements and the tandem fusions of ancestral chromosome blocks, for example, chromosome I in modern poplar is indicated to be derived from multiple rearrangements involving three major tandem fusions (Tuskan et al. 2006). Comparative genetic mapping indicates that fission or fusion of chromosome members has occurred in willow and poplar (Berlin et al. 2010). Moreover, multiple lines of evidence indicate that different autosomes have evolved into sex chromosomes in the two lineages subsequent to their divergence (Hou et al. 2015; Pucholt et al. 2015). However, a genome-wide comparison of sequence divergence between Populus and Salix has not been conducted previously.
Using the sequenced individual as the maternal parent, we previously established a large mapping pedigree for Salix suchowensis, and constructed highly saturated genetic maps for the maternal and paternal parents separately (Hou et al. 2015). Based on the established maps, sequence scaffolds of S. suchowensis were anchored along each chromosome for the female and male willow (Hou et al. 2015). In the present study, we integrated the sequence scaffolds anchored along each chromosome in the female and male S. suchowensis. Together with the anchored sequence scaffolds in the P. trichocarpa genome (Tuskan et al. 2006; Yin et al. 2008), it is feasible to perform genome-wide syntenic comparison of the corresponding chromosomes in willow and poplar. Using the most up-to-date genome assemblies for willow and poplar, we conducted intragenome and intergenome syntenic comparisons among the 19 chromosome members to elucidate the genomic mechanism that contributed to the divergence of Populus and Salix. The results contribute valuable information to improve our understanding of genome dynamics in closely related plant lineages subsequent to ancient genome duplication.
Materials and Methods
Integration of Sequence Scaffolds of S. suchowensis
In a previous study, highly saturated genetic maps for the maternal and paternal parents were built separately for a full-sib mapping pedigree of S. suchowensis by using single-nucleotide polymorphism (SNP) markers genotyped by Illumina resequencing and amplified fragment length polymorphism (AFLP) markers, in which the sequenced individual was the maternal parent (Hou et al. 2015). Willow species are outbreeding, and heterozygosity varies among loci on each chromosome. In map construction, if the genotype of a SNP marker is lm × ll (heterozygous in the female, homozygous in the male), the marker will be mapped on the maternal map; if the genotype is nn × np (heterozygous in the male, homozygous in the female), the corresponding marker will be mapped on the paternal map. In mapping the sequence scaffolds, if a scaffold only contains mapped lm × ll loci, it will be anchored on a chromosome in the female willow (female-mapped scaffold); if a scaffold merely contains mapped nn × np loci, it will be anchored on a chromosome in the male willow (male-mapped scaffold); if a scaffold contains both the mapped lm × ll and the mapped nn × np loci, it will be simultaneously anchored on chromosomes in the female and male willows (female-male-mapped scaffold). By using female-male-mapped scaffolds as references, we integrated female-mapped scaffolds and male-mapped scaffolds on each chromosome based on their relative genetic distances from the shared female-male-mapped scaffolds. Given the recombination heterogeneity between the female and male willows, the relative genetic distances in the male were scaled according to those in the female. Willow sequence assemblies were generated by Dai et al. (2014), which were conducted independently without taking poplar genome as reference. In the chromosome reconstructions, the non-capture gaps between the anchored sequence scaffolds were represented by 100 letters of “N”. Chromosome identities of willow were designated based on sequence homology to chromosome sequences of P. trichocarpa (http://genome.jgi.doe.gov/pages/dynamicOrganismDownload.jsf?organism=Ptrichocarpa, last accessed June 8, 2016).
