

Abstract
Background
Persistent thromboxane (TX) generation while receiving aspirin therapy is associated with an increased risk of cardiovascular events. The Reduction in Graft Occlusion Rates (RIGOR) study found that aspirin‐insensitive TXA 2 generation, indicated by elevated urine 11‐dehydro‐TXB 2 (UTXB 2) 6 months after coronary artery bypass graft surgery, was a potent risk factor for vein graft thrombosis and originated predominantly from nonplatelet sources. Our goal was to identify risks factors for nonplatelet TXA 2 generation.
Methods and Results
Multivariable modeling was performed by using clinical and laboratory variables obtained from 260 RIGOR subjects with verified aspirin‐mediated inhibition of platelet TXA 2 generation. The strongest variable associated with UTXB 2 6 months after surgery, accounting for 47.2% of the modeled effect, was urine 8‐iso‐prostaglandin (PG)F 2α, an arachidonic acid metabolite generated nonenzymatically by oxidative stress (standardized coefficient 0.442, P<0.001). Age, sex, race, lipid therapy, creatinine, left ventricular ejection fraction, and aspirin dose were also significantly associated with UTXB 2 (P<0.03), although they accounted for only 4.8% to 10.2% of the modeled effect. Urine 8‐iso‐PGF 2α correlated with risk of vein graft occlusion (odds ratio 1.67, P=0.001) but was not independent of UTXB 2. In vitro studies revealed that endothelial cells generate TXA 2 in response to oxidative stress and direct exposure to 8‐iso‐PGF 2α.
Conclusions
Oxidative stress–induced formation of 8‐iso‐PGF 2α is strongly associated with nonplatelet thromboxane formation and early vein graft thrombosis after coronary artery bypass graft surgery. The endothelium is potentially an important source of oxidative stress–induced thromboxane generation. These findings suggest therapies that reduce oxidative stress could be useful in reducing cardiovascular risks associated with aspirin‐insensitive thromboxane generation.
Keywords: aspirin, isoprostane, oxidative stress, thrombosis, thromboxane
Subject Categories: Thrombosis, Pathophysiology, Oxidant Stress, Cardiovascular Surgery, Platelets
Introduction
The cardioprotective property of aspirin derives principally from its antiplatelet effect resulting from the irreversible inhibition of the cyclooxygenase (COX)‐1 enzyme and consequent suppression of platelet thromboxane A2 (TXA2) generation. In addition to mediating the activation of the platelet in which it is formed, TXA2 is secreted and directly activates adjacent quiescent platelets and stimulates vasoconstriction via binding to surface thromboxane receptors.1 Several clinical studies have shown that patients with persistent TXA2 generation while taking aspirin therapy are at increased risk for atherothrombotic events, including death.2, 3
Aspirin therapy is standard of care after coronary artery bypass graft surgery (CABG) in large part because of its ability to decrease the rate of vein graft thrombosis by half during the first postoperative year.4 The Reductions in Graft Occlusion Rates (RIGOR) study investigated the hypothesis that failure of aspirin to adequately inhibit platelet activation would increase the incidence of early vein graft thrombosis after first‐time CABG.5 With the use of arachidonic acid platelet aggregometry, a specific indicator of platelet COX‐1 activity, aspirin therapy was found to suppress platelet TXA2 generation and inhibit aggregation in >95% and >99% of subjects at 3 days and 6 months after surgery, respectively. There was also no observable association between the rare failure to suppress platelet TXA2 generation and vein graft occlusion assessed 6 months after surgery. Despite effective suppression of platelet‐derived TXA2 generation by the use of aspirin, measurement of urine levels of the stable TXA2 metabolite 11‐dehydroTXB2 (UTXB2) revealed persistent total‐body TXA2 generation in 73% and 31% of subjects at 3 days and 6 months after surgery, respectively. Further, UTXB2 ≥450 pg/mg creatinine measured 6 months after CABG was associated with a 2.6‐fold increased risk of vein graft thrombosis compared with levels of <450 pg/mg creatinine.
These data indicate that a substantial percentage of patients taking aspirin continue to generate TXA2 6 months after CABG that originates from predominantly nonplatelet pathways and is associated with an increased risk of early vein graft thrombosis. The source and stimuli for nonplatelet TXA2 generation in patients with cardiovascular disease are largely unknown. The goal of this study was to use multivariable modeling to identify factors associated with nonplatelet TXA2 generation.
Materials and Methods
Subjects
The Reduction in Graft Occlusion Rates (RIGOR) study was a multicenter observational study of 368 subjects undergoing first‐time CABG between 2003 and 2006 that was designed to investigate the association between thrombotic risk factors and early saphenous vein graft occlusion. Patients were enrolled between October 2003 and October 2006 at 4 participating institutions: Johns Hopkins Hospital, Baltimore, MD; Christiana Hospital, Christiana, DE; Peninsula Regional Medical Center, Salisbury, MD; and Walter Reed Army Hospital, Washington, DC. Institutional human subject research review board approval was obtained at all participating sites, and all subjects provided written consent. A detailed description of the study design, patient characteristics, and principal findings has been previously published.5, 6, 7 Patients ≥18 years of age undergoing first‐time CABG with implantation of at least 1 saphenous vein graft were eligible for enrollment. Those with an anticipated requirement for postoperative oral anticoagulation or antiplatelet therapy other than aspirin were excluded, although those prescribed these agents for unforeseen postoperative conditions (eg, atrial fibrillation) continued in the study. All patients were administered aspirin (300–325 mg) within 24 hours of surgery. At hospital discharge, patients were given a supply of 325 mg enteric‐coated aspirin and instructed to take 1 tablet daily for 6 months unless directed otherwise by their physician. Pill counts were performed at each postoperative encounter. Demographic, historical, procedural, clinical, and laboratory data were recorded for all patients.
