

Abstract
Background
The regulator of G‐protein signaling 5 (Rgs5), which functions as the regulator of G‐protein‐coupled receptor (GPCR) including muscarinic receptors, has a potential effect on atrial muscarinic receptor‐activated IKA ch current.
Methods and Results
In the present study, hearts of Rgs5 knockout (KO) mice had decreased low‐frequency/high‐frequency ratio in spectral measures of heart rate variability. Loss of Rgs5 provoked dramatically exaggerated bradycardia and significantly (P<0.05) prolonged sinus nodal recovery time in response to carbachol (0.1 mg/kg, intraperitoneally). Compared to those from wild‐type (WT) mice, Langendorff perfused hearts from Rgs5 KO mice had significantly (P<0.01) abbreviated atrial effective refractory periods and increased dominant frequency after administration of acetylcholine (ACh; 1 μmol/L). In addition, whole patch clamp analyses of single atrial myocytes revealed that the ACh‐regulated potassium current (IKA ch) was significant increased in the time course of activation and deactivation (P<0.01) in Rgs5 KO, compared to those in WT, mice. To further determine the effect of Rgs5, transgenic mice with cardiac‐specific overexpression of human Rgs5 were found to be resistant to ACh‐related effects in bradycardia, atrial electrophysiology, and atrial tachyarrhythmia (AT).
Conclusion
The results of this study indicate that, as a critical regulator of parasympathetic activation in the heart, Rgs5 prevents vagal‐related bradycardia and AT through negatively regulating the IKA ch current.
Keywords: atrial tachyarrhythmia, bradycardia, IKAch, regulator of G‐protein signaling 5
Subject Categories: Electrophysiology, Arrhythmias, Basic Science Research
Introduction
The autonomic nervous system (ANS) plays a critical role in the control of heart rate (HR) and atrial tachyarrhythmia (AT).1 Enhancement of parasympathetic activity or activation of acetylcholine (ACh)‐regulated potassium current (IKAch) slows sinus rate and creates an arrhythmogenic substrate for AT.2, 3, 4 Since the discovery that ACh release from the vagus nerve is associated with arrhythmias, key agents and mechanisms underlying this action of ACh have been identified. ACh released from the vagus nerve binds to the muscarinic (M2) receptor, leading to activation and dissociation of inhibitory G‐protein heterotrimers. The resulting βγ‐dimer directly activates the IKAch, slowing pacemaker depolarization and shortening the atrial effective refractory period (AERP).5
The regulator of G‐protein signaling 5 (Rgs5) functions as a GTPase‐activating protein for Gα subunits and plays an important role in negatively regulating G‐protein‐coupled receptor (GPCR)‐mediated signaling. In cardiovascular tissue, interaction of Rgs5 with Gα(q) and Gα(i) subunits inhibits activity of the M2 receptor.6, 7 Muscarinic receptors are required to reconstitute the normal activation and deactivation kinetics of IKAch channel; thus, Rgs5 is potentially an important regulator of atrial muscarinic receptor‐activated IKAch current. The present study attempts to link autonomic function with Rgs5 in the control of HR and rhythm by comparing in vivo and in vitro electrophysiological properties in Rgs5 knockout (KO) and cardiac transgenic (TG) mice.
Materials and Methods
Experimental Animals
All protocols conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85‐23, revised 1996). Experiments were approved by the animal care and use committee of Wuhan University (Wuhan, China). Male experimental mice ages 8 to 10 weeks were used in the studies. A human RGS5 cDNA construct that contained full‐length human RGS5 cDNA was cloned downstream of the human cardiac alpha‐myosin heavy chain (α‐MHC) promoter. TG mice were produced by microinjecting the α‐MHC‐RGS5 construct into fertilized mouse embryos. Male Rgs5 KO mice (C57 background) and wild‐type (WT) littermates ages 8 to 10 weeks were used in the studies.8 Genotyping was performed by polymerase chain reaction (PCR). WT (n=28), Rgs5 KO (n=28), and Rgs5 TG (n=23) mice were provided with food and water and held on standard 12‐hour light and dark cycles in a temperature‐ and humidity‐controlled house.
