

Abstract
Background
Patients with stable coronary artery disease (CAD) constitute a heterogeneous group in which the treatment benefits by angiotensin‐converting enzyme (ACE)‐inhibitor therapy vary between individuals. Our objective was to integrate clinical and pharmacogenetic determinants in an ultimate combined risk prediction model.
Methods and Results
Clinical, genetic, and outcomes data were used from 8726 stable CAD patients participating in the EUROPA/PERGENE trial of perindopril versus placebo. Multivariable analysis of phenotype data resulted in a clinical risk score (range, 0–21 points). Three single‐nucleotide polymorphisms (rs275651 and rs5182 in the angiotensin‐II type I‐receptor gene and rs12050217 in the bradykinin type I‐receptor gene) were used to construct a pharmacogenetic risk score (PGXscore; range, 0–6 points). Seven hundred eighty‐five patients (9.0%) experienced the primary endpoint of cardiovascular mortality, nonfatal myocardial infarction or resuscitated cardiac arrest, during 4.2 years of follow‐up. Absolute risk reductions ranged from 1.2% to 7.5% in the 73.5% of patients with PGXscore of 0 to 2. As a consequence, estimated annual numbers needed to treat ranged from as low as 29 (clinical risk score ≥10 and PGXscore of 0) to 521 (clinical risk score ≤6 and PGXscore of 2). Furthermore, our data suggest that long‐term perindopril prescription in patients with a PGXscore of 0 to 2 is cost‐effective.
Conclusions
Both baseline clinical phenotype, as well as genotype determine the efficacy of widely prescribed ACE inhibition in stable CAD. Integration of clinical and pharmacogenetic determinants in a combined risk prediction model demonstrated a very wide range of gradients of absolute treatment benefit.
Keywords: angiotensin‐converting enzyme inhibitor, coronary artery disease, individualized therapy, pharmacogenetics, risk model
Subject Categories: ACE/Angiotension Receptors/Renin Angiotensin System, Coronary Artery Disease, Atherosclerosis, Genetics, Pharmacology
Introduction
The European Trial on Reduction of Cardiac Events with Perindopril in Stable Coronary Artery Disease (EUROPA) and the Heart Outcomes Prevention Evaluation (HOPE) have demonstrated the effectiveness of angiotensin‐converting enzyme (ACE) inhibitors perindopril and ramipril, respectively, by reduction of mortality and morbidity from cardiovascular events among patients with stable coronary artery disease (CAD).1, 2 Consequently, ACE inhibitors are recommended in clinical guidelines on secondary prevention in patients with stable CAD and hence widely used in this population.3, 4, 5
However, patients with stable CAD constitute a heterogeneous group in which the absolute risk of cardiovascular complications varies between individuals.6, 7
Several approaches toward identification of those patients that are most likely to benefit from ACE‐inhibitor therapy have previously been reported. A previously published post‐hoc analysis of the EUROPA trial studied baseline clinical risk factors such as age, sex, smoking, cholesterol, and blood pressure levels.6 A risk score founded on such baseline clinical risk factors was able to identify patients at high, medium, and relatively low absolute risk (>3%, 1–3% and 1% per annum, respectively) of experiencing cardiovascular death, nonfatal myocardial infarction (MI), and resuscitated cardiac arrest.6 In contrast to the absolute treatment benefit, the relative treatment effect of perindopril, however, was not modified by the baseline level of risk.6 Similar conclusions were drawn after investigation of the relation between treatment benefit by perindopril and baseline renal function or the degree of blood pressure reduction.8, 9, 10
A novel approach toward selection of those that are likely to respond (or not) to ACE‐inhibitor therapy is to identify information on genetic variation among patients.11 A recent publication by our group demonstrated that genetic variation in the renin‐angiotensin‐aldosterone system (RAAS) and the kallikrein‐bradykinin (KB) pathway is associated with the treatment benefit of perindopril.12 Three single‐nucleotide polymorphisms (SNPs), 2 of which in the angiotensin‐II type I (AT1) receptor gene and one in the bradykinin type I (BK1) receptor gene, were used to construct an integer‐based pharmacogenetic risk score (PGXscore), ranging from 0 to 6 points.12 We were able to identify 2 distinct subgroups within the overall study population of 8726 patients on the basis of this PGXscore.12 One subgroup (73.5% of the patients) was characterized by a more pronounced treatment benefit, whereas no treatment benefit was apparent in the remaining 26.5% of patients.
This current analysis is an ultimate extension of both the previously published clinical risk model6 and pharmacogenetic risk profile.12 Its purpose is 2‐fold: (1) to investigate the relation between identified genetic determinants of treatment benefit and different levels of baseline clinical risk and (2) to integrate clinical and pharmacogenetic determinants in an ultimate combined risk prediction model.
Methods
Study Population and Design
The PERindopril GENEtic association study (PERGENE) is a substudy of the EUROPA trial. The designs of both studies have been reported previously.1, 12 In brief, the EUROPA trial was a randomized, double‐blinded, placebo‐controlled study designed to assess the effect of perindopril (8 mg daily) on the combined primary endpoint of cardiovascular mortality, nonfatal myocardial infarction (MI), and resuscitated cardiac arrest in 12 218 patients with stable CAD, but without overt heart failure or uncontrolled hypertension. Use of perindopril resulted in a 20% relative risk reduction (adjusted hazard ratio [HR]: 0.80; 95% CI: 0.71–0.91) in the rate of the primary endpoint during a mean follow‐up of 4.2 years.1
A DNA biobank was established within the EUROPA trial for the purpose of the PERGENE substudy, which investigates whether genetic variation is a determinant of the risk of future adverse cardiovascular outcome and/or treatment benefit by use of perindopril.11 DNA was successfully isolated in 9454 patients, using an automated isolation process.11 Comprehensive coverage of genetic variation in both the RAAS and KB pathways was ensured by a haplotype‐tagging SNP procedure in 12 candidate genes, as described in detail previously.11, 12
Our study was approved by the institutional review board of every participating center and written informed consent for genetic association analyses was obtained from all patients.
Clinical Risk Score
Uni‐ and multivariable Cox proportional hazard regression analyses were performed to study the relation between the primary endpoint (cardiovascular mortality, nonfatal MI, and resuscitated cardiac arrest) and baseline clinical patient characteristics, such as demographic and clinical variables, medical history, laboratory tests, and concomitant medication. Interaction by treatment was investigated for each clinical characteristic. A final multivariable clinical risk model was constructed using a backward step‐wise elimination procedure in which removal testing was based on the probability of the likelihood ratio statistic based on the maximum partial likelihood estimates. In order to develop a clinical risk scoring system, the log HRs from the final multivariable model were converted to an estimated risk score.6, 13 Clinical risk scores were calculated for each of the patients of the currently described population (only those trial participants of whom both baseline clinical characteristics and [pharmaco]genetic profile were complete). The study population was divided into tertiles in order to distinguish low, medium, and high clinical risk profiles.