Platelet Studies
Platelet‐rich plasma was prepared from blood collected in 3.2% citrate by centrifugation at 100 rpm for 10 minutes, and the platelet count was adjusted to 180 000/mm3 by the addition of platelet‐poor plasma. Undiluted samples with a platelet count of <100 000/mm3 were excluded from analysis. Impedance platelet aggregometry was performed by stimulation with arachidonic acid (0.5 mmol/L), ADP (20 μmol/L), epinephrine (50 μmol/L), and collagen (1 μg/mL) with use of a Chrono‐Log Model 560CA aggregometer. The maximum aggregation response within 5 minutes was recorded in ohms. Subjects were considered to have aspirin‐induced suppression of significant platelet COX‐1 activity and TXA2 generation if arachidonic acid–induced platelet aggregation was absent as indicated by a value of ≤1 Ω (normal range in our laboratory for aspirin‐naïve subjects: 5–17 Ω) based on prior data demonstrating that suppression of platelet TXA2 generation by >99% is required to suppress arachidonic acid–induced aggregation by 95%.8 Shear‐dependent platelet aggregation was measured by using the Platelet Function Analyzer‐100® (PFA‐100) device (Siemens Healthcare Diagnostics) as previously described9 in whole blood collected in 3.8% citrate. Samples were tested with the collagen/ADP agonist cartridge, which assesses global platelet reactivity but is not affected by aspirin. Samples from subjects with a platelet count <50 000/m3 were excluded from analysis. Samples with nonclosure were assigned a closure time (CT) value of 300 seconds, the maximum measurable by the device.
Measurement of Urine Prostanoids
11‐Dehydro‐thromboxane B2 (TXB2) was measured in urine (UTXB2) with ELISA and expressed as a ratio to urine creatinine as previously described.9 Aspirin responsiveness based on this assay was defined as UTXB2 <400 pg/mg creatinine according to established criteria.10
Assessment of Saphenous Vein Graft Patency
Vein graft patency was assessed 6 months after CABG by the use of multidetector computed tomography coronary–angiography as previously described.6 Data from clinically driven invasive coronary angiograms could be used for the primary end point analysis if performed within 6 weeks of the anticipated 6‐month follow‐up visit or if it was the only assessment of vein graft patency before an adverse clinical end point. Multisegmented grafts were statistically considered as separate vein grafts according to the Society of Thoracic Surgeons criteria. Reconstructed images were analyzed by 2 blinded reviewers and classified as patent (containing stenoses of 0–75%), significantly diseased (containing stenoses of 76–99%), or occluded (containing a 100% stenosis). There was 98% concordance in assessment of vein graft patency between reviewers. In cases of discordance, a third reviewer adjudicated all vein grafts in that patient.
In Vitro Prostanoid Generation
Human umbilical vein endothelial cells were maintained in EGM‐2 medium (Lonza) at 37°C under 5% CO2. Confluent cells in 10‐cm plates were stimulated with hydrogen peroxide (Sigma‐Aldrich) and 8‐iso‐prostaglandin (PG)F2α (Cayman Chemical) for 1 hour at the indicated concentrations. Conditioned media were spiked with tetradeuterated 11‐dehydro‐TXB2, TXB2, and 8‐iso‐PGF2α (Cayman Chemical) as internal standards before solid phase extraction by using 50‐mg BondElut C18 reverse phase cartridges (Agilent Technologies) preconditioned with ethanol and water to concentrate eicosanoid species. Acidified samples (2% formic acid) were loaded and washed sequentially with water, 15% ethanol, and hexane and then eluted with ethyl acetate, dried, and resuspended in 15% acetonitrile. Calibrants were prepared in the same way over a 0.5‐ to 500‐ng/mL concentration range. Liquid chromatography/mass spectrometry (MS)–MS was performed by using a Dionex UltiMate 3000 UHPLC system in line with a TSQ Quantiva triple quadruple mass spectrometer (Thermo Fisher Scientific). Chromatographic separation was performed with a Kinetex C18 (1.7 μm, 100 Å) 50×100‐mm column maintained at 40°C. A multistep gradient with (A) water with 0.005% (v/v) acetic acid, pH 5.7, and (B) 5% methanol/95% acetonitrile with 0.005% acetic acid, at a flow rate of 0.6 mL/min was used. After a 15‐μL injection, the gradient started at 15% B (0–0.6 minutes), increased to 40% B (0.6–2 minutes), increased to 95% B (2–4 minutes), was maintained at 95% B (4–4.5 minutes), decreased to 15% B (4.5–4.7 minutes), and was maintained at 15% B (4.7–8 minutes). Tandem MS was performed in negative ion mode with spray voltage set at 3.3 kV, ion transfer tube temperature at 356°C, and vaporizer temperature at 420°C. The sheath, auxillary, and sweep gases were set at 52, 16, and 2 AU, respectively. The following m/z transitions were monitored for quantification: m/z 353.2→193.1 (CE 26 eV) and 353.2→309.1 (CE 20 eV) for 8‐iso‐PGF2α; 357.2→197.1 (CE 26 eV) and 357.2→313.1 (CE 20 eV) for d4‐iso‐PGF2α, m/z 367.2→243.1 (CE 20 eV) and 367.2→305.1 (CE 16 eV) for 11‐dehydro‐TXB2; m/z 371.2→247.1 (CE 20 eV) and 371.2→309.1 (CE 16 eV) for d4‐11‐dehydro‐TXB2; 369.2→169.1 (CE 18 eV) and 369.2→195.0 (CE 15 eV) for TXB2; and 373.2→173.1 (CE 18 eV) and 373.2→199.0 (CE 15 eV) for d4‐TXB2. Area ratios of analyte to internal standard were calculated, and concentrations of the samples were determined from the standard curve. All data were processed and integrated in Xcalibur, version 3.0 (Thermo).
Statistical Analysis
UTXB2 values were normalized by using the natural logarithmic transform. Univariate analyses were performed by using those variables deemed biologically plausible or supported by the literature. Colinearity of covariates was tested by using Fisher exact and Pearson correlation for categorical and continuous variables, eliminating highly collinear covariates based on clinical significance. All predictors with P≤0.15 on univariate analysis were included in a multivariable model that was optimized by using the corrected Akaike Information Criterion. To facilitate comparison of the contribution of independent variables in the multivariable model, the coefficient estimates are reported for independent variables standardized to a variance of 1 (β coefficients). The relative importance of the independent variables was further assessed by dominance analysis.11 The regression was performed for all possible combinations of the identified predictors, the incremental contribution of each variable to the resulting models was averaged to obtain general dominance (additive decomposition), and conditional dominance evaluations were performed. The independent variables were ranked for their contribution to the multivariable model based on their dominance weights. For vein graft analysis, grafts classified as severely diseased were considered as patent. Univariate analyses were performed on a per‐graft basis for the odds of occlusion versus patency for UTXB2 and urinary (U)8‐iso‐PGF2α. Proportions were compared by using a χ2 or Fisher's exact test, and comparisons among groups were made with ANOVA, McNemar, or Kruskal–Wallis testing, as appropriate. Analyses were performed by using Stata/MP 10.0 for Windows (StatCorp). Differences were considered significant when P<0.05.