Intragenome and Intergenome Syntenic Comparisons
Genome sequences and protein sequences of S. suchowensis were downloaded from the website (http://115.29.234.170/willow, last accessed June 8, 2016). The corresponding information for P. trichocarpa was retrieved from the Joint Genome Institute, United States Department of Energy website (http://genome.jgi.doe.gov/pages/dynamicOrganismDownload.jsf?organism=Ptrichocarpa, last accessed June 8, 2016). Intragenome and intergenome syntenic comparisons were performed with MCScanX (Wang et al. 2012) and conducted according to the pipeline as described by Hou et al. (2015). Syntelogs related to the “salicoid” duplication were further screened based on the Ks values, which were calculated by using KaKs_Calculator 2.0 (Wang et al. 2010). Range of Ks values related to the “salicoid” duplication were determined based on the Ks values distribution for willow and poplar separately. According to Cui et al. (2006), Ks values that were <0.005 were discarded. Collinearity charts were drawn using the software package VGSC (a web-based Vector Graph toolkit for displaying genome Synteny and Collinearity; available at http://bio.njfu.edu.cn/vgsc-web/, last accessed June 8, 2016) (Xu et al. 2016).
Experimental Verification of Interchromosomal Rearrangements Between Willow and Poplar
The chromosomes in Salicaceae species are typically metacentric and small (Blackburn and Harrison 1924; Nakajima 1937). Divergence took place over 50 Ma, therefore fluorescent in situ hybridization (FISH) analysis was unsuitable to obtain cytological evidence for interchromosomal rearrangements between willow and poplar. Based on the homology between chromosomes I and XVI of willow and poplar, we divided poplar chromosome I into regions A and B (boundary at 21,383,037 bp), and designated poplar chromosome XVI as region C (fig. 3). The boundary between homologous regions A and C in willow was at 11,107,087 bp on willow chromosome I, and the homologous region B in willow corresponded to willow chromosome XVI (fig. 3). For verification of the interchromosomal rearrangements between these two genomes, we developed simple sequence repeat (SSR) markers in regions A, B, and C on poplar chromosomes I and XVI (supplementary table S1, Supplementary Material online), and mapped these markers to the genetic map of willow to verify their correspondence in the willow genome. First, SSRs on poplar chromosomes I and XVI obtained from the SSR primers database established by Yin et al. (2009) were searched against the willow genome sequence. The SSR primer pairs with sequences conserved in willow and poplar were identified, and a subset of primer pairs were selected and synthesized by the Generay Biotechnology Company, Shanghai, China. Marker segregation was tested using DNA isolated from the two parents and six progeny in the S. suchowensis mapping population of Hou et al. (2015). The SSR primer pairs that generated segregating loci were genotyped by using 92 progeny from the same pedigree. For SSR genotyping, GeneMapper version 3.7 Software, (Applied Biosystems, Foster City, CA, USA) was used to score the amplicons. The segregating SSR markers were linked to the willow linkage map built by Hou et al. (2015) with MapMaker version 3.0 (Lander et al. 1987). Examination of whether poplar-origin SSRs were located in the corresponding regions on the genetic map and reconstructed pseudomolecules of willow enabled validation of major interchromosomal rearrangements between poplar and willow.