Telemetry Electrocardiogram Recording
Telemetry electrocardiogram (ECG) measurements (lead II) were recorded in mice under pentobarbital sodium anesthesia (60 mg/kg, IP), as described previously.9 Signals were digitized continuously at 1 kHz and recorded by using a data acquisition system (Dynamic Systems Inc., Poestenkill, NY). The software (P3) was used to analyze the telemetry ECG‐recorded data. For each mouse, analysis of cardiac rhythm and measurements of HR were performed using 24‐hour continuous experimental recording. For heart rate variability (HRV) analysis, the parameters of frequency domain, including low‐frequency (LF; 0.4–1.5 Hz) and high‐frequency (HF; 1.5–4.0 Hz) powers were analyzed.10 LF and HF powers were normalized (nLF and nHF) to account for differences in total power (TP) among animals by multiplying the power region of interest by 100 and dividing by the difference between TP and very LF power (0.0–0.4 Hz).
Preparation of Langendorff‐Perfused Hearts
Mice were heparinized (100 U, IP) and anaesthetized with pentobarbital sodium (60 mg/kg, IP), and the heart was then quickly excised and transferred to ice‐cold HEPES‐buffered Tyrode solution. The heart was then rapidly transferred and fixed to the Langendorff‐perfusion system (AD Instruments, Bella Vista, NSW, Australia). Perfusion was commenced in a retrograde manner through the aorta at 2.0 to 2.5 mL/min by a peristaltic pump (AD Instruments). In this way, the heart was perfused by the HEPES‐buffered Tyrode solution passing through the aorta into coronary arteries. After initiation of perfusion, hearts regained a pink color and spontaneous rhythmic contractions. Isolated hearts were perfused for 20 minutes before further experimental testing. Hearts that did not recover to regular spontaneous rhythm or had irreversible myocardial ischemia were discarded.
Electrical Stimulated Protocol
The programmed electrical stimulation (PES) protocol was used for AERP examinations. PES consisted of an 8‐stimuli (S1) drive train followed by a ninth extrastimulus (S2); cycle length of the S1 train was under 125 and 100 ms, respectively. AERP was defined as the longest S1 to S2 interval that could not elicit an atrial deflection. Sinus node recovery time (SNRT) was measured after a 2‐second pacing train at a basic cycle length of 100 ms and defined as the interval between the last stimulus of the pacing train and onset of the first sinus return beat. Maximal SNRT (SNRTmax) was defined as the interval between end of the stimuli and recovered sinus rhythm. Corrected SNRT (cSNRT) was calculated by subtracting SNRTmax from the RR interval before stimulation.
Arrhythmia Induction
Inducibility of AT was tested by using both PES and burst pacing (2‐ms pulses at 50 Hz, 2‐second burst duration) up to 3 minutes of pacing in both atrial locations. AT in isolated mouse hearts was defined as rapid atrial waveforms and irregular ventricular responses recorded by electrodes in atria and ventricle, respectively.
Electrogram Spectrum Analysis
To quantitate characteristics of bipolar electrograms recorded during AT, power spectrum analysis was applied by a 4096‐point fast Fourier transformation with Lab Chrat7.0 software. A dominant frequency (DF) corresponded to the highest peak in the power spectrum in the range of 5 to 40 Hz and with a resolution of 0.24 Hz.
Isolation of Atrial Cardiac Myocytes
Cardiomyocytes from Rgs5−/− and WT mice were isolated as previously described.11 Briefly, hearts were removed and retrogradely perfused on a Langendorff system with the following solutions: HEPES‐buffered Tyrode solution (5 minutes); Ca2+‐free HEPES‐buffered Tyrode solution (5 minutes); enzyme solution (15 minutes); and KB solution (5 minutes). At the end of perfusion, the whole ventricle was dissected from the heart, and atria were placed in ice‐cold KB solution. Temperature of the perfusion was maintained at 37°C. Isolated cardiac myocytes were stored in KB solution at 4°C until needed.
Cellular Electrophysiology Recording
Whole cell voltage clamp was carried out using an EPC‐9 amplifier (List Instruments, Darmstadt, Germany), and data were recorded and analyzed with Pulse‐pulsefit software interface (version 8.31; HEKA Co, Lambrecht (Pfalz), Germany). During the experiments, myocytes were continuously superfused with extracellular solution (2 mL/min) and 1 μmol/L of ACh (Sigma‐Aldrich, St. Louis, MO). Current signals were filtered at 3 kHz by an 8‐pole Bessel filter, digitized at a sampling rate of 1 kHz. All experiments were carried out at room temperature (20–22°C). IKAch was recorded with the application of 1 μmol/L of ACh for 10 to 15 seconds at a holding potential of −120 mV. Amplitude of IKAch was measured by excluding the inward rectified potassium currents (IK1) in myocytes after superfusion with 100 μmol/L of BaCl2. Current densities (pA/pF) were obtained by normalizing current amplitudes (pA) by the Cm (pF).