Results
Study Population Characteristics
Of the 368 subjects undergoing first‐time CABG enrolled in the RIGOR study, 299 had measurement of UTXB2 and platelet reactivity at the time of assessment of vein graft patency 6 months after surgery. Thirty‐nine subjects were excluded from the primary analyses: 2 because they had discontinued aspirin, 32 because they were taking additional nonaspirin antiplatelet agents, and 5 because of arachidonic acid–induced platelet aggregation ≥1 Ω despite aspirin therapy. Therefore, 260 subjects taking aspirin monotherapy with verified suppression of platelet COX‐1 activity and TXA2 generation by >99%8 were used for the primary analyses. UTXB2 in this study cohort was non–normally distributed (Figure 1) with a median of 328 pg/mg creatinine (IQR 232–451 pg/mg creatinine). Despite confirmed aspirin‐induced suppression of arachidonic acid–induced platelet activation, 82 (31.5%) subjects had UTXB2 ≥400 pg/mg creatinine, the accepted threshold with this assay for defining putative aspirin nonresponsiveness.10 Table 1 shows the clinical characteristics of the study cohort as a whole, in subjects stratified by UTXB2, and in subjects excluded from the primary analyses.
Figure 1.
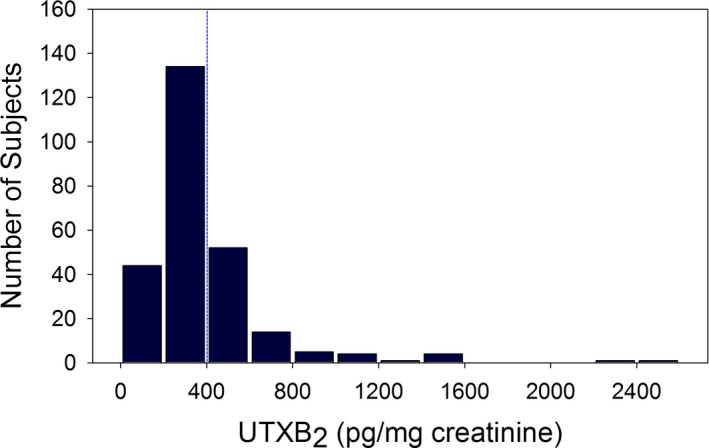
Distribution of urine 11‐dehydroTXB 2 (UTXB2) in the study cohort of 260 subjects. The blue dashed line indicates the putative threshold for defining aspirin responsiveness with this assay.10
Table 1.
Baseline, Operative, and Postoperative Characteristics of the 260 Study Subjects Stratified by UTXB2 and the 39 Excluded Subjects
| Characteristic | <400 pg/mg Creatinine | ≥400 pg/mg Creatinine | P Value | Total Included | Total Excluded | P Value |
|---|---|---|---|---|---|---|
| No. of patients | 178 | 82 | 260 | 39 | ||
| Age, y | 63 (55–69) | 66 (57–73) | 0.07 | 63 (56–71) | 63 (59–72) | 0.44 |
| Male sex | 153 (86%) | 58 (71%) | 0.006 | 211 (81%) | 25 (64%) | 0.02 |
| White race | 17 (10%) | 18 (22%) | 0.01 | 35 (13%) | 5 (13%) | 1.0 |
| Body mass index, kg/m2 | 29 (26–33) | 28 (26–33) | 0.87 | 29 (26–33) | 26 (24–30) | <0.001 |
| Medical history, n | ||||||
| Hypertension | 148 (83%) | 65 (79%) | 0.49 | 213 (82%) | 32 (84%) | 0.82 |
| Dyslipidemia | 150 (85%) | 65 (79%) | 0.29 | 215 (83%) | 34 (89%) | 0.48 |
| Diabetes | 56 (32%) | 39 (48%) | 0.018 | 95 (37%) | 11 (29%) | 0.47 |
| Heart failure | 16 (9%) | 18 (22%) | 0.006 | 34 (13%) | 4 (10%) | 0.80 |
| Peripheral/cerebrovascular disease | 28 (16%) | 18 (22%) | 0.226 | 46 (18%) | 8 (21%) | 0.66 |
| Atrial fibrillation | 5 (3%) | 3 (4%) | 0.71 | 8 (3%) | 4 (10%) | 0.06 |
| Current tobacco use | 33 (19%) | 27 (33%) | 0.017 | 60 (23%) | 11 (28%) | 0.55 |
| Myocardial infarction | 64 (36%) | 39 (48%) | 0.08 | 103 (40%) | 18 (46%) | 0.49 |
| Prior PCI | 40 (22%) | 12 (15%) | 0.18 | 52 (20%) | 9 (23%) | 0.67 |
| Preoperative LVEF | 0.78 | 0.75 | ||||
| ≤30% | 14 (8%) | 8 (10%) | 22 (8%) | 3 (8%) | ||
| 30–50% | 59 (33%) | 29 (35%) | 88 (34%) | 11 (28%) | ||
| >50% | 105 (59%) | 45 (55%) | 150 (58%) | 25 (64%) | ||
| Urgent/emergent surgery | 100 (56%) | 57 (70%) | 0.06 | 157 (60%) | 30 (77%) | 0.052 |
| Euroscore | 3 (1–5) | 4 (3–6) | 0.004 | 3 (2–5) | 4 (2–5) | 0.25 |
| Arterial graft implanted | 175 (98%) | 79 (96%) | 0.38 | 254 (98%) | 36 (92%) | 0.10 |
| No. of SVGs per subject | 0.2 | 0.03 | ||||
| 1 | 48 (27%) | 20 (24%) | 68 (26%) | 17 (44%) | ||
| 2 | 70 (39%) | 41 (50%) | 111 (43%) | 12 (31%) | ||
| 3 | 44 (25%) | 12 (15%) | 56 (22%) | 10 (26%) | ||
| ≥4 | 16 (9%) | 9 (11%) | 25 (10%) | 0 (0%) | ||
| Medications at the time of SVG patency assessment | ||||||
| Aspirin | 178 (100%) | 82 (100%) | 1.0 | 260 (100%) | 37 (95%) | <0.001 |
| Nonaspirin antiplatelet | 0 (0%) | 0 (0%) | 1.0 | 0 (0%) | 33 (85%) | <0.001 |
| Aspirin low dose (<325 mg) | 11 (6%) | 10 (12%) | 0.14 | 21 (8%) | 12 (31%) | <0.001 |
| Oral anticoagulation | 6 (3%) | 7 (9%) | 0.12 | 13 (5%) | 3 (8%) | 0.49 |
| β‐Blocker | 153 (86%) | 61 (75%) | 0.035 | 214 (82%) | 35 (90%) | 0.36 |
| ACE inhibitor/ARB | 113 (63%) | 47 (57%) | 0.4 | 160 (62%) | 26 (67%) | 0.60 |
| Lipid‐lowering agent | 162 (91%) | 65 (79%) | 0.015 | 227 (87%) | 37 (95%) | 0.28 |
Values are median (IQR) or n (%). ACE indicates angiotensin‐converting enzyme; ARB, angiotensin II receptor blocker; LVEF, left ventricular ejection fraction; PCI, percutaneous coronary intervention; SVG, saphenous vein graft; UTXB2, urinary thromboxane B2.