Pharmacogenetic Risk Profile and Replication
The PERGENE substudy assessed 52 SNPs with the use of Taqman allelic discrimination assays (Applied Biosystems, Foster City, CA) and Sequenom (San Diego, CA) mass spectrometric genotyping. Quality control for the accuracy of genotyping involved testing duplicates from a randomly selected group of samples (5%) for concordance between samples (always >99% replication). Individual SNP call rates ranged between 95% and 98%. To ensure DNA quality, only patients who were successfully genotyped for more than 90% of the selected 52 SNPs were included in the PERGENE analyses (n=8907).12
Seven SNPs have previously been reported to significantly modify the treatment effect of perindopril in univariate analyses.12 After multivariate adjustment and correction for multiple testing, 3 SNPs remained significant modifiers of the perindopril treatment effect: rs275651 and rs5182 in the AT1 receptor gene and rs12050217 in the BK1 receptor gene. These 3 SNPs formed the foundation of a previously published PGXscore, ranging from 0 to 6 points, which was constructed by counting the number of alleles that were associated with a decreased benefit of perindopril treatment.12 The association between the PGXscore and treatment benefit by perindopril, as found in PERGENE,12 was replicated in the PROGRESS study, which investigated the treatment effect of perindopril in patients with cerebrovascular disease.14
Statistical Analysis
Differences in baseline clinical characteristics between low, medium, and high clinical risk groups were assessed by chi‐square tests in the case of categorical data or 1‐way ANOVA in the case of continuous data. A multivariate Cox proportional hazards regression model was fitted with the following covariates: clinical risk score; PGXscore; treatment; and treatment×PGXscore interaction (full model). The baseline hazard function, H0(t), was estimated by dividing the cumulative hazard at the end of follow‐up through the exponential function of the mean of the covariates. The cumulative survival under perindopril treatment versus placebo at the median follow‐up of 4.2 years was calculated for each clinical risk score within the separate pharmacogenetic risk strata as follows: S (4.2 years)=0.033975×exp (0.196×clinical risk score−0.203×PGXscore−0.793×treatment−0.318×interaction term). With respect to “treatment,” placebo was defined as 0 and perindopril treatment as 1. The “interaction term” was the multiplication of the PGXscore×treatment. Absolute and relative risks, as well as crude and adjusted HRs, are presented with 95% CIs. Numbers needed to treat (NNT) in order to prevent 1 event per annum were calculated as the inverse of the absolute risk reduction at the mean clinical risk scores per stratum.
The performance of the model consisting of clinical risk score only was compared by 2 different methods with the full model with respect to discrimination. First, the c‐index and areas under the 2 receiver operating characteristic curves were compared by a nonparametric method, as previously described by de Long et al.15 Secondly, the difference in model‐based discrimination slopes was evaluated through integrated discrimination improvement (IDI).16 Calibration of both the model consisting of clinical risk score only and the full model was tested with the Hosmer‐Lemeshow (H‐L) goodness‐of‐fit test. All statistical tests were 2‐sided with a type I error level of 0.05, except for the IDI, for which a conservative significance level of 0.01 was maintainted.16
We performed a cross‐validation within our own data set by bootstrap methods, as suggested by Harrell et al.17 We constructed 300 bootstrap samples (training) from the full original sample with the same size as the original (test). Models were built in the training sets. C‐indices were then obtained in these training sets (Ctraining) and compared with the c‐indices of the models when applied to the test set (Ctest). The optimism in the fit from bootstrap sample i is defined as Oi=Ci,training−Ci,test. We report the mean O of these optimism estimates. Analyses were performed with IBM SPSS statistics (version 23.0; IBM Corp, Armonk, NY) and STATA software (version 12; StataCorp LP, College Station, TX).
Cost‐Effectiveness Analysis
We examined the potential cost‐effectiveness of the combined clinical risk score and PGXscore. The time horizon was restricted to the duration of the EUROPA trial/PERGENE study (mean follow‐up of 4.2 years). Costs were set at 15 euros for the analysis of the 3 SNPs of the PGXscore, 50 euros for perindopril (based on the current price of perindopril 8‐mg tablets in The Netherlands), and 3000 euros for a clinical event (a weighted average of the costs of treating MI and the costs of cardiac death). The health loss of a clinical event within the trial duration was set at 0.6 years (a weighted average of the relative frequency and life‐years lost from MI [0 years] and cardiac death [2 years]).
The following patient management strategies were examined, against the strategy of no perindopril treatment (as the comparator):
Pharmacogenetic testing only in patients with a high clinical risk score and perindopril treatment only if PGXscore=0 to 2
Pharmacogenetic testing only in patients with a medium or high clinical risk score and perindopril treatment only if PGXscore=0 to 2
Pharmacogenetic testing in all patients and perindopril treatment only if PGXscore=0 to 2
Perindopril treatment in all patients irrespective of PGX score.
Results
Complete data on baseline clinical patient characteristics and (pharmaco)genetic profile were obtained for 8726 patients (of which 4338 were allocated to perindopril and 4388 to placebo). Median follow‐up was 4.2 years (interquartile range: 4.0–4.5), during which 785 patients (9.0%) experienced the primary endpoint of cardiovascular mortality, nonfatal MI or resuscitated cardiac arrest. Treatment with perindopril was protective in the overall study population; the number of patients on perindopril treatment that experienced the primary endpoint was 346 (8.0%) versus 439 (10.0%) on placebo (adjusted HR: 0.80; 95% CI: 0.68–0.92). Baseline characteristics of the overall study population and various subgroups according to the clinical risk level are provided in Table 1. Interaction between study treatment and clinical characteristics (including concomitant medication) was not found.
Table 1.