Fig. 3.—
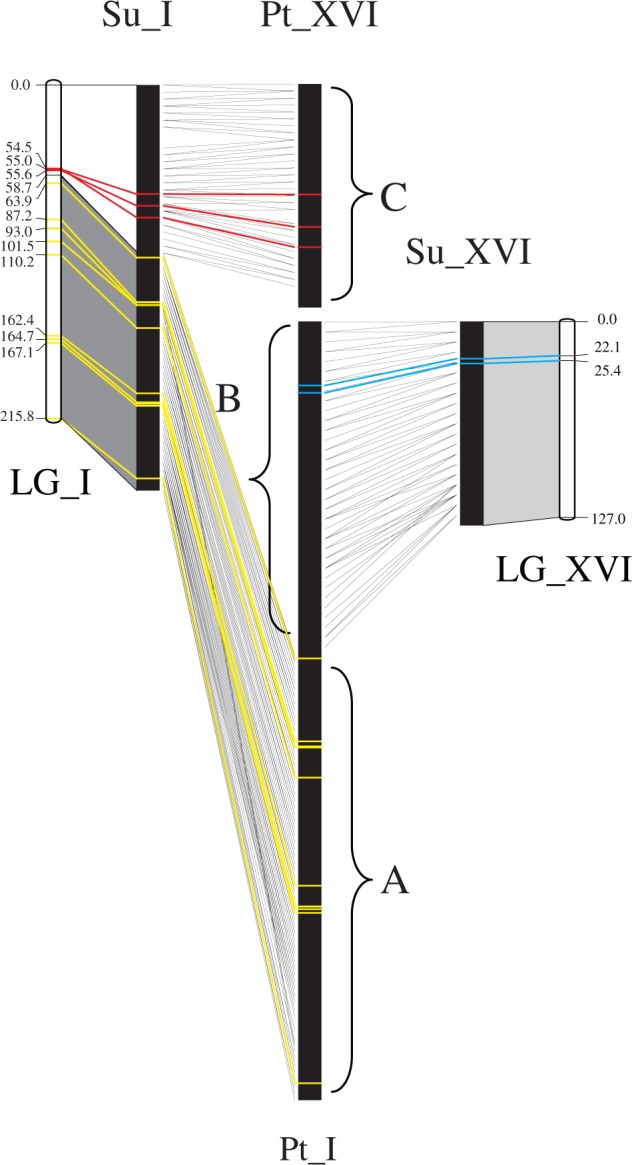
Interchromosomal rearrangements associated with willow and poplar chromosomes I and XVI. Note: Su: Salix suchowensis; Pt: Populus trichocarpa; LG: linkage group; A, B, and C represent regions A, B, and C in poplar, respectively; dark gray, light gray, and white regions correspond to homologous regions A, B, and C in willow, respectively; yellow, blue, and red lines represent the universal SSR markers located in regions A, B, and C, respectively.
Results
Integration of Sequence Scaffolds of S. suchowensis
Integration analysis enabled a total of 1,025 sequence scaffolds to be anchored on the 19 chromosomes (Assembly ver2.0; http://bio.njfu.edu.cn/willow_chromosome, last accessed June 8, 2016). The anchored sequence scaffolds covered a total physical distance of 229.2 Mb, which accounted for about 75.4% (229.2/303.8 Mb) of the total sequence scaffold assemblies of S. suchowensis generated by Dai et al. (2014). The number of sequence scaffolds on each chromosome and their physical spans varied greatly among the chromosome members. At the extremes, chromosome XII contained the least number of anchored sequence scaffolds (44) and spanned the shortest physical length (7.3 Mb). By contrast, chromosome I contained the highest number of sequence scaffolds (118) and covered the longest physical distance (26.3 Mb), which was about double the average physical length of the chromosome reconstructions. This biggest chromosome was commonly observed in many previous cytological studies. Size of this gigantic chromosome is reported to be almost double the size of the other chromosomes in various Saliaceae species (Blackburn and Harrison 1924; Erlanson and Hermann 1927; Müntzing 1936; Nakajima 1937; Johnsson 1940). According to Dai et al. (2014), the number of genes contained in each chromosome reconstruction ranged from 684 to 2,642, with a total number of 24,931 genes (table 1), which covered 93.7% of the total number of genes in the willow genome.
Table 1.