Relationship Between Clinical and Laboratory Variables and UTXB2
To identify potential stimuli and sources for nonplatelet thromboxane generation, univariate analyses were used to explore associations of a wide array of demographic, clinical, and laboratory variables to UTXB2 expressed as a continuous variable (Tables 2 and 3). A multivariable model (fit statistic of 0.44, P<0.0001) was then constructed to identify independent predictors of UTXB2 (Table 4). The strongest predictor of UTXB2, accounting for nearly half of the modeled effect, was U8‐iso‐PGF2α, an isoprostane formed by nonenzymatic metabolism of arachidonic acid under conditions of oxidative stress.12 Figure 2 shows the high degree of correlation between normalized values of U8‐iso‐PGF2α and UTXB2 in the study population. Age, race, and sex were also independently associated with UTXB2 and accounted for ≈10% of the modeled effect, while lipid therapy (predominantly statins), renal function, left ventricular function, and aspirin dose contributed to lesser degrees.
Table 2.
Univariate Analyses of the Associations of Subject Demographics, Past Medical History and Medication Use With UTXB2 (Normalized by Natural Log Transformation of pg/mg Creatinine)
| Characteristic | Standardized Coefficienta | P Value |
|---|---|---|
| Female sex | 0.225 | <0.001 |
| Age, y | 0.203 | 0.001 |
| White race (versus nonwhite) | −0.188 | 0.006 |
| Obesity (BMI ≥30 kg/m2) | −0.101 | 0.093 |
| Medical history | ||
| Hypertension | −0.031 | 0.579 |
| Dyslipidemia | −0.141 | 0.030 |
| Diabetes | 0.110 | 0.091 |
| Current tobacco use | 0.155 | 0.011 |
| Former tobacco use | 0.097 | 0.111 |
| Myocardial infarction | 0.052 | 0.394 |
| Percutaneous coronary intervention | −0.054 | 0.272 |
| Congestive heart failure | 0.140 | 0.015 |
| Cerebrovascular disease | 0.044 | 0.524 |
| Deep venous thrombosis/pulmonary embolus | −0.027 | 0.436 |
| Peripheral vascular disease | 0.167 | 0.010 |
| Chronic obstructive pulmonary disease | 0.139 | 0.016 |
| Atrial fibrillation | 0.038 | 0.581 |
| Preoperative LVEF: <30% vs 30–50% | −0.110 | 0.360 |
| Preoperative LVEF: <30% vs ≥50% | −0.148 | 0.211 |
| Left ventricular ejection fraction (%) | −0.157 | 0.011 |
| Euroscore: 0–2 vs 3–5 | 0.227 | 0.001 |
| Euroscore: 0–2 vs ≥6 | 0.281 | <0.001 |
| CABG urgency: elective vs urgent or emergent | 0.044 | 0.501 |
| Medications | ||
| Aspirin dose (81 mg vs higher) | −0.187 | 0.003 |
| Oral anticoagulation | 0.124 | 0.088 |
| β‐Blocker | −0.096 | 0.112 |
| Angiotensin II receptor blocker | −0.046 | 0.488 |
| Angiotensin‐converting enzyme inhibitor | −0.029 | 0.636 |
| Lipid therapy | −0.195 | 0.001 |
| Diuretic | 0.046 | 0.457 |
| Insulin | 0.020 | 0.726 |
| Insulin sensitizer | −0.004 | 0.954 |
| Insulin secretagogue | 0.064 | 0.346 |
BMI indicates body mass index; CABG, coronary artery bypass graft surgery; LVEF, left ventricular ejection fraction; UTXB2, urinary thromboxane B2.
Coefficients are standardized to 1 SD of the predictor. Huber–White sandwich estimates were used to produce robust estimates of variance.
Table 3.
Univariate Analyses of the Associations of Laboratory Variables to UTXB2 (Normalized by Natural Log Transformation of pg/mg Creatinine)
| Characteristic | Standardized Coefficienta | P Value |
|---|---|---|
| Hematologic parameter | ||
| Leukocyte count: 4.5–11×103 mm−3 | Reference | |
| Leukocyte count: ≤4.5×103 mm−3 | 0.023 | 0.702 |
| Leukocyte count: ≥11×103 mm−3 | 0.078 | <0.001 |
| MCV 80–100 fL | Reference | |
| MCV <80 fL | 0.067 | 0.354 |
| MCV >100 fL | 0.003 | 0.925 |
| Hematocrit (%) | −0.089 | 0.183 |
| Red cell distribution width (≤14.5 vs >14.5%) | 0.130 | 0.042 |
| RDW, %−3 | 0.141 | 0.024 |
| Platelet count (<150 vs ≥150×103 mm−3) | −0.032 | 0.582 |
| Reticulocyte (ln %) | −0.083 | 0.273 |
| Mean platelet volume (%−1) | −0.050 | 0.401 |
| Immature platelet fraction (ln %) | 0.055 | 0.390 |
| Blood group: O vs other | −0.015 | 0.805 |
| Rh positivity | 0.071 | 0.341 |
| Creatinine (−[mg/dL]−½) | −0.166 | 0.002 |
| C‐reactive protein (<5 vs ≥5 mg/L) | 0.139 | 0.027 |
| Fibrinogen <390 vs ≥390 mg/dL | 0.124 | 0.049 |
| vonWillebrand factor (>150% vs ≤150%) | 0.192 | 0.002 |
| Urine 8‐iso‐PGF2α (ln pg/mg creatinine) | 0.500 | <0.0001 |
| Urine 8‐iso‐PGF2α (<1061 vs ≥1061 pg/mg creatinine) | 0.353 | <0.001 |
| Fasting serum insulin (ln μU/mL) | −0.008 | 0.91 |
| Impedance platelet aggregation in ohms to | ||
| ADP (20 μmol/L) | 0.041 | 0.452 |
| Collagen (1 μg/mL) | −0.006 | 0.910 |
| Epinephrine (50 μmol/L) | 0.078 | 0.188 |
| PFA‐100 collagen/ADP (closure time in s) | −0.014 | 0.811 |
| PFA‐100 collagen‐epinephrine (closure time in s) | −0.097 | 0.147 |
MCV indicates ; PG, prostaglandin; RDW, ; UTXB2, urinary thromboxane B2..