Baseline Study Population Characteristics
| Total Population | Clinical Risk Level | P Valuea | |||
|---|---|---|---|---|---|
| Low | Medium | High | |||
| N (%) | 8726 | 3167 (36.3) | 3474 (39.8) | 2085 (23.9) | |
| Age, y | 59.8 (9.3) | 57.7 (8.0) | 59.4 (9.2) | 63.8 (10.0) | <0.001 |
| Male sex (%) | 85.5 | 81.5 | 87.6 | 88.2 | <0.001 |
| Hypertension (%)b | 29.0 | 23.0 | 28.0 | 39.0 | <0.001 |
| Diabetes mellitus (%) | 13.0 | 4.0 | 11.0 | 30.0 | <0.001 |
| Hypercholesterolemia (%)c | 63.0 | 69.0 | 60.0 | 58.0 | <0.001 |
| Current smoking (%)d | 15.0 | 6.0 | 16.0 | 25.0 | <0.001 |
| Obesity (BMI >30 kg/m2) (%) | 21.3 | 8.2 | 24.0 | 36.7 | <0.001 |
| Symptomatic CAD (%)e | 25.4 | 9.5 | 25.9 | 48.9 | <0.001 |
| Family history of CAD (%) | 27.0 | 22.0 | 29.0 | 32.0 | <0.001 |
| Previous MI (%) | 65.0 | 44.0 | 73.0 | 84.0 | <0.001 |
| Previous revascularisation (%) | 55.0 | 75.0 | 49.0 | 33.0 | <0.001 |
| Previous stroke or PAD (%) | 8.9 | 0.8 | 5.2 | 27.5 | <0.001 |
| Concommitant medication | |||||
| Platelet inhibitors (%) | 92.0 | 94.0 | 92.0 | 89.0 | <0.001 |
| Beta‐blockers (%) | 63.0 | 62.0 | 65.0 | 63.0 | 0.104 |
| Lipid‐lowering agents (%) | 55.0 | 64.0 | 53.0 | 46.0 | <0.001 |
| Calcium antagonists (%) | 32.0 | 29.0 | 31.0 | 37.0 | <0.001 |
| Systolic blood pressure, mm Hg | 136.9 (15.2) | 132.7 (13.9) | 137.7 (15.1) | 142.1 (15.5) | <0.001 |
| Diastolic blood pressure, mm Hg | 81.8 (8.1) | 80.6 (7.9) | 82.4 (8.1) | 82.7 (8.3) | <0.001 |
| Creatinine clearance, μmol/Lf | 86.5 (25.7) | 88.9 (22.2) | 87.7 (26.6) | 80.9 (28.3) | <0.001 |
| Total cholesterol, mmol/L | 5.4 (1.0) | 5.1 (0.9) | 5.5 (1.0) | 5.7 (1.1) | <0.001 |
| Outcome | |||||
| Randomization, allocation to perindopril (%) | 49.7 | 51.0 | 48.9 | 49.5 | 0.296 |
| Primary endpoint (%) | 9.0 | 4.6 | 8.8 | 16.2 | <0.001 |
| Systolic/diastolic blood pressure reduction by perindopril, mm Hgg | 8.6/4.0 | 7.3/3.9 | 9.2/4.1 | 9.6/4.1 | <0.001/0.416 |
| Risk score | 0 to 6 | 7 to 9 | 10 to 21 | ||
| Mean clinical risk score | 7.67 (2.83) | 4.84 (1.20) | 7.93 (0.80) | 11.53 (1.74) | N.A. |
| Mean pharmacogenetic risk score | 1.82 (1.13) | 1.82 (1.12) | 1.82 (1.12) | 1.86 (1.11) | 0.435 |
Summary statistics for continuous variables are presented as mean (SD). Categorical data are summarized as percentages. BMI indicates body mass index; CAD, coronary artery disease; MI, myocardial infarction; N.A., not applicable; PAD, peripheral artery disease.
For differences between low, medium, and high clinical risk levels.
Blood pressure >140/90 mm Hg or receiving antihypertensive treatment.
Previously known total cholesterol >6.5 mmol/L or receiving lipid‐lowering treatment.
Use of tobacco within the last month.
Stable angina pectoris or history of congestive heart failure.
Estimation by Cockroft‐Gault equation.
Blood pressure reduction was calculated as the mean difference in blood pressure from screening visit 1 to randomization after the 4‐week run‐in period of the Europa trial in which all patients were treated with perindopril.
Significant baseline clinical risk predictors and the point‐scoring system, which derived from backward elimination, are presented in Table 2. The log HRs from the final multivariable model that were converted to the clinical risk score are provided in Table S1. The clinical risk score could theoretically range from 0 to 32, yet calculated individual scores within our study population ranged from 0 to 21 with a mean value of 7.67±2.83 (Figure 1). The 33rd and 67th percentiles were at 6.00 and 9.00 points, respectively, and used as cutoffs in order to distinguish low, medium, and high clinical risk levels. The skewness of the distribution (Figure 1) prevented formation of 3 groups of similar size. It should be noted that the high‐risk group consists of 23.9% of the overall study population (Tables 1 and 3). Incidences of all known baseline cardiovascular risk factors were highest in the higher clinical risk groups (Table 1), with the exception of previously diagnosed hypercholesterolemia, which actually was lowest in the high‐risk subgroup. In accord, high‐risk patients also presented with the lowest rate of statin use. These findings, however, were counterbalanced by the fact that patients in the high‐risk subgroup did have the highest total cholesterol levels (Table 1).
Table 2.
Clinical Risk Scores of Baseline Risk Parameters
| Continuous Clinical Risk Parameters | Clinical Risk Score Points | |||
|---|---|---|---|---|
| Age, y | Systolic Blood Pressure | Creatinine Clearance | Total Cholesterol, mmol/L (mg/dL) | |
| <67 | ≤130 | >70 | ≤3.5 (≤135) | 0 |
| 67 to 69 | >130 to ≤160 | >55 to ≤70 | >3.5 to ≤5.0 (>135–≤193) | 1 |
| 70 to 72 | >160 | >35 to ≤55 | >5.0 to ≤6.5 (>193–≤251) | 2 |
| 73 to 76 | ≤35 | >6.5 to ≤8.0 (>251–≤309) | 3 | |
| 77 to 79 | >8.0 (>309) | 4 | ||
| 80 to 82 | 5 | |||
| 83 to 85 | 6 | |||
| >85 | 7 | |||
| Dichotomous Clinical Risk Parameters | Clinical Risk Score Points |
|---|---|
| Previous stroke or PAD | 3 |
| Male sex | 2 |
| Obesity (BMI >30 kg/m2) | 2 |
| Current smoking | 2 |
| Symptomatic CAD | 2 |
| Diabetes mellitus | 2 |
| Previous MI | 2 |
| Family history of CAD | 1 |
| Previous revascularisation | −1 |
The range of clinical risk scores=0 to 32 and points for each of applicable variables need to be added to each other. BMI indicates body mass index; CAD, coronary artery disease; MI, myocardial infarction; PAD, peripheral artery disease.
Figure 1.
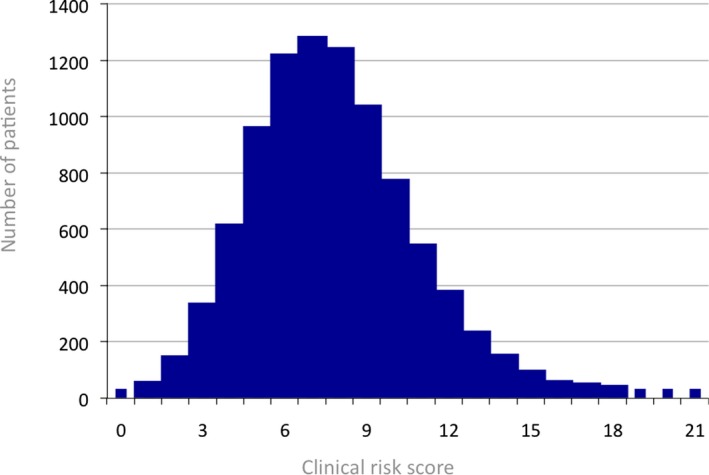
Clinical risk score distribution. The mean value of the clininal risk score (N=8726) was of 7.67±2.83.
Table 3.
Distribution of Patients Over Clinical and Pharmacogenetic Risk Strata
| Clinical risk level | Low | Medium | High |
| Clinical risk score | 0 to 6 | 7 to 9 | 10 to 21 |
| Pharmacogenetic risk score N (%) | |||
| 0 | 362 (4.1) | 390 (4.5) | 232 (2.7) |
| 1 | 945 (10.8) | 1037 (11.9) | 618 (7.1) |
| 2 | 1027 (11.8) | 1144 (13.1) | 655 (7.5) |
| ≥3 | 833 (9.5) | 903 (10.3) | 580 (6.6) |
Treatment benefit of perindopril was only demonstrated within the group of patients with pharmacogenetic risk scores <3 (N=6410, 73.5% of the total study population). The linear‐by‐linear association P value for the entire table is 0.43.