Summary of the Sequence Scaffolds Anchored on Each Salix suchowensis Chromosome
| Chromosome No. | Number of Anchored Scaffolds | Length of the Chromosome Reconstructions (bp) | Average Length of Anchored Scaffolds (bp) | Number of Genes Contained in Each Chromosome |
|---|---|---|---|---|
| I | 118 | 26,293,657 | 222,828 | 2,642 |
| II | 93 | 16,228,781 | 174,503 | 1,844 |
| III | 24 | 12,249,699 | 510,404 | 1,399 |
| IV | 46 | 13,408,292 | 291,485 | 1,422 |
| V | 44 | 13,917,003 | 316,296 | 1,581 |
| VI | 25 | 16,519,934 | 660,797 | 1,884 |
| VII | 48 | 8,250,254 | 171,880 | 856 |
| VIII | 23 | 12,035,713 | 523,292 | 1,444 |
| IX | 13 | 9,584,840 | 737,295 | 1,174 |
| X | 30 | 14,178,701 | 472,623 | 1,725 |
| XI | 99 | 11,081,354 | 111,933 | 1,011 |
| XII | 44 | 7,280,141 | 165,458 | 684 |
| XIII | 100 | 10,740,571 | 107,406 | 1,061 |
| XIV | 20 | 10,550,056 | 527,503 | 1,260 |
| XV | 71 | 8,695,248 | 122,468 | 964 |
| XVI | 29 | 13,231,175 | 456,247 | 1,606 |
| XVII | 70 | 8,535,887 | 121,941 | 841 |
| XVIII | 44 | 8,151,547 | 185,262 | 797 |
| XIX | 84 | 8,222,739 | 97,890 | 736 |
| Total | 1,025 | 229,155,592 | 5,977,511 | 24,931 |
Intragenome and Intergenome Syntenic Comparisons
According to Tang et al. (2008), the overall median Ks value of the duplicated genes related to the γ triplication event is 1.54, and that associated with the “salicoid” duplication is 0.27 for Populus. By plotting the Ks values of syntelogs, the highly peaks appear between 0.00 and 1.00 both in willow and poplar, and the distributions can model as normals (supplementary fig. S1, Supplementary Material online). We consider Ks values in range of are associated with the “salicoid” duplication. It was determined that syntelogs with Ks values in range of 0.037–0.734 and 0.005–0.607 were associated with the “salicoid” duplication in willow and poplar, respectively (supplementary table S2, Supplementary Material online). Collinearity charts were drawn by using syntelogs with Ks values in the corresponding range for each genome. According to the pinwheel charts of Populus, the distribution of syntenic blocks among chromosome members are generally consistent with that revealed in the previous report (Tuskan et al. 2006), only with a few fine-scale inconsistencies, which might due to the changes in the updated Populus genome assembly. Similar as that in Populus, many chromosomal segments are also found to have a parallel paralogous segment in Salix genome. Comparison of the pinwheel charts of the corresponding chromosomes between poplar and willow revealed that the two genomes shared almost identical pinwheel patterns for 15 chromosomes, consisting of chromosomes II, IV, V, VII, VIII, IX, X, XI, XII, XIII, XIV, XV, XVII, XVIII, and XIX (supplementary fig. S2, Supplementary Material online). However, the pinwheel patterns differed between the genomes for four chromosomes, namely chromosomes I, III, VI, and XVI (supplementary fig. S2, Supplementary Material online). In a counterclockwise direction of the pinwheel charts, the upper portion of poplar chromosome I had a large paralogous segment on poplar chromosome III, and vice versa. By contrast, the upper portion of willow chromosome I contained a large paralogous segment on willow chromosome VI, and vice versa (fig. 1). Poplar chromosome XVI contained a major paralogous segment located on poplar chromosome VI, and vice versa. Willow chromosome XVI was revealed to have a major paralogous segment on willow chromosome III, and vice versa (fig. 1). The above differences in the genomes of willow and poplar can be hypothesized to be caused by rearrangements involving chromosomes I and XVI. To verify this hypothesis, we conducted syntenic comparisons for the corresponding chromosomes in the willow and poplar genome (fig. 2). This analysis revealed macrosynteny for most of the corresponding chromosome members. However, no homology was detected between poplar chromosome XVI and willow chromosome XVI, and similarly between the upper portion of poplar chromosome I and the upper portion of willow chromosome I. Referring to the pinwheel charts of these chromosomes, poplar chromosome XVI showed high homology with the upper portion of willow chromosome I, and willow chromosome XVI showed high homology with the upper portion of poplar chromosome I (fig. 2). This finding provided evidence for chromosome fission and fusion involving chromosomes I and XVI between the two genomes. In addition to these major interchromosomal rearrangements, both the intragenome and intergenome syntenic comparisons indicated that several chromosomes have experienced minor reorganizational exchanges between these two lineages (fig. 2 and supplementary fig. S2, Supplementary Material online).