Coefficients are standardized to 1 SD of the predictor. Huber–White sandwich estimates were used to produce robust estimates of variance.
Table 4.
Independent Risk Factors for UTXB2 a After Adjustment of Other Variables by Multivariable Regression Analysis
| Characteristic | Standardized Coefficient | P Value | Dominance Weight | Dominance Ranking |
|---|---|---|---|---|
| Urine 8‐iso‐PGF2α (ln pg/mg creatinine) | 0.442 | <0.001 | 0.472 | 1 |
| Age, y | 0.239 | <0.001 | 0.102 | 2 |
| Female sex | 0.129 | 0.015 | 0.093 | 3 |
| White race (versus nonwhite) | −0.172 | 0.009 | 0.085 | 4 |
| Lipid therapy | −0.161 | 0.004 | 0.077 | 5 |
| Creatinine (−[mg/dL]−½) | −0.152 | 0.002 | 0.072 | 6 |
| Aspirin dose (81 mg vs higher) | −0.145 | 0.004 | 0.052 | 7 |
| Left ventricular ejection fraction (%) | −0.113 | 0.032 | 0.048 | 8 |
PG indicates prostaglandin; UTXB2, urinary 11‐dehydro thromboxane B2 (pg/mg creatinine).
Normalized using natural log transform.
Figure 2.
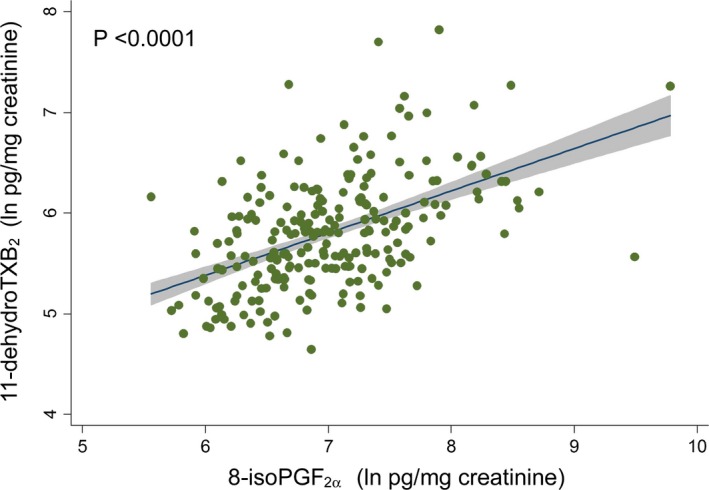
Linear regression of normalized levels of urine 11‐dehydro‐thromboxane (TX)B 2 to 8‐iso‐prostglandin (PG)F 2α in 228 subjects.
Relationship of U8‐Iso‐PGF2α to Early Vein Graft Failure
We previously found that aspirin‐insensitive thromboxane generation, defined by elevated UTXB2, was a novel and independent risk factor for vein graft thrombosis 6 months after CABG.5 Given that U8‐iso‐PGF2α is a major determinant of UTXB2, we investigated whether there was a direct relationship of the former to early graft thrombosis. Stratification of RIGOR subjects, regardless of antiplatelet use or aspirin responsiveness, by tertile of U8‐iso‐PGF2α, revealed a proportional increase in the prevalence of vein graft occlusion (Figure 3), and normalized U8‐iso‐PGF2α correlated with graft occlusion when considered on a per‐graft basis (odds ratio 1.67, P=0.001). U8‐iso‐PGF2α was not, however, an independent predictor of vein graft occlusion in multivariable modeling when UTXB2 was included as a variable (data not shown), indicating the primacy of the latter.
Figure 3.
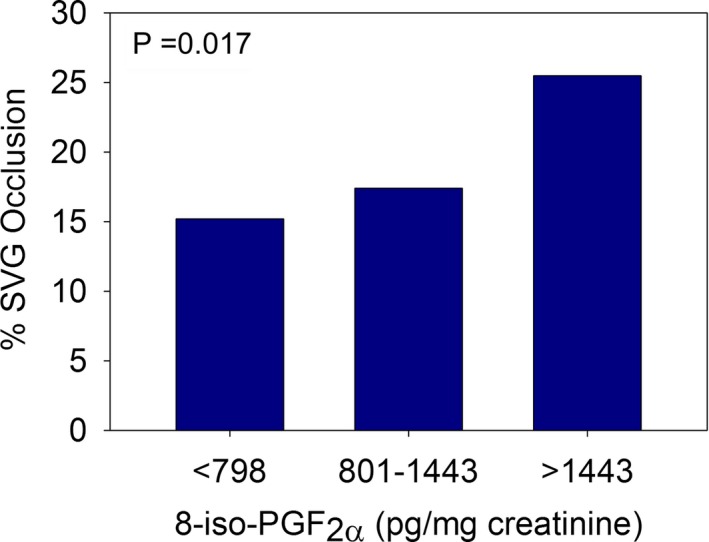
Prevalence of saphenous vein graft (SVG) occlusion in 225 subjects stratified by tertile of urine 8‐iso‐prostaglandin (PG)F 2α.
Relationship Between Endothelial Thromboxane Generation and Oxidative Stress
The strong association between U8‐iso‐PGF2α and UTXB2 highly suggests, but does not in itself prove, a causal relationship between oxidative stress and nonplatelet TXA2 generation. Because the endothelium is a potential major source of nonplatelet TXA2 generation in vivo, we determined the effect of oxidative stress and direct stimulation with 8‐iso‐PGF2α on endothelial TXA2 generation. Exposure of human umbilical vein endothelial cells to hydrogen peroxide resulted in a dose‐dependent increase in the concentration of 8‐iso‐PGF2α, TXB2, and 11‐dehydro‐TXB2 in the conditioned media, indicative of cellular TXA2 generation (Figure 4A). Further, direct stimulation of human umbilical vein endothelial cells with 8‐iso‐PGF2α results in endothelial TXA2 production (Figure 4B), establishing a mechanistic link among oxidative stress, 8‐iso‐PGF2α formation, and nonplatelet TXA2 generation.