The primary endpoint rates in the low, medium, and high clinical risk groups were 4.6%, 8.8%, and 16.2%, respectively (P<0.001; Table 1). These differences in event rate can be explained by the observed differences in baseline clinical risk factors, but not by confounding attributed to study drug allocation, given that the latter was similar over the 3 clinical risk strata (P=0.296; Table 1).
Adjusted HRs for the treatment effect of perindopril were 0.72, 0.70, and 0.91 for the lowest to highest clinical risk tertiles, respectively. Heterogeneity of treatment effect was tested and ruled out (P=0.31). Thus, the relative treatment benefit was not modified by the baseline clinical risk level. However, baseline clinical risk level did modify absolute risk reductions. Use of perindopril in the overall study population (n=8726) resulted in a 2.23% risk reduction of the primary endpoint (95% CI: 1.03–3.44; annual NNT: 189; 95% CI: 122–401). However, absolute risk reductions varied from 1.24% to 2.17% and 3.97% in the lowest, medium, and highest clinical risk tertiles. As a consequence, NNTs were inversely related to increasing clinical risk scores (Table 4).
Table 4.
Numbers Needed to Treat (Per Annum)
| Clinical risk level | Low | Medium | High |
| Clinical risk score | 0 to 6 | 7 to 9 | 10 to 21 |
| NNT per clinical risk stratum | 382 | 218 | 119 |
| NNT per pharmacogenetic risk stratum | |||
| 0 | 93 | 54 | 29 |
| 1 | 164 | 92 | 50 |
| 2 | 521 | 298 | 164 |
| ≥3a | −529 | −302 | −164 |
Stratum with nonsignificant risk increase attributed to use of perindopril. NNT indicates numbers needed to treat.
The pharmacogenetic risk scoring system, based on the previously identified 3 SNPs, is presented in Table S2. Risk alleles for lack of treatment benefit were T, C, and G for Rs275651, Rs5182 and Rs12050217, respectively. The individual PGXscores range from 0 to 6 points with a mean value of 1.82±1.13 (Table 1). Significant heterogeneity of treatment effect across pharmacogenetic profiles was observed. A pronounced treatment benefit was observed in 6410 patients (73.5%), with PGXscore <3 (adjusted HR: 0.67; 95% CI: 0.56–0.79), whereas no benefit was observed in the remaining subgroup of 2316 patients (26.5%) with PGXscore ≥3 (adjusted HR: 1.26; 95% CI: 0.97–1.67; Table 3 for patient distribution).
Use of perindopril in patients with PGXscores of 0 and 1 point resulted in absolute risk reductions of 7.50% (95% CI: 3.69–11.73) and 4.30% (95% CI: 2.00–6.53), respectively. Consequently, annual NNTs were 55 (95% CI: 113–38) for patients with a PGXscore of 0 and 97 (95% CI: 210–63) for patients with a PGXscore of 1. The point estimate of the absolute risk reduction associated with use of perindopril in the subgroup of PGXscore of 2 was in the same positive direction, yet nonsignificant (1.34%; 95% CI: −0.77 to 3.47; and NNT [per annum]=311; 95% CI: −546 to 122).
In contrast, a nonsignificant estimated absolute risk increase of 1.32% was observed in patients with a PGXscore ≥3 using perindopril (95% CI for risk increase: −0.97 to 3.67 and NNT [per annum]=−315; 95% CI: −118 to 433).
Combined Baseline Clinical and Pharmacogenetic Risk Profiles
Mean pharmacogenetic risk scores were identical over all 3 clinical risk strata (P=0.435; Table 1), and formal testing did not trace interaction between clinical and PGXscores. The Distribution of patients over the various clinical and pharmacogenetic risk strata is given in Table 3. Figures 2A through 2D describe the relation between absolute risks of the primary endpoint, clinical risk profile, and treatment for each of the separate pharmacogenetic risk strata. Lack of treatment benefit was observed across the entire spectrum of clinical risk in patients with a PGXscore ≥3 (Figures 2D and 3).
Figure 2.
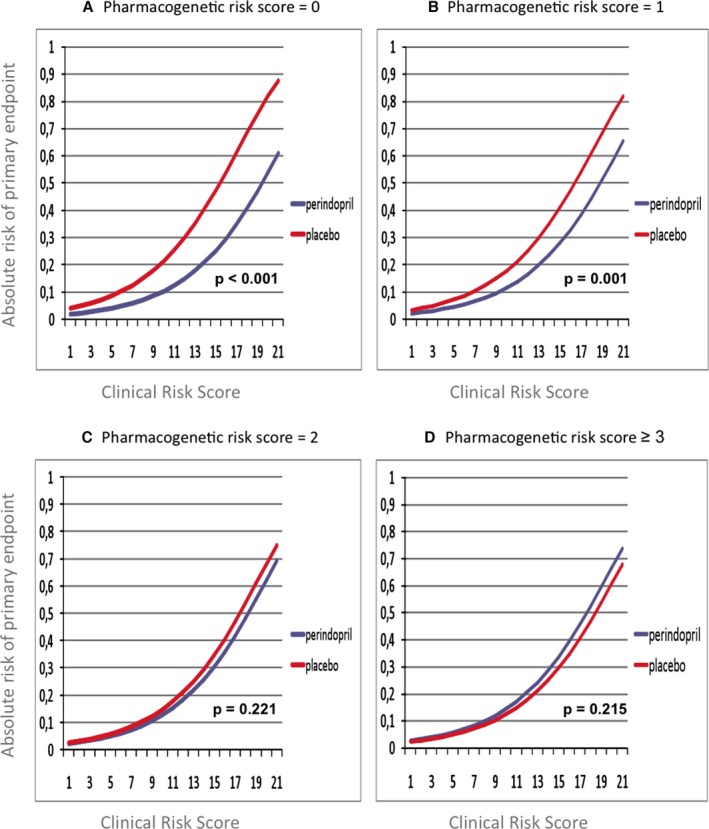
Absolute risks of the primary endpoint across different clinical (X‐axis) and pharmocogenetic risk strata (Panels A through D). P values were derived from multivariate Cox proportional hazards regression models fitted with the following covariates: clinical risk score; PGXscore; treatment; and treatment×PGXscore interaction. PGXscore indicates pharmacogenetic risk score.
Figure 3.