Solutions
(1) HEPES‐buffered Tyrode solution (in mmol/L): NaCl 130; KCl 5.4; CaCl2 1.8; MgCl2 1; Na2HPO4 0.3; HEPES 10; glucose 10; pH adjusted to 7.4 with NaOH; (2) KB solution (in mmol/L): taurine 10; glutamic acid 70; KCl 25; KH2PO4 10; glucose 22; EGTA 0.5; pH adjusted to 7.2 with KOH; (3) enzyme solution: Ca2+‐free HEPES‐buffered Tyrode containing 0.6 mg/mL of collagenase type II (Invitrogen, Carlsbad, CA), 0.1% BSA, 20 mmol/L of taurine, and 30 μmol/L of CaCl2; (4) extracellular solution containing (in mmol/L): NaCl 130; KCl 5.4; CaCl2 1; MgCl2 1; Na2HPO4 0.3; HEPES 10; glucose 10; pH adjusted to 7.4 with NaOH; and (5) pipette solution (in mmol/L): K‐aspartate 110; KCl 20; MgC12 1; MgATP 4; GTP 0.2; EGTA 0.1; and 10 HEPES; pH 7.2 with KOH.
Real‐Time PCR Analysis
mRNA levels of Kir3.1 and Kir3.4 were determined by real‐time PCR in mouse atria. The real‐time PCR procedure was carried out as previously described. Forward and reverse primers for Kir3.1 and Kir3.4 were:
| Kir3.1 | F | TCAAATCTCGGCAGACACCT | 282 bp |
| R | AACGATGACCCCAAAGCAC | ||
| Kir3.4 | F | CCTTGAACCAGACCGACATT | 187 bp |
| R | CCTGTTGCTTCTACCATTCCTT |
Western Blot Analysis
Cardiac tissue and cultured neonatal rat cardiac myocytes were lysed in RIPA lysis buffer. Protein was measured using a protein assay kit (Thermo Fisher Scientific, Waltham, MA), and equal amounts of samples (50 mg/lane) were separated using PAGE with 4% to 12% Bis‐Tris gels (Invirogen) and subsequently transferred to PVDF membranes. Immunoblots were incubated overnight at 4°C with the antibody (CST; Bioworld, St. Louis Park, MN). After washing, immunoblots were incubated with secondary immunoglobulin G antibodies. Immunoblots were scanned using the Odyssey Infrared Imaging System (LI‐COR Biosciences, Lincoln, NE), and specific protein expression levels were normalized to GAPDH for the total cell lysate.
Statistical Analysis
All data are expressed as mean±SEM. One‐way ANOVA was used for multiple comparisons of normally distributed data, whereas for comparison of 2 groups of normally distributed data, unpaired 2‐tailed Student t tests were used. A value of P<0.05 was considered significant. Patch‐clamp data were analyzed using Origin 6.0.
Results
RGS5 Expression
To examine the impact of RGS5 protein on atrial physiology, expression of Rgs5 was detected in mouse heart in vivo. Compared to WT mice, murine RGS5 expression is absent in RGS5KO mouse atria. Human Rgs5 protein levels were analyzed in various tissues by Western blot analysis. We found a robust expression of human Rgs5 protein in atria and ventricle, but did not detect it in other organs (Figure 1).
Figure 1.
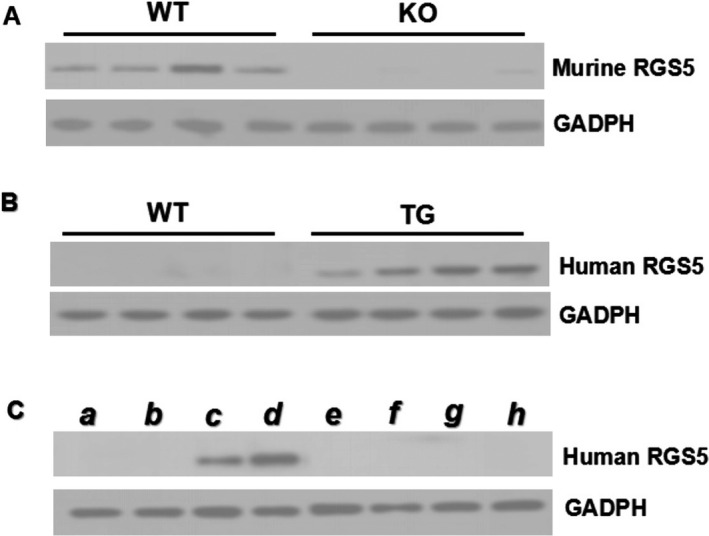
A, Representative Western blots of murine Rgs5 protein in atrial tissue from WT and RGS5 KO mice; (B) Representative Western blots of human Rgs5 protein in atrial tissue from 4 lines of TG and WT mice. C, Representative Western blot of human Rgs5 protein from different tissue of TG mice as indicated (a, lung; b, muscle; c, atria; d, ventricle; e, kidney; f, spleen; g, liver; h, brain). KO indicates knockout mice; TG, transgenic mice; WT, wild‐type mice.