Figure 4.
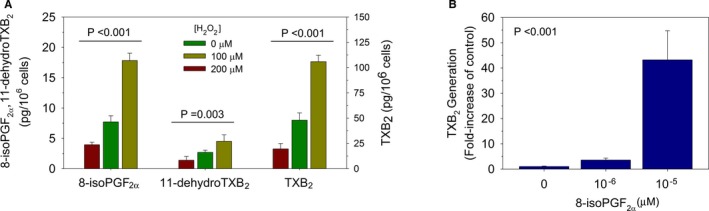
A, Human umbilical vein endothelial cells (HUVECs) under oxidative stress by exposure to hydrogen peroxide (H2O2) for 1 hour generate thromboxane (TX) and isoprostanes in a dose‐dependent manner. B, HUVECs exposed to 8‐iso‐prostaglandin (PG)F 2α for 1 hour generate thromboxane in a dose‐dependent manner. Values shown are the mean of n=3 ±SEM.
Discussion
The major findings of this study are that (1) oxidative stress–induced formation of 8‐iso‐PGF2α is the strongest variable associated with nonplatelet TXA2 generation in patients 6 months after CABG; (2) age, sex, race, lipid therapy, aspirin dose, and kidney and left ventricular function are also independently associated with nonplatelet TXA2 generation, though to a much lesser degree; (3) U8‐iso‐PGF2α directly correlates with incidence of early vein graft thrombosis, but its predictive power is not independent of UTXB2; and (4) endothelial cells are capable of generating TXA2 in response to both oxidative stress and direct stimulation with 8‐iso‐PGF2α, thus representing a potential source of nonplatelet TXA2 generation in vivo.
Platelets are the predominant source of TXA2 generation in healthy individuals, and measurement of stable TXA2 metabolites in the urine has been used clinically as an indicator of the antiplatelet effects of aspirin. Substudies from the Heart Outcomes Prevention Evaluation (HOPE) and the Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management and Avoidance (CHARISMA) trials found that in patients with either established or at high risk for cardiovascular disease who are receiving aspirin therapy, those with UTXB2 in the highest quartile had a 1.66‐ to 1.80‐fold increased risk of death, myocardial infarction, and stroke compared those in the lowest quartile.2, 3 An early interpretation of these results was that aspirin failed to adequately inhibit platelet COX‐1 activity in a substantial number of subjects, leading to persistent TXA2 generation, increased platelet reactivity, and elevated cardiovascular risk. The RIGOR study also found that elevated UTXB2 was associated with increased cardiovascular risk, being a potent and independent risk factor for early vein graft thrombosis.5 Unlike the HOPE and CHARISMA data, which did not measure platelet‐specific TXA2 generation, the RIGOR data revealed that failure of aspirin to inhibit platelet COX‐1 and TXA2 generation was in fact rare, occurring in <1% of subjects 6 months after surgery. This provided compelling evidence that aspirin‐insensitive TXA2 generation in patients with cardiovascular disease, unlike in healthy individuals, predominantly originates from nonplatelet sources.
The current analysis extends our previous results and identifies oxidative stress as a potentially major stimulus for nonplatelet TXA2 generation by showing a strong correlation with 8‐iso‐PGF2α, an arachidonic acid metabolite formed nonenzymatically in a variety of cell types by free radical oxidation.12 While frequently used as a marker of oxidative stress, 8‐iso‐PGF2α is also a biologically active prostanoid that can bind to and activate cellular thromboxane receptors (see reviews13 and 14). Although it is not as potent a platelet agonist as TXA2, 8‐iso‐PGF2α can potentiate platelet activation in response to collagen and ADP as well as directly stimulate vasoconstriction. In 2 small prior studies involving patients with unstable angina and diabetes who were taking aspirin, a correlation between UTXB2 and 8‐iso‐PGF2α was identified, though in neither was the source of TXA2 generation specifically evaluated.15, 16 Our analysis not only establishes a strong association between 8‐iso‐PGF2α and aspirin‐insensitive TXA2 generation in a larger study cohort but also conclusively demonstrates that the latter originates predominantly from nonplatelet sources.
Mounting evidence suggests that oxidative stress―induced generation of 8‐iso‐PGF2α and nonplatelet TXA2 is more than just linearly associated but is causally linked. Treatment with the antioxidant vitamin E has been shown to reduce both U8‐iso‐PGF2α and UTXB2 levels in aspirin‐naïve smokers.17 More definitive are the findings that fetal porcine cerebral and retinal microvessels generate TXA2 when incubated with 8‐iso‐PGF2α, an effect that is blocked by indomethacin.18, 19 Our data not only confirm this finding in macrovascular endothelial cells but also reveal that oxidative stress itself can be a primary stimulus for endothelial TXA2 generation. Whether this effect is mediated by the endothelial generation of 8‐so‐PGF2α and autocrine stimulation of cellular thromboxane receptors or involves additional intracellular pathways is an area of active investigation.
Because 8‐iso‐PGF2α is a stable prostanoid that freely circulates, its biological effects can be widespread and distinct from sources of origin. Inflammatory cells are capable of directly generating TXA2 and 8‐iso‐PGF2α, and patients undergoing cardiac surgery with cardiopulmonary bypass have a marked inflammatory response with an observable increase in U8‐iso‐PGF2α in the early postoperative period.20, 21, 22 In the RIGOR study, UTXB2 was significantly higher 3 days after CABG than at 6 months, though only the latter correlated with risk of vein graft occlusion.5 While we did find significant correlations of UTXB2 with white blood cell count, C‐reactive protein, or fibrinogen on univariate analyses, none of these variables was independently associated with UTXB2, suggesting that inflammation was not a major stimulus for nonplatelet TXA2 generation 6 months after CABG. It is conceivable that the vein grafts themselves contribute to TXA2 generation given that surgical preparation and pressure‐induced distention are known to increase oxidative stress in vein segments.23, 24 However, we did not observe any correlation between the number of vein graft segments implanted at the time of surgery and either UTXB2 or U8‐iso‐PGF2α 6 months later (data not shown). The elevated UTXB2 and U8‐iso‐PGF2α observed in a substantial percentage of the RIGOR study cohort are, therefore, likely due predominantly to underlying cardiovascular disease or its risk factors, rather than effects of the CABG per se. UTXB2 has previously been shown to be elevated in diabetics compared with nondiabetics.16 While this was also true in our analysis, diabetes was not found to be an independent predictor of UTXB2 when 8‐iso‐PGF2α was considered as a variable, suggesting that they are different manifestations of the same pathologic process.