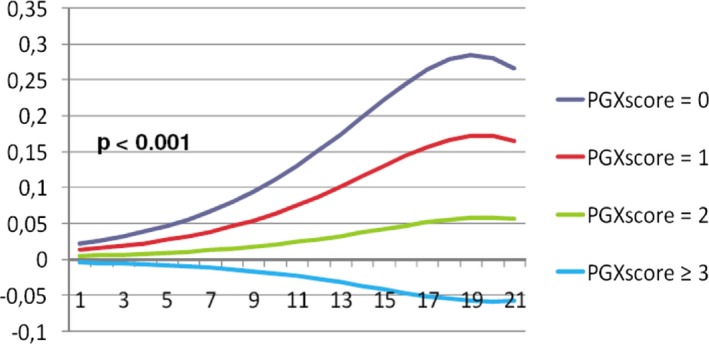
Absolute risk reduction (Y‐axis on a 0 to 1 scale) by perindopril across different levels of clinical (X‐axis) and pharmacogenetic risk. PGXscore indicates pharmacogenetic risk score.
Increasing clinical risk scores led to increasingly pronounced risk differences between perindopril and placebo in all pharmacogenetic strata. Hence, extremes of treatment effect were found in patients with high clinical risk profiles. For example, use of perindopril in patients with a clinical risk score of 19 resulted in an estimated absolute risk reduction of 28.42% (95% CI: 22.46–34.09) in the case of a PGXscore of 0 versus an estimated risk increase of 5.82% (95% CI: 1.78–9.83) in the case of a PGXscore ≥3 (Figure 3). Concordantly, NNTs decreased in subgroups with higher clinical risk profiles and lower PGXscores, both of which were associated with more pronounced treatment effects (Table 4). Estimated NNTs were as low as 29 (95% CI: 17–113) in patients with a high clinical risk profile and a PGXscore of 0, whereas those with a low clinical risk profile and a PGXscore of ≥3 did not experience any benefit (NNT=−529; 95% CI: −105 to 189).
In separate analyses with only cardiovascular mortality or nonfatal MI as sole endpoint, we observed directional concordance, compared to the presented analysis of the combined primary endpoint, with respect to NNTs over the various clinical and pharmacogenetic risk strata.
Discrimination and Calibration of the Clinical and Combined Risk Models
Calibration and discrimination were assessed for 2 models: (1) the model consisting of clinical risk score only and (2) the full model consisting of clinical risk score, PGXscore, and treatment×PGXscore interaction.
Addition of pharmacogenetic information on top of clinical risk profile resulted in better discrimination. The c‐index for the full model (0.68; 95% CI: 0.66–0.70) was significantly higher than the c‐index for the model consisting of the clinical risk score only (0.66; 95% CI: 0.64–0.68; P=0.0015). The full model also resulted in a significantly better discrimination when assessed with integrated discrimination improvement (magnitude of increase in IDI: 0.00472; P=0.0002). Validation of both models by bootstrap methods showed that the bias in the estimated discrimination performance (c‐index) is likely to be small, because the mean optimism estimates were only 0.006 and 0.007 for the clinical risk score and the full model, respectively. Finally, the H‐L goodness‐of‐fit tests were nonsignificant (P=0.43 for the model with the clinical risk score only and P=0.94 for the full model), indicating adequate calibration for both models (Figure 4).
Figure 4.
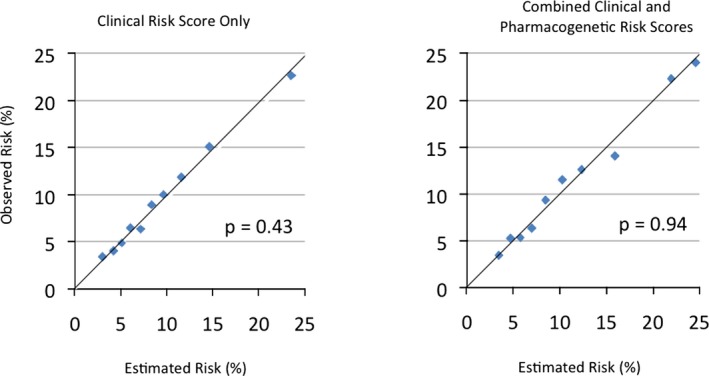
Observed versus estimated risks according to the clinical and combined (full) risk prediction models. P values were derived from Hosmer‐Lemeshow goodness‐of‐fit test.
Cost‐Effectiveness of Tailored Perindopril Treatment on the Basis of Pharmacogenetic Testing
The results of the cost‐effectiveness analysis, against the strategy of no treatment with perindopril as comparator, are displayed in Table 5.
Table 5.
Costs, Gained Life‐Years, and Incremental Cost‐Effectiveness Ratio of Various Treatment Strategies, Against the Strategy of No Treatment With Perindopril as Comparator
| Strategy | No. of Patients Treated With Perindopril, N (%) | Incremental Costs (Weighted) | Life‐Years Gained (Weighted) | ICER |
|---|---|---|---|---|
| 1. Pharmacogenetic testing only in patients with a high clinical risk score (≥10) and perindopril treatment only if PGXscore=0 to 2 | 1505/8726 (17.2) | 30.38 | 0.0017 | 18 139 |
| 2. Pharmacogenetic testing only in patients with a medium or high clinical risk score (≥7) and perindopril treatment only if PGXscore=0 to 2 | 4076/8726 (46.7) | 90 | 0.0032 | 27 987 |
| 3. Pharmacogenetic testing in all patients and perindopril treatment only if GXscore=0 to 2 | 6410/8726 (73.5) | 147 | 0.0040 | 36 743 |
| 4. Perindopril treatment in all patients irrespective of PGXscore | 8726 (100) | 232 | 0.0035 | 67 230 |
The time horizon was restricted to the duration of the EUROPA trial/PERGENE study (mean follow‐up of 4.2 years). Costs are in euros. ICER indicates incremental cost‐effectiveness ratio; PGXscore, pharmacogenetic risk score.
The highest number of gained life‐years is observed in strategies 3 and 4. Strategy 3 implies that all patients are genetically tested and only those with a PGXscore of 0 to 2 are treated with perindopril. Strategy 4 implies that none of the patients are genetically tested and all are treated with perindopril. Strategy 4, however, is dominated by strategy 3. The lower incremental cost‐effectiveness ratio (ICER) of strategy 3 indicates that tailored perindopril therapy on the basis of the PGXscore will ultimately reduce costs, with a similar effectiveness in terms of gained life‐years.
Strategy 1 results in the least life‐years gained, but also in the least costs and the lowest ICER and therefore may be an option when strictly reasoning from the cost perspective alone.
Discussion
The present study highlights that clinical as well as pharmacogenetic determinants independently modify absolute treatment benefit by ACE inhibitor perindopril in a population of patients with stable CAD. Moreover, both clinical and pharmacogenetic profiles could be expressed in risk scores that are fairly simple to use for clinical decision making. We propose the use of a PGXscore on top of known clinical risk factors for better risk stratification and more concrete estimation of absolute treatment benefits of ACE‐inhibitor therapy in daily clinical practice. Increasing clinical risk scores and decreasing PGXscores were consistently and positively related to the absolute treatment benefit by ACE‐inhibitor perindopril. Impressive risk gradients and, as a consequence, important differences in NNT were found across various subgroups. The annual NNT in the overall study population was 189, whereas estimates as low as 106 for the entire clinical high‐risk subgroup and even 29, in case of a combined high clinical risk profile and a PGXscore of 0, were observed. On the other hand, the entire subgroup of patients with a PGXscore ≥3 (26.5% of the overall study cohort) was characterized by a lack of treatment benefit, which was consistent across all 3 clinical risk levels.