Heart Rate Variability Analysis
ECG analyses showed that basal heart rates of conscious Rgs5 KO (n=8), Rgs5 TG (n=8), and WT (n=8) mice were similar over a 24‐hour period. However, to determine the autonomic effect of Rgs5, we performed spectral analysis of HRV. In Rgs5 KO mice, changes in HRV predominantly reflect changes in the vagal nervous system, and the HF power was significantly higher (P<0.05) and the LF/HF ratio was lower compared to WT mice (0.66±0.09 vs 0.88±0.05; P<0.05). However, cardiac overexpression of Rgs5 appeared to confer autonomic balance in terms of LF/HF ratio (1.01±0.17). Furthermore, administration of carbachol (0.1 mg/kg, IP) induced a dramatic bradycardia in Rgs5 KO hearts; RR interval in Rgs5 KO mice was significantly longer than that in WT mice (P<0.05). However, Rgs5 TG mice had minimum reaction to carbachol administration, indicating that serious vagus‐related dysfunction of the sinus node was induced in the absence of Rgs5 (Figure 2).
Figure 2.
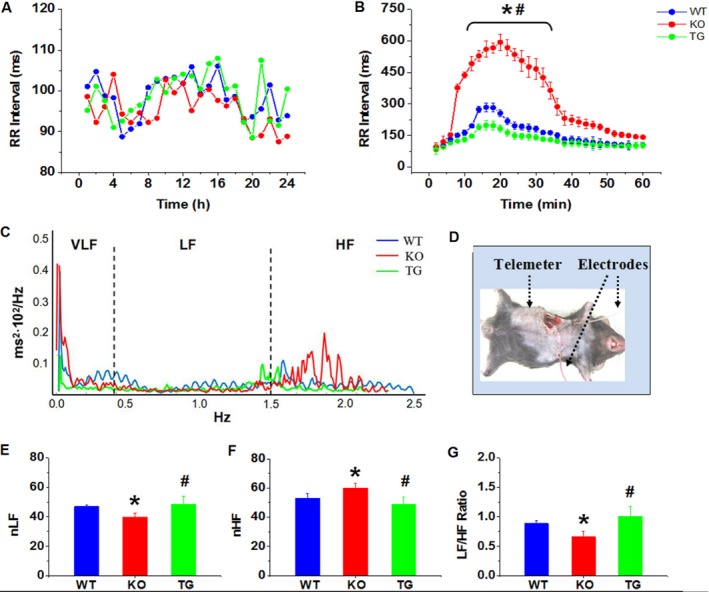
A, Mean RR interval of conscious mice were assessed by 24‐hour ECG recording. (B) Effect of CCh (0.1mg/kg, IP) on heart rate in conscious mice. C, Representative power spectrum density plot of heart rate variability (HRV). Cut‐off frequencies divided power spectrum into 3 main parts: very‐low‐frequency (VLF) power between 0.0 and 0.4 Hz; low‐frequency (LF) power between 0.4 and 1.5 Hz; and high‐frequency (HF) power between 1.5 and 4.0 Hz. D, Photograph for implantation of telemetry ECG. E through G, Frequency‐domain measures of HRV by normalized (n) HF, nLF, and LF/HF ratio. *P<0.05 vs WT and Rgs5 TG by ANOVA; # P<0.05 vs WT and Rgs5 KO by ANOVA. Eight conscious mice were used in each group. CCh indicates carbachol; ECG, electrocardiogram; KO, knockout mice; TG, transgenic mice; WT, wild‐type mice.