The mechanism by which nonplatelet TXA2 generation could adversely affect cardiovascular risk is currently unknown. TXA2 necessarily acts locally because of a short half‐life (≈30 seconds) due to degradation to biologically inert TXB2. Although aspirin‐inhibited platelets cannot generate appreciable amounts of TXA2, they could potentially still aggregate in response to locally generated nonplatelet TXA2 to cause thrombosis. However, several pieces of evidence argue against platelet hyperreactivity being a major mediator of cardiovascular risk by nonplatelet TXA2 generation. First, the addition of clopidogrel did not reduce cardiovascular risk in CHARISMA subjects with elevated UTXB2.3 Second, UTXB2 in the RIGOR study cohort was independent of multiple parameters of platelet reactivity, including shear‐dependent platelet aggregation and whole blood aggregation performed in response to multiple different agonists (Table 3). Third, we found no correlation between U8‐iso‐PGF2α and platelet reactivity, suggesting that circulating 8‐iso‐PGF2α did not significantly “prime” or potentiate platelet aggregation in response to more physiologic agonists (data not shown). Rather than potentiating platelet reactivity, local TXA2 generation could predispose to thrombus formation by altering endothelial thromboresistance. Consistent with this concept are the recent findings that thromboxane receptor activation stimulates tissue factor expression in both endothelial cells and monocytes.25, 26
A significant finding of our analysis was that U8‐iso‐PGF2α correlated directly with the incidence of early vein graft thrombosis. This suggests that therapies aimed at reducing oxidative stress might be a viable strategy to reduce nonplatelet TXA2 generation and improve outcomes after cardiac surgery. Antioxidants have been shown to be efficacious at reducing the incidence of postoperative atrial fibrillation.27, 28 While they have not been evaluated for efficacy at reducing graft failure, this is an eminently testable hypothesis.
Our study has several potential limitations. Although we examined a wide array of clinical and laboratory variables associated with UTXB2 generation in subjects 6 months after CABG, presumably at a time when the effects of surgery have subsided, it is possible that the relative contributions of oxidative stress and other identified factors differ in populations with various forms or severity of cardiovascular disease. Although the RIGOR cohort was extensively phenotyped and we evaluated numerous potential variables, the 8 variables identified in the multivariable modeling account for only slightly less than half of the modeled effect on UTXB2. Thus, additional risk factors for nonplatelet TXA2 generation remain to be identified.
In summary, we identified several risk factors for nonplatelet TXA2 generation in subjects 6 months after CABG. Not only was oxidative stress‐induced formation of 8‐iso‐PGF2α the strongest identified risk factor, but it also directly correlated with risk of early vein graft thrombosis. In vitro studies revealed that macrovascular endothelial cells are capable of generating TXA2 under conditions of oxidative stress and with direct stimulation with 8‐iso‐PGF2α, not only establishing a mechanistic link between oxidative stress and nonplatelet TXA2 generation but pointing to dysfunctional endothelial cells as a potentially major source. These findings provide valuable insights into the pathobiology of nonplatelet TXA2 generation and identify potential therapeutic strategies for its suppression.
Sources of Funding
The study was supported by the Johns Hopkins Institute for Clinical and Translational Research (funded by UL1 RR025005 from the National Center for Research Resources, National Institutes of Health), funding from the Flight Attendant Medical Research Foundation (Dr Rade), and National Institutes of Health 1KL2RR025006‐01 (Dr Nazarian), with material support from Corgenix Inc and Esoterix, Inc. In addition, the parent RIGOR study received funding support from The Medicines Company, AstraZeneca Pharmaceuticals, and Sanofi‐BMS as well as material support from Siemens Healthcare Diagnostics, Inc and GlaxoSmithKline.
Disclosures
None.
(J Am Heart Assoc. 2016;5:e002615 doi: 10.1161/JAHA.115.002615)
References
- 1. Capra V, Back M, Angiolillo DJ, Cattaneo M, Sakariassen KS. Impact of vascular thromboxane prostanoid receptor activation on hemostasis, thrombosis, oxidative stress, and inflammation. J Thromb Haemost. 2014;12:126–137. [DOI] [PubMed] [Google Scholar]
- 2. Eikelboom JW, Hirsh J, Weitz JI, Johnston M, Yi Q, Yusuf S. Aspirin‐resistant thromboxane biosynthesis and the risk of myocardial infarction, stroke, or cardiovascular death in patients at high risk for cardiovascular events. Circulation. 2002;105:1650–1655. [DOI] [PubMed] [Google Scholar]
- 3. Eikelboom JW, Hankey GJ, Thom J, Bhatt DL, Steg PG, Montalescot G, Johnston SC, Steinhubl SR, Mak KH, Easton JD, Hamm C, Hu T, Fox KAA, Topol EJ. Incomplete inhibition of thromboxane biosynthesis by acetylsalicylic acid. Determinants and effect on cardiovascular Risk. Circulation. 2008;118:1690. [DOI] [PubMed] [Google Scholar]
- 4. Antiplatelet Trialists’ Collaboration . Collaborative overview of randomised trials of antiplatelet therapy‐II: maintenance of vascular graft or arterial patency by antiplatelet therapy. BMJ. 1994;308:168. [PMC free article] [PubMed] [Google Scholar]