The clinical risk score in our study was based upon easily obtainable traditional risk factors that have repeatedly proven to be valuable predictors.18, 19, 20 The full model predicted the highest absolute risk reductions in patients with higher clinical risk profiles. In this regard, it remains important to emphasize that formally no heterogeneity of relative treatment effect was found across the various clinical risk levels. Furthermore, the mean clinical risk score in the high‐risk level was 11.53. Scores of, for example, 19 can therefore be regarded as extremely high. Such extreme risk scores were under‐represented in our randomized, clinical trial data, but nevertheless such patients do present themselves in clinical practice. It is plausible that in such extremely high‐risk individuals, the risk is largely determined by the aforementioned risk factors, and that an ACE inhibitor alone will have relatively less effect on survival. In other words, the magnitude of both controllable and uncontrollable clinical risk factors in such a patient could have a relatively more profound effect on the risk of reaching the primary endpoint than the potential relative treatment benefit by an ACE inhibitor alone. Obviously, the absolute risk benefit will remain high in such patients, and treatment with an ACE‐inhibitor should therefore be warranted. This finding, however, once again emphasizes the necessity of proper management of all controllable risk factors in patients with stable CAD.
With this in mind, it is remarkable that a history of coronary revascularization was associated with a modestly reduced risk for the primary endpoint (−1 point) in the presented risk model. This particular observation should be interpreted with some reservation, given that several specifically designed trials, such as RITA‐2,21 COURAGE,22 and BARI 2D,23 failed to demonstrate survival benefit of coronary revascularization over optimal medical therapy.
The pharmacogenetic risk score in our study was based upon 3 SNPs that have previously emerged after comprehensive coverage of the RAAS and KB systems and subsequent correction for multiple testing.12 Furthermore, the pharmacogentic risk score has previously been replicated in participants of the PROGRESS‐trial.12 Clinical risk factors,6 renal function,8 degree of blood pressure reduction,9 and a number of biomarkers24, 25 have been explored within the EUROPA trial, yet only pharmacogenetic information has permitted to distinguish responders to perindopril from nonresponders (26.5% of all patients). Furthermore, the PGXscore accentuated striking differences in absolute treatment benefits of ACE‐inhibitor therapy within each of the separate clinical risk strata.
The data that are presented here are unique. Although it is widely recognized that both phenotype as well as genotype play a fundamental role in health and disease outcome, very few reports exist that actually combine both for prognostication. To our best knowledge, this is the first and only article that combines clinical and genetic information in patients with CAD. Pharmacogenetic information is successfully translated into a potential clinical utility to study the gradients of treatment effect by an ACE inhibitor. The sample size is large and various additional qualities of a well‐designed placebo‐controlled, double‐blinded RCT, such as high‐quality phenotypical data and independent event adjudication, are apparent. Previous studies that have investigated the relation between genetic variation and treatment benefit by ACE‐inhibitor therapy usually were characterized by small sample sizes and nonrandomized designs without placebo controls.26 Only 2 studies with large sample sizes have been reported. Harrap et al. studied macrovascular events, dementia, and cognitive decline in 5688 patients with a history of cerebrovascular disease in the PROGRESS study and found no interaction between genetic variation and treatment benefit by perindopril.27
Negative findings were also published by the GenHAT investigators, who studied cardiovascular mortality and nonfatal MI in 7528 patients on a lisinopril‐based regimen in the setting of an active‐controlled RCT.28 These 2 studies obviously differ from our present study in the type of study population, endpoints, and study drug. The most remarkable difference with our study, however, is the fact that both studies solely focussed on a single ACE insertion/deletion polymorphism, thus not taking account of the full complexity of the RAAS and KB systems. Furthermore, our PGXscore was replicated in the PROGRESS study, in which a similar direction and magnitude of pharmacogenetic interaction was observed.12
Our findings also have some limitations. This study describes differences in treatment benefit across a range of clinical and genetic subgroups. The constituents of the clinical scoring system are all well‐established cardiovascular risk factors, and the PGXscore has been replicated. Still, it is important to realize that, in general, any post‐hoc analysis based on subgroups should primarily be regarded as hypothesis generating. Confirmation of our findings in other large data sets would invigorate the presented conclusions and derived clinical implications. The EUROPA trial was powered for detection of treatment benefit for the entire study population irrespective of clinical or pharmacogenetic risk categories. Thus, lack of power cannot be excluded as an explanation for the observed nonsignificant treatment benefit in the higher PGX scores. On the other hand, it must be noted that the absolute numbers of study participants in PGXscores ≥2 are higher than those below (Table 3).
Patients enrolled in the EUROPA trial primarily consisted of Caucasian males without overt heart failure, who were randomized to placebo or perindopril 8 mg daily. The generalizability of the presented results toward other patient groups, for example, those with a higher proportion of women, heart failure, patients of other ethnicities, or those using other ACE inhibitors or lower dosages of perindopril, may therefore be limited. Testing of these particular genetic variants in a large, randomized heart failure trial would be required before suggesting that the same phenomenon exists in that very different patient group.
Our combined primary endpoint consisted of cardiovascular mortality, nonfatal MI, and resuscitated cardiac arrest. Resuscitated cardiac arrest, however, only occurred in very few instances. Therefore, our results with respect to clinical and pharmacogenetic determinants of treatment benefit are primarily associated with incidence of cardiovascular mortality and nonfatal MI.
In order to facilitate clinical utility and ease of use, we specifically chose to develop an integer‐based risk score. Disadvantages of integer‐based risk scores in general include the fact that not all variables have exactly the same contribution to the model. Furthermore, certain combinations of risk factors may act synergistically to increase risk in a manner that is more than additive. Such synergy may be underestimated in a purely additive, integer‐based risk score.29
Replication of the 3 SNPs that formed the PGXscore in the PROGRESS trial14 demonstrated concordant associations between the risk score and treatment benefit by perindopril.12 The individual interaction terms of the 3 SNPs, however, did not reach statistical significance in that particular trial because of limited statistical power (replication could take place in 1051 patients only). Unfortunately, larger replication cohorts are not available.
Although the clinical risk model consists of established cardiovascular risk factors, formally the combined clinical and pharmacogenetic risk score has not been independently validated on a separate data set.
Finally, our risk model does not contain data on circulating serum biomarkers other than total cholesterol and creatinine levels. A prespecified substudy of the EUROPA trial, called PERTINENT, actually did study bradykinin and angiotensin II, but also markers of endothelial function (nitric oxide synthase) and inflammation (C‐reactive protein, tumour necrosis factor alpha, and von Willebrand factor). Use of perindopril was reflected in various circulating biomarker levels, which were interpreted as a biochemical indication of normalization of the angiotensin II/bradykinin balance, reduction of inflammation, and prevention of endothelial apoptosis.24, 25 Unfortunately, the cohort in which these biomarkers were assessed was too small in order to properly study the interaction between the various serum biomarkers and treatment effect by perindopril on clinical endpoints.