Function of Sinoatrial Node
To confirm sinoatrial node (SAN) disruption, sinoatrial conduction was assessed by analyzing the SNRT in isolated heart preparations. Atrial pacing revealed that the maximum SNRT and corrected SNRT of Rgs5 KO hearts (n=15) were longer than those of WT hearts (n=15) with or without ACh perfusion. However, Rgs5 TG (n=10) mice showed well recovery property in SAN after rapid atrial pacing; the SNRTmax and cSNRT were shortest among the 3 groups in either ACh condition (Table 1).
Table 1.
Electrophysiological Values Recorded in the Absence and Presence of Ach in Rgs5−/− and WT Mouse Heart
| Baseline | Ach | Washout | |||||||
|---|---|---|---|---|---|---|---|---|---|
| WT (n=15) | KO (n=15) | TG (n=10) | WT (n=15) | KO (n=15) | TG (n=10) | WT (n=15) | KO (n=15) | TG (n=10) | |
| RR | 225.3±48.0 | 267.3±57.8a | 142.2±23.7b | 262.9±38.5 | 351.7±61.9a | 166.2±28.7b | 232.1±35.0 | 265.4±42.1 | 150.1±30.1b |
| AERP125 | 35.5±6.4 | 36.3±6.7 | 33.2±4.6 | 24.9±4.6 | 12.3±3.4a | 29.1±4.5b | 33.6±7.8 | 36.2±6.1a | 33.2±4.0 |
| AERP100 | 36.0±4.5 | 35.9±4.8 | 33.6±3.8 | 23.8±4.0 | 11.8±3.6a | 30.1±3.8b | 35.9±9.1 | 35.5±4.2 | 33.6±3.0 |
| SNRTmax | 286.5±61.3 | 356.4±78.3a | 200.5±40.9b | 416.9±50.7 | 555.7±71.5a | 209.8±34.3b | 306.0±78.9 | 350.0±67.7a | 210.4±39.2b |
| cSNRT | 61.2±11.7 | 89.1±20.9a | 58.2±9.7 | 154.0±25.0 | 204.0±26.4a | 43.6±6.2b | 74.2±19.6 | 80.6±39.3 | 60.1±10.4b |
| SNRTi | 1.3±0.2 | 1.3±0.3 | 1.4±0.2 | 1.6±0.2 | 1.7±0.3 | 1.2±0.2b | 1.3±0.2 | 1.3±0.2 | 1.4±0.3 |
Values are means±SE. Subscripts 125 and 100 refer to drive cycle lengths of 125 and 100 ms, respectively. Ach indicates acetylcholine; AERP, atrial effective refractory periods; cSNRT, corrected sinus nodal recovery time; KO, knockout mice; RR, RR interval; SNRTi, sinus nodal recovery time index; SNRTmax, maximal sinus nodal recovery time; TG, transgenic mice; WT, wild‐type mice.
P<0.05 vs. WT and Rgs5TG by ANOVA.
P<0.05 vs WT and Rgs5KO by ANOVA.
Atrial Effective Refractory Periods
PES was used to examine AERPs in isolated hearts. AERPs were substantially shorter after addition of 1 μmol/L of ACh to perfusate solution. As shown in Table 1, under this condition, significant differences between AERPs in Rgs5 KO (n=15) and WT mice (n=15) were observed during drive cycle lengths of 125 and 100 ms (P<0.01). Notably, the AERP in Rgs5 TG (n=10) showed longer than WT atria (P<0.05). To determine whether shortening of AERPs was dependent on ACh, AERPs were remeasured after eliminating the drug by perfusion of HEPES‐buffered Tyrode solution for 30 minutes; results were similar before or after application of ACh.
Susceptibility to AT
Atrial burst pacing and PES induced AT in both groups of mice. Although burst pacing is thought to be a nonphysiological provocation, this process reliably creates cardiac electrical instability and induces AT. By contrast, PES causes premature atrial beats, which often precede physiological atrial arrhythmias. Duration of atrial arrhythmias is an important parameter in assessing risk, and sustained atrial arrhythmias have been defined in large animal and human electrophysiological studies as those lasting longer than 30 seconds. Obviously, Rgs5 KO hearts (n=15) were mostly (60%) insensitive to the single extrastimulus and burst stimuli in the baseline situation. Application of ACh increased susceptibility to PES (80.0%) and burst pacing (93.3%) in Rgs5 KO hearts (Table 2). Hearts from Rgs5 TG mice (n=10) showed less susceptibility to stimulation than those from WT (n=15) or Rgs5 KO mice (P<0.05). In Rgs5 KO mice (n=15), 20% of PES‐induced and 66.7% of burst‐induced hearts displayed sustained AT; however, sustained ATs were induced by PES and burst stimuli in 7.1% and 28.7% of WT hearts, respectively (Figure 3).