Fig. 1.—

Intragenome syntenic pinwheel charts for willow and poplar chromosomes I, III, VI and XVI. Note: Su: Salix suchowensis; Pt: Populus trichocarpa; Roman numerals represent the chromosome number; red ellipses indicate the major syntenic inconsistencies in the corresponding willow and poplar chromosomes.
Fig. 2.—

Syntenic comparison of the homologous chromosomes between Salix suchowensis and Populus trichocarpa. Note: Su: S. suchowensis; Pt: P. trichocarpa; Roman numerals represent the chromosome number.
Experimental Verification of Interchromosomal Rearrangements Between Willow and Poplar
By blasting a total of 2,000 poplar SSR primers, which were selected from regions A, B, and C in the poplar SSR primers database (Yin et al. 2009), against the willow genome sequence, we detected 56 universal SSR primers that were transferable between willow and poplar. Twenty-five universal SSR primers were located in poplar region A and all were physically mapped to the homologous region A in willow; 16 universal SSR primers were located in poplar region B and all were physically mapped to the homologous region B in willow; the remaining 15 universal SSR primers were located in poplar region C and all were physically mapped to the homologous region C in willow (fig. 3 and supplementary table S2, Supplementary Material online).
The universal SSR primer pairs were synthesized and subjected to a segregation test in the pedigree mapping population of willow. Ten universal primers from region A, two universal primers from region B and three universal primers from region C generated segregating loci in willow. The SSR primer pairs that generated segregating loci were genotyped with 92 progeny from the willow mapping pedigree, and the obtained SSR markers were integrated into the genetic map of willow (Hou et al. 2015). On the integrated map, the boundary between the genetic intervals corresponding to homologous region A and homologous region C in willow was at 58.7cM on LG_I, while LG_XVI covered the complete homologous region B in willow (fig. 3). Mapping results showed that the 10 segregating primers from poplar region A were mapped between 63.9 and 215.8 cM on LG_I of willow, the two segregating primers from poplar region B were mapped between 22.1and 25.4 cM on LG_XVI of willow, and the three segregating primers from poplar region C were mapped between 54.5and 55.6 cM on LG_I of willow (fig. 3). Therefore, all of the segregating SSR markers that originated from the target region in poplar were mapped in the expected region on the genetic map of willow. This finding verified that the detected major interchromosomal rearrangements did not result from misconnection of chromosomal blocks across the relevant chromosome members in either of the two genomes.
Discussion
Modern poplars and willows are generally diploids, and possess a basic haploid chromosome number of n = 19. Analysis of the poplar genome sequence reveals that nearly every chromosomal segment in Populus genome have a parallel paralogous segment elsewhere in the genome as a result of the “salicoid” duplication event according to Tuskan et al. (2006). The collinearity of genetic maps among multiple Populus species suggests that genome reorganization occurred before the evolution of extant Populus species (Tuskan et al. 2006). Alignment of Salix linkage maps with the Populus genome sequence reveals macrosynteny between the willow and poplar genomes (Hanley et al. 2006; Berlin et al. 2010). A number of studies have confirmed that Populus and Salix share this lineage-specific “salicoid” duplication (Hanley et al. 2006; Tuskan et al. 2006; Berlin et al. 2010; Dai et al. 2014). Thus these two lineages originate from a common tetraploid ancestor. By contrast, modern Salicaceae species generally appear as diploids. After the “salicoid” duplication, genome diploidization must have occurred through a genome reorganization process in the genome of their progenitor. If genome reorganization occurs independently after the divergence of willow and poplar, the distribution of syntenic blocks among chromosome members between these two lineages should be different by large. However, similar distribution pattern was observed on the majority of chromosome members between these two genomes (supplementary fig. S2, Supplementary Material online), which indicated that divergence of Salix and Populus took place after the genome reorganization in their common progenitor. On the pinwheel charts of both willow and poplar, it was noticeable that most of the duplicated segments were distributed in either the central or the telomeric region of the other chromosome. Luo et al. (2009) reported that in Aegilops tauschii major translocation breakpoints occurred in the telomeric or centromeric regions, and resulted in fusion of telomeric with telomeric, telomeric with centromeric, or centromeric with centromeric breakpoints. Therefore, we speculate that a similar mechanism also trigger the genome reorganization in the progenitor of modern willows and poplars.