In conclusion, our results show that a combination of phenotypical and genetic information can be used to demonstrate a range of gradients of absolute treatment benefit by ACE‐inhibitor therapy in an otherwise seemingly homogeneous population of patients with stable CAD. Clinical and pharmacogenetic profiling in individual patients may both clarify their distinct level of absolute risk of adverse events and furthermore also the degree of risk reduction by an ACE‐inhibitor regimen. Refraining from ACE‐inhibitor therapy in those patients that are expected to lack any treatment benefit may avoid unnecessary side effects, reduce health care costs, and increase overall efficacy of the drug. Future RCTs could advance the field of individualized medicine by incorporation of a similar combined clinical and pharmacogenetic approach in their study design.
Sources of Funding
The PERGENE study is funded by a Dutch noncommercial charity: the Netherlands Heart Foundation (Grant No.: NHS2005B219). Oemrawsingh is supported by the Netherlands Heart Foundation (Grant No.: NHS2007B012). Prof Kim Fox is a National Institute for Health Research (NIHR) senior investigator, supported by the NIHR Cardiovascular Biomedical Medical Research Unit at the Royal Brompton Hospital.
Disclosures
There is no commercial association that might pose a conflict of interest in connection with this manuscript. The sponsor of the EUROPA trial, Servier, had no role in the design, conduct, analysis, or interpretation of this substudy, nor in the preparation, review, or approval of the manuscript.
Supporting information
Table S1. Regression Coefficients From the Final Multivariable Model That Were Converted to the Clinical Risk Score
Table S2. Pharmacogenetic Risk Score on the Basis of Allele Distribution Appendix S1. EUROPA/PERGENE Investigators.
Acknowledgments
Oemrawsingh, Brugts, and Prof Boersma were instrumental in the conception and drafting of the manuscript. All other co‐authors have reviewed, critically revised the intellectual content of, and finally approved submission of the manuscript. Oemrawsingh, Brugts, and Boersma had full access to all study data and take responsibility for the integrity of the data and the accuracy of the analysis.
(J Am Heart Assoc. 2016;5:e002688 doi: 10.1161/JAHA.115.002688)
Accompanying Tables S1, S2, and Appendix S1 (which lists the members of the EUROPA/PERGENE Investigators) are available at http://jaha.ahajournals.org/content/5/3/e002688/suppl/DC1
References
- 1. Fox KM. Efficacy of perindopril in reduction of cardiovascular events among patients with stable coronary artery disease: randomised, double‐blind, placebo‐controlled, multicentre trial (the EUROPA study). Lancet. 2003;362:782–788. [DOI] [PubMed] [Google Scholar]
- 2. Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G. Effects of an angiotensin‐converting‐enzyme inhibitor, ramipril, on cardiovascular events in high‐risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342:145–153. [DOI] [PubMed] [Google Scholar]
- 3. Fox K, Garcia MAA, Ardissino D, Buszman P, Camici PG, Crea F, Daly C, De Backer G, Hjemdahl P, Lopez‐Sendon J, Marco J, Morais J, Pepper J, Sechtem U, Simoons M, Thygesen K, Priori SG, Blanc J‐J, Budaj A, Camm J, Dean V, Deckers J, Dickstein K, Lekakis J, McGregor K, Metra M, Morais J, Osterspey A, Tamargo J, Zamorano JL. Guidelines on the management of stable angina pectoris: executive summary: the task force on the management of stable angina pectoris of the European Society of Cardiology. Eur Heart J. 2006;27:1341–1381. [DOI] [PubMed] [Google Scholar]
- 4. Fraker TD, Fihn SD; Writing on behalf of the 2002 Chronic Stable Angina Writing Committee, 2002 WRITING COMMITTEE MEMBERS , Gibbons RJ, Abrams J, Chatterjee K, Daley J, Deedwania PC, Douglas JS, Ferguson TB, Fihn SD, Fraker TD, Gardin JM, O'Rourke RA, Pasternak RC, Williams SV, Smith SC, Jacobs AK, Adams CD, Anderson JL, Buller CE, Creager MA, Ettinger SM, Halperin JL, Hunt SA, Krumholz HM, Kushner FG, Lytle BW, Nishimura R, Page RL, Riegel B, Tarkington LG, Yancy CW. 2007 chronic angina focused update of the ACC/AHA 2002 guidelines for the management of patients with chronic stable angina: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines Writing Group to develop the focused update of the 2002 guidelines for the management of patients with chronic stable angina. Circulation. 2007;116:2762–2772. [DOI] [PubMed] [Google Scholar]
- 5. De Backer G, Ambrosioni E, Borch‐Johnsen K, Brotons C, Cifkova R, Dallongeville J, Ebrahim S, Faergeman O, Graham I, Mancia G, Cats VM, Orth‐Gomér K, Perk J, Pyörälä K, Rodicio JL, Sans S, Sansoy V, Sechtem U, Silber S, Thomsen T, Wood D, Albus C, Bages N, Burell G, Conroy R, Christian Deter H, Hermann‐Lingen C, Humphries S, Fitzgerald A, Oldenburg B, Schneiderman N, Uutela A, Williams R, Yarnell J, Priori SG, Angeles Alonso Garcia M, Blanc J‐J, Budaj A, Cowie M, Dean V, Deckers J, Fernández Burgos E, Lekakis J, Lindahl B, Mazzotta G, McGregor K, Morais J, Oto A, Smiseth O, Trappe H‐J, Budaj A, Agardh C‐D, Bassand J‐P, Deckers J, Godycki‐Cwirko M, Heagerty A, Heine R, Home P, Priori S, Puska P, Rayner M, Rosengren A, Sammut M, Shepherd J, Siegrist J, Simoons M, Tendera M, Zanchetti A. European guidelines on cardiovascular disease prevention in clinical practice. Eur Heart J. 2003;24:1601–1610. [DOI] [PubMed] [Google Scholar]
- 6. Deckers JW, Goedhart DM, Boersma E, Briggs A, Bertrand M, Ferrari R, Remme WJ, Fox K, Simoons ML; on behalf of the EUROPA Investigators . Treatment benefit by perindopril in patients with stable coronary artery disease at different levels of risk. Eur Heart J. 2006;27:796–801. [DOI] [PubMed] [Google Scholar]
- 7. Pitt B. ACE inhibitors for patients with vascular disease without left ventricular dysfunction—may they rest in PEACE? N Engl J Med. 2004;351:2115–2117. [DOI] [PubMed] [Google Scholar]
- 8. Brugts JJ, Boersma E, Chonchol M, Deckers JW, Bertrand M, Remme WJ, Ferrari R, Fox K, Simoons ML. The cardioprotective effects of the angiotensin‐converting enzyme inhibitor perindopril in patients with stable coronary artery disease are not modified by mild to moderate renal insufficiency: insights from the EUROPA trial. J Am Coll Cardiol. 2007;50:2148–2155. [DOI] [PubMed] [Google Scholar]