- 5. Gluckman TJ, McLean RC, Schulman SP, Kickler TS, Shapiro EP, Conte JV, McNicholas KW, Segal JB, Rade JJ. Effects of aspirin responsiveness and platelet reactivity on early vein graft thrombosis after coronary artery bypass graft surgery. J Am Coll Cardiol. 2011;57:1069–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gluckman TJ, Segal JB, Schulman SP, Shapiro EP, Kickler TS, Prechel MM, Conte JV, Walenga JM, Shafique I, Rade JJ. Effect of anti‐platelet factor‐4/heparin antibody induction on early saphenous vein graft occlusion after coronary artery bypass surgery. J Thromb Haemost. 2009;7:1457–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McLean RC, Nazarian SM, Gluckman TJ, Schulman SP, Thiemann DR, Shapiro EP, Conte JV, Thompson JB, Shafique I, McNicholas KW, Villines TC, Laws KM, Rade JJ. Relative importance of patient, procedural and anatomic risk factors for early vein graft thrombosis after coronary artery bypass graft surgery. J Cardiovasc Surg (Torino). 2011;52:877–885. [PMC free article] [PubMed] [Google Scholar]
- 8. Pulcinelli FM, Riondino S, Celestini A, Pignatelli P, Trifiro E, Di Renzo L, Violi F. Persistent production of platelet thromboxane A2 in patients chronically treated with aspirin. J Thromb Haemost. 2005;3:2784–2789. [DOI] [PubMed] [Google Scholar]
- 9. Nazarian SM, Thompson JB, Gluckman TJ, Laws K, Jani JT, Kickler TS, Rade JJ. Clinical and laboratory factors associated with shear‐dependent platelet hyper‐reactivity in patients on chronic aspirin therapy. Thromb Res. 2009;126:379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fritsma GA, Ens GE, Alvord MA, Carroll AA, Jensen R. Monitoring the antiplatelet action of aspirin. JAAPA. 2001;14:57–62. [PubMed] [Google Scholar]
- 11. Grömping U. Estimators of relative importance in linear regression based on variance decomposition. Am Stat. 2007;61:139–147. [Google Scholar]
- 12. Davi G, Falco A, Patrono C. Determinants of F2‐isoprostane biosynthesis and inhibition in man. Chem Phys Lipids. 2004;128:149–163. [DOI] [PubMed] [Google Scholar]
- 13. Milne GL, Yin H, Hardy KD, Davies SS, Roberts LJ. Isoprostane generation and function. Chem Rev. 2011;111:5973–5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bauer J, Ripperger A, Frantz S, Ergun S, Schwedhelm E, Benndorf RA. Pathophysiology of isoprostanes in the cardiovascular system: implications of isoprostane‐mediated thromboxane A2 receptor activation. Br J Pharmacol. 2014;171:3115–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cipollone F, Ciabattoni G, Patrignani P, Pasquale M, Di Gregorio D, Bucciarelli T, Davi G, Cuccurullo F, Patrono C. Oxidant stress and aspirin‐insensitive thromboxane biosynthesis in severe unstable angina. Circulation. 2000;102:1007–1013. [DOI] [PubMed] [Google Scholar]
- 16. Ames PR, Batuca JR, Muncy IJ, De La Torre IG, Pascoe‐Gonzales S, Guyer K, Matsuura E, Lopez LR. Aspirin insensitive thromboxane generation is associated with oxidative stress in type 2 diabetes mellitus. Thromb Res. 2012;130:350–354. [DOI] [PubMed] [Google Scholar]
- 17. Patrignani P, Panara MR, Tacconelli S, Seta F, Bucciarelli T, Ciabattoni G, Alessandrini P, Mezzetti A, Santini G, Sciulli MG, Cipollone F, Davi G, Gallina P, Bon GB, Patrono C. Effects of vitamin E supplementation on F(2)‐isoprostane and thromboxane biosynthesis in healthy cigarette smokers. Circulation. 2000;102:539–545. [DOI] [PubMed] [Google Scholar]
- 18. Hou X, Gobeil F Jr, Peri K, Speranza G, Marrache AM, Lachapelle P, Roberts J, Varma DR, Chemtob S, Ellis EF. Augmented vasoconstriction and thromboxane formation by 15‐F(2t)‐isoprostane (8‐iso‐prostaglandin F(2alpha)) in immature pig periventricular brain microvessels. Stroke. 2000;31:516–524. [DOI] [PubMed] [Google Scholar]
- 19. Lahaie I, Hardy P, Hou X, Hassessian H, Asselin P, Lachapelle P, Almazan G, Varma DR, Morrow JD, Roberts LJ, Chemtob S. A novel mechanism for vasoconstrictor action of 8‐isoprostaglandin F2 alpha on retinal vessels. Am J Physiol. 1998;274:R1406–R1416. [DOI] [PubMed] [Google Scholar]
- 20. Yang CW, Unanue ER. Neutrophils control the magnitude and spread of the immune response in a thromboxane A2‐mediated process. J Exp Med. 2013;210:375–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schmitt D, Shen Z, Zhang R, Colles SM, Wu W, Salomon RG, Chen Y, Chisolm GM, Hazen SL. Leukocytes utilize myeloperoxidase‐generated nitrating intermediates as physiological catalysts for the generation of biologically active oxidized lipids and sterols in serum. Biochemistry. 1999;38:16904–16915. [DOI] [PubMed] [Google Scholar]
- 22. Cavalca V, Sisillo E, Veglia F, Tremoli E, Cighetti G, Salvi L, Sola A, Mussoni L, Biglioli P, Folco G, Sala A, Parolari A. Isoprostanes and oxidative stress in off‐pump and on‐pump coronary bypass surgery. Ann Thorac Surg. 2006;81:562–567. [DOI] [PubMed] [Google Scholar]
- 23. West NEJ, Guzik TJ, Black E, Channon KM. Enhanced superoxide production in experimental venous bypass graft intimal hyperplasia: role of NAD(P)H oxidase. Arterioscler Thromb Vasc Biol. 2001;21:189–194. [DOI] [PubMed] [Google Scholar]
- 24. Shi Y, Patel S, Davenpeck KL, Niculescu R, Rodriguez E, Magno MG, Ormont ML, Mannion JD, Zalewski A. Oxidative stress and lipid retention in vascular grafts: comparison between venous and arterial conduits. Circulation. 2001;103:2408–2413. [DOI] [PubMed] [Google Scholar]
- 25. Bode M, Mackman N. Regulation of tissue factor gene expression in monocytes and endothelial cells: thromboxane A2 as a new player. Vascul Pharmacol. 2014;62:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Del TS, Basta G, Lazzerini G, Chancharme L, Lerond L, De CR. Involvement of the TP receptor in TNF‐alpha‐induced endothelial tissue factor expression. Vascul Pharmacol. 2014;62:49–56. [DOI] [PubMed] [Google Scholar]
- 27. Rodrigo R, Korantzopoulos P, Cereceda M, Asenjo R, Zamorano J, Villalabeitia E, Baeza C, Aguayo R, Castillo R, Carrasco R, Gormaz JG. A randomized controlled trial to prevent post‐operative atrial fibrillation by antioxidant reinforcement. J Am Coll Cardiol. 2013;62:1457–1465. [DOI] [PubMed] [Google Scholar]
- 28. Guo XY, Yan XL, Chen YW, Tang RB, Du X, Dong JZ, Ma CS. Omega‐3 fatty acids for postoperative atrial fibrillation: alone or in combination with antioxidant vitamins? Heart Lung Circ. 2014;23:743–750. [DOI] [PubMed] [Google Scholar]