Apart from the similarities between the two genomes, we detected two major interchromosomal rearrangements associated with chromosomes I and XVI of willow and poplar, which indicated that chromosomal fission and fusion had occurred involving the associated chromosomes between the two genomes (fig. 3). The rearrangements might have arisen through a process (“poplar-to-willow”) that involved poplar chromosome I breaking into two parts; subsequently, the lower portion (region A) was terminally joined with poplar chromosome XVI (region C), giving rise to willow chromosome I (fig. 2), whereas the upper portion (region B) of poplar chromosome I gave rise to willow chromosome XVI. Alternatively, the rearrangements might have resulted from a process (“willow-to poplar”) that involved willow chromosome I breaking into two parts; the lower portion (homologous region A) was terminally joined with willow chromosome XVI (homologous region B), giving rise to chromosome I in poplar (fig. 2), and the upper portion (homologous region C) of willow chromosome I gave rise to poplar chromosome XVI. Fusion of chromosomes has occurred frequently during the evolution of higher plants, such as in Arabidopsis thaliana, in which the n = 5 genome is considered to be derived from an ancestral n = 8 genome. This process involved at least three chromosomal fusions that resulted from reciprocal translocations between two ancestral chromosomes, after a pericentric inversion converted one chromosome into an acrocentric chromosome (Lysak et al. 2006). An additional striking example among grasses has been reported, in which chromosomal fusions are proposed to have occurred through a process in which an entire chromosome was inserted by its telomeres into the centromeric region of another chromosome (Srinivasachary et al. 2007; Luo et al. 2009). Such a process also led to the transition from nine ancestral chromosomes to the n = 17 karyotype of extant Pyreae. In that instance, chromosome XV was proposed to have arisen from the translocation of an entire copy of chromosome IX into the centromeric region of chromosome VIII after a relatively recent genome-wide duplication in the apple genome (Velasco et al. 2010). In the present study, we propose that a major translocation breakpoint occurred in the centromeric region of poplar chromosome I, and subsequently the lower portion fused with a telomeric breakpoint of poplar chromosome XVI, which gave rise to willow chromosome I. Thus, the proposed mechanism of chromosomal fusion that contributed to the divergence of Salix and Populus differed from those responsible in Arabidopsis, extant Pyreae and grasses.
Although genome comparisons reveal that Salix and Populus share the same large-scale genomic history, the phylogenetic history of Salicaceae is not altogether clear. Dorn (1976) and Skvortsov (1999) suggested that Populus is evolutionarily more primitive than Salix, and certain morphological features point towards willows having arisen from the most advanced group in section Populus (Dickmann and Kuzovkina 2008). Therefore, we propose that the “poplar-to-willow” process is more likely to underlie the divergence of these sister genera. As envisaged in the “poplar-to-willow” process, fission and fusion of the ancestral chromosomes first gave rise to the diploid progenitor of modern Populus after the “salicoid” duplication event. Subsequently, chromosomal fission and fusion involving poplar chromosomes I and XVI was manifested in the progenitor of modern Salix. According to Dai et al. (2014), this additional round of chromosomal rearrangements causing Salix diverged from Populus was estimated to have occurred approximately 6 Ma after the “salicoid” duplication event based on the 4DTv values of the orthologous pairs between these two lineages. In addition to the major interchromosomal rearrangements, several chromosomes are indicated to have experienced minor reorganizational exchanges within and between the two genomes, which might have resulted from fine-scale chromosomal rearrangements or be associated with the activities of transposons. However, the observed fine-scale rearrangements might be artifacts due to limitation in the precision of the genetic maps.