- 9. Remme WJ, Deckers JW, Fox KM, Ferrari R, Bertrand M, Simoons ML. Secondary prevention of coronary disease with ACE inhibition–does blood pressure reduction with perindopril explain the benefits in EUROPA? Cardiovasc Drugs Ther. 2009;23:161–170. [DOI] [PubMed] [Google Scholar]
- 10. Brugts JJ, Ninomiya T, Boersma E, Remme WJ, Bertrand M, Ferrari R, Fox K, MacMahon S, Chalmers J, Simoons ML. The consistency of the treatment effect of an ACE‐inhibitor based treatment regimen in patients with vascular disease or high risk of vascular disease: a combined analysis of individual data of ADVANCE, EUROPA, and PROGRESS trials. Eur Heart J. 2009;30:1385–1394. [DOI] [PubMed] [Google Scholar]
- 11. Brugts JJ, de Maat MPM, Boersma E, Witteman JCM, van Duijn C, Uitterlinden AG, Bertrand M, Remme W, Fox K, Ferrari R, Danser AHJ, Simoons ML. The rationale and design of the PERindopril GENEtic association study (PERGENE): a pharmacogenetic analysis of angiotensin‐converting enzyme inhibitor therapy in patients with stable coronary artery disease. Cardiovasc Drugs Ther. 2009;23:171–181. [DOI] [PubMed] [Google Scholar]
- 12. Brugts JJ, Isaacs A, Boersma E, van Duijn CM, Uitterlinden AG, Remme W, Bertrand M, Ninomiya T, Ceconi C, Chalmers J, MacMahon S, Fox K, Ferrari R, Witteman JCM, Danser AHJ, Simoons ML, de Maat MPM. Genetic determinants of treatment benefit of the angiotensin‐converting enzyme‐inhibitor perindopril in patients with stable coronary artery disease. Eur Heart J. 2010;31:1854–1864. [DOI] [PubMed] [Google Scholar]
- 13. Sullivan LM, Massaro JM, D'Agostino RB. Presentation of multivariate data for clinical use: the Framingham Study risk score functions. Stat Med. 2004;23:1631–1660. [DOI] [PubMed] [Google Scholar]
- 14. Randomised trial of a perindopril‐based blood‐pressure‐lowering regimen among 6,105 individuals with previous stroke or transient ischaemic attack. Lancet. 2001;358:1033–1041. [DOI] [PubMed] [Google Scholar]
- 15. DeLong ER, DeLong DM, Clarke‐Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44:837–845. [PubMed] [Google Scholar]
- 16. Pencina MJ, D'Agostino RB, D'Agostino RB, Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med. 2008;27:157–172; discussion 207–12. [DOI] [PubMed] [Google Scholar]
- 17. Harrell FE, Lee KL, Mark DB. Multivariable prognostic models: issues in developing models, evaluating assumptions and adequacy, and measuring and reducing errors. Stat Med. 1996;15:361–387. [DOI] [PubMed] [Google Scholar]
- 18. Conroy RM, Pyörälä K, Fitzgerald AP, Sans S, Menotti A, De Backer G, De Bacquer D, Ducimetière P, Jousilahti P, Keil U, Njølstad I, Oganov RG, Thomsen T, Tunstall‐Pedoe H, Tverdal A, Wedel H, Whincup P, Wilhelmsen L, Graham IM. Estimation of ten‐year risk of fatal cardiovascular disease in Europe: the SCORE project. Eur Heart J. 2003;24:987–1003. [DOI] [PubMed] [Google Scholar]
- 19. Clayton TC, Lubsen J, Pocock SJ, Voko Z, Kirwan B‐A, Fox KAA, Poole‐Wilson PA; on behalf of the ACTION investigators . Risk score for predicting death, myocardial infarction, and stroke in patients with stable angina, based on a large randomised trial cohort of patients. BMJ. 2005;331:869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boersma E, Pieper KS, Steyerberg EW, Wilcox RG, Chang W‐C, Lee KL, Akkerhuis KM, Harrington RA, Deckers JW, Armstrong PW, Lincoff AM, Califf RM, Topol EJ, Simoons ML. Predictors of outcome in patients with acute coronary syndromes without persistent ST‐segment elevation: results from an international trial of 9461 patients. Circulation. 2000;101:2557–2567. [DOI] [PubMed] [Google Scholar]
- 21. Coronary angioplasty versus medical therapy for angina: the second Randomised Intervention Treatment of Angina (RITA‐2) trial. RITA‐2 trial participants. Lancet. 1997;350:461–468. [PubMed] [Google Scholar]
- 22. Boden WE, O'Rourke RA, Teo KK, Hartigan PM, Maron DJ, Kostuk WJ, Knudtson M, Dada M, Casperson P, Harris CL, Chaitman BR, Shaw L, Gosselin G, Nawaz S, Title LM, Gau G, Blaustein AS, Booth DC, Bates ER, Spertus JA, Berman DS, Mancini GBJ, Weintraub WS. Optimal medical therapy with or without PCI for stable coronary disease. N Engl J Med. 2007;356:1503–1516. [DOI] [PubMed] [Google Scholar]
- 23. Frye RL, August P, Brooks MM, Hardison RM, Kelsey SF, MacGregor JM, Orchard TJ, Chaitman BR, Genuth SM, Goldberg SH, Hlatky MA, Jones TLZ, Molitch ME, Nesto RW, Sako EY, Sobel BE. A randomized trial of therapies for type 2 diabetes and coronary artery disease. N Engl J Med. 2009;360:2503–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ceconi C, Fox K, Remme W, Simoons M, Bertrand M, Parrinello G, Kluft C, Blann A, Cokkinos D, Ferrari R. ACE inhibition with perindopril and endothelial function. Results of a substudy of the EUROPA study: PERTINENT. Cardiovasc Res. 2007;73:237–246. [DOI] [PubMed] [Google Scholar]
- 25. Ferrari R, Fox K. Insight into the mode of action of ACE inhibition in coronary artery disease. Drugs. 2009;69:265–277. [DOI] [PubMed] [Google Scholar]
- 26. Danser AHJ, Batenburg WW, van den Meiracker AH, Danilov SM. ACE phenotyping as a first step toward personalized medicine for ACE inhibitors. Why does ACE genotyping not predict the therapeutic efficacy of ACE inhibition? Pharmacol Ther. 2007;113:607–618. [DOI] [PubMed] [Google Scholar]
- 27. Harrap SB, The ACE. Gene I/D polymorphism is not associated with the blood pressure and cardiovascular benefits of ACE inhibition. Hypertension. 2003;42:297–303. [DOI] [PubMed] [Google Scholar]
- 28. Arnett DK. Pharmacogenetic association of the angiotensin‐converting enzyme insertion/deletion polymorphism on blood pressure and cardiovascular risk in relation to antihypertensive treatment: the Genetics of Hypertension‐Associated Treatment (GenHAT) study. Circulation. 2005;111:3374–3383. [DOI] [PubMed] [Google Scholar]
- 29. Cooney MT, Dudina AL, Graham IM. Value and limitations of existing scores for the assessment of cardiovascular risk: a review for clinicians. J Am Coll Cardiol. 2009;54:1209–1227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Regression Coefficients From the Final Multivariable Model That Were Converted to the Clinical Risk Score
Table S2. Pharmacogenetic Risk Score on the Basis of Allele Distribution Appendix S1. EUROPA/PERGENE Investigators.