In conclusion, in this study we obtained evidence for the genomic mechanism that contributed to the divergence of the willow and poplar lineages after the “salicoid” duplication event. The results contribute to an improved understanding of how chromosomal rearrangements may promote the divergence of closely related lineages of higher plants.
Supplementary Material
Supplementary figures S1 and S2 and tables S1 and S2 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
This work is supported by the National Basic Research Project [2012CB114505], and the Natural Science Foundation of China [31561123001, 31570662]. It is also enabled by the Program for Innovative Research Team of the Educational Department of China and in Universities of Jiangsu Province, and the Priority Academic Program Development (PAPD) program and the Doctorate Fellowship Foundation of Nanjing Forestry University.
Literature Cited
- Berlin S, Lagercrantz U, von Arnold S, Öst T, Rönnberg-Wästljung AC. 2010. High-density linkage mapping and evolution of paralogs and orthologs in Salix and Populus. BMC Genom. 11:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn KB, Harrison JH. 1924. A preliminary account of the chromosomes and chromosome behaviour in the Salicaceae. Ann Bot-Lond. 38:361–378. [Google Scholar]
- Blanc G, Hokamp K, Wolfe KH. 2003. A recent polyploidy superimposed on older large-scale duplications in the Arabidopsis genome. Genome Res. 13:137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon SB, et al. 2006. Legume genome evolution viewed through the Medicago truncatula and Lotus japonicus genomes. Proc Natl Acad Sci U S A. 103:14959–14964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon SB, et al. 2014. Multiple polyploidy events in the early radiation of nodulating and nonnodulating legumes. Mol Biol Evol. 32:193–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F, et al. 2013. Deciphering the diploid ancestral genome of the Mesohexaploid Brassica rapa. Plant Cell 25:1541–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L, et al. 2006. Widespread genome duplications throughout the history of flowering plants. Genome Res. 16:738–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, et al. 2014. The willow genome and divergent evolution from poplar after the common genome duplication. Cell Res. 24:1274–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bodt S, Maere S, Van de Peer Y. 2005. Genome duplication and the origin of angiosperms. Trends Ecol Evol. 20:591–597. [DOI] [PubMed] [Google Scholar]
- Dickmann DI, Kuzovkina J. 2008. Poplars and willows in the world In: Isebrands JG, Richardson J, editors. Poplars and willows in the world, meeting the needs of society and the environment. Rome, Italy: Food and Agricultural Organization of the United States/International Poplar Commission, Working paper IPC/9-2. [Google Scholar]
- Dorn RD. 1976. A synopsis of American Salix. Can J Bot. 54:2769–2789. [Google Scholar]
- Erlanson EW, Hermann FJ. 1927. The morphology and cytology of perfect flowers in Populus tremuloides Michx. Mich Acad Sci Arts Lett. 8:97–110. [Google Scholar]
- Friis EM, Pedersen KR, Crane PR. 2006. Cretaceous angiosperm flowers: innovation and evolution in plant reproduction. Palaeogeogr Palaeoclimatol. 232:251–293. [Google Scholar]
- Govaerts R. 2001. How many species of seed plants are there? Taxon 50:1085–1090. [Google Scholar]
- Hanley SJ, Mallott MD, Karp A. 2006. Alignment of a Salix linkage map to the Populus genomic sequence reveals macrosynteny between willow and poplar genomes. Tree Genet Genomes. 3:35–48. [Google Scholar]
- Heywood VH. 1993. Flowering plants of the world. London: B.T. Batsford Ltd. [Google Scholar]
- Hou J, et al. 2015. Different autosomes evolved into sex chromosomes in the sister genera of Salix and Populus. Sci Rep-UK. 5:9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaillon O, et al. 2007. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449:463–467. [DOI] [PubMed] [Google Scholar]
- Johnsson H. 1940. Cytological studies of diploid and triploid Populus tremula and of crosses between them. Hereditas 26:321–352. [Google Scholar]
- Lander ES, et al. 1987. MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:174–181. [DOI] [PubMed] [Google Scholar]
- Liu S, et al. 2014. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat Commun. 5:3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo MC, et al. 2009. Genome comparisons reveal a dominant mechanism of chromosome number reduction in grasses and accelerated genome evolution in Triticeae. Proc Natl Acad Sci U S A. 106:15780–15785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lysak MA, et al. 2006. Mechanisms of chromosome number reduction in Arabidopsis thaliana and related Brassicaceae species. Proc Natl Acad Sci U S A. 103:5224–5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müntzing A. 1936. The chromosomes of a giant Populus tremula. Hereditas 21:383–393. [Google Scholar]
- Nakajima G. 1937. Cytological studies in some dioecious plants. Cytologia 1:282–292. [Google Scholar]
- Pucholt P, Rönnberg-Wästljung AC, Berlin S. 2015. Single locus sex determination and female heterogamety in the basket willow (Salix viminalis L.). Heredity 114:575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simillion C, Vandepoele K, Van Montagu MCE, Zabeau M, de Peer YV. 2002. The hidden duplication past of Arabidopsis thaliana. Proc Natl Acad Sci U S A. 99:13627–13632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skvortsov AK. 1999. Willows of Russia and adjacent countries Taxonomical and Geographical Revision (English translation of 1968 Russian edition). Joensuu, Finland: University of Joensuu. [Google Scholar]
- Soltis DE, Bell CD, Kim S, Soltis PS. 2008. Origin and early evolution of angiosperms. Ann N Y Acad Sci. 1133:3–25. [DOI] [PubMed] [Google Scholar]
- Srinivasachary Dida MM, Gale MD, Devos KM. 2007. Comparative analyses reveal high levels of conserved colinearity between the finger millet and rice genomes. Theor Appl Genet. 115:489–499. [DOI] [PubMed] [Google Scholar]
- Tang H, et al. 2008. Unraveling ancient hexaploidy through multiply-aligned angiosperm gene maps. Genome Res. 18:1944–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate JA, Soltis DE, Soltis PS. 2005. Polyploidy in plants In: Gregory TR, editor. The evolution of the genome. San Diego: Elsevier Academic Press; p. 371-426. [Google Scholar]
- Town CD, et al. 2006. Comparative genomics of Brassica oleracea and Arabidopsis thaliana reveal gene loss, fragmentation, and dispersal after polyploidy. Plant Cell 18:1348–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuskan GA, et al. 2006. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313:1596–1604. [DOI] [PubMed] [Google Scholar]
- Velasco R, et al. 2010. The genome of the domesticated apple (Malus×domestica Borkh.). Nat Genet. 42:833–839. [DOI] [PubMed] [Google Scholar]
- Wang D, Zhang Y, Zhang Z, Zhu J, Yu J. 2010. KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteom Bioinformatics 8:77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, et al. 2012. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40:e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, et al. 2016. VGSC: a web-based Vector Graph toolkit of genome Synteny and Collinearity. Biomed Res Int. 2016 http://dx.doi.org/10.1155/2016/7823429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin T, et al. 2008. Genome structure and emerging evidence of an incipient sex chromosome in Populus. Genome Res. 18:422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin T, et al. 2009. Microsatellite primer resource for Populus developed from the mapped sequence scaffolds of the Nisqually-1 genome. New Phytol. 181:498–503. [DOI] [PubMed] [Google Scholar]