Table 2.
Incidence and Duration of Induced Atrial Tachyarrhythmia
| Baseline | Ach | Washout | |||||||
|---|---|---|---|---|---|---|---|---|---|
| WT (n=15) | KO (n=15) | TG (n=10) | WT (n=15) | KO (n=15) | TG (n=10) | WT (n=15) | KO (n=15) | TG (n=10) | |
| Inducibility | |||||||||
| PES8 Hz | 0 | 33.3% (5/15) | 10% (1/10) | 21.4% (3/14) | 73.3% (11/15)a | 20% (2/10) | 7.1% (1/14) | 33.3% (5/15)a | 20% (2/10) |
| PES10 Hz | 6.7% (1/15) | 26.7% (4/15)a | 0 | 42.9% (6/14) | 60.0% (9/15)a | 20% (2/10)b | 7.1% (1/14) | 6.7% (1/15) | 0 |
| Burst | 7.1% (1/14) | 33.3% (5/15)a | 10% (1/10) | 50.0% (7/14) | 93.3% (14/15)a | 20% (2/10)b | 7.1% (1/14) | 26.7% (4/15)a | 20% (2/10) |
| No response | 85.7% (12/14) | 40.0% (6/15)a | 80% (8/10) | 42.9% (6/14) | 0 | 70% (7/10)b | 92.9% (13/14) | 46.7% (7/15)a | 70% (7/10) |
| Duration | |||||||||
| <10 s | 7.1% (1/14) | 40.0% (6/15)a | 10% (2/10) | 14.3% (2/14) | 6.7% (1/15) | 10% (1/10) | 0 | 40.0% (6/15)a | 10% (1/10) |
| 10 to 30 s | 7.1% (1/14) | 13.3% (2/15) | 0 | 21.4% (3/14) | 20.0% (3/15) | 20% (2/10) | 7.1% (1/14) | 13.3% (2/15) | 20% (2/10) |
| >30 s | 0 | 6.7% (1/15) | 0 | 21.4% (3/14) | 73.3% (11/15)a | 0 | 0 | 0 | 0 |
Ach indicates acetylcholine; KO, knockout mice; PES, programmed electric stimuli; TG, transgenic mice; WT, wild‐type mice.
P<0.05 vs. WT and Rgs5TG by ANOVA.
P<0.05 vs WT and Rgs5KO by ANOVA.
Figure 3.
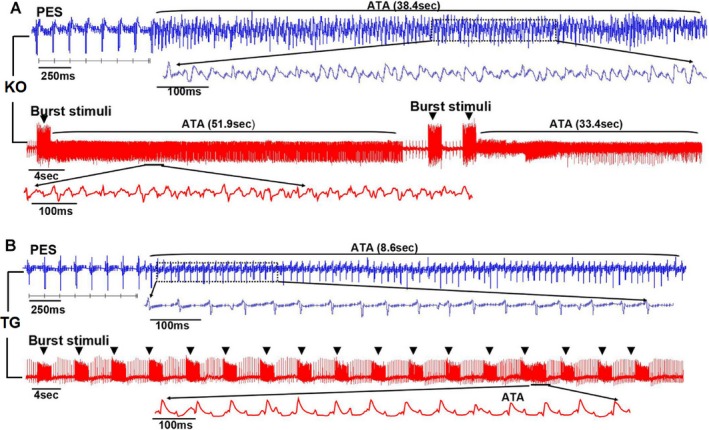
PES and burst pacing induced atrial tachyarrhythmias (ATs) in isolated hearts from WT (n=15), Rgs5 KO (n=15), and Rgs5 TG (n=10) mice during administration of 1 μmol/L of ACh. A, More‐sustained rapid atrial activity (duration >30 seconds) were observed in Rgs5 KO hearts. B, Rgs5 TG hearts showed nonsustained rapid atrial activity. ACh indicates acetylcholine; ATA, atrial tachyarrhythmia; KO, knockout mice; PES, programmed electric stimuli; TG, transgenic mice; WT, wild‐type mice.
Effect of ACh on AT on DF
Frequency analysis of bipolar electrograms showed that application of ACh increased the DF in the WT (n=15) and Rgs5 KO (n=15) groups (P<0.05). At the concentration of 1 μmol/L of ACh, the DF of the Rgs5 KO (n=15) mice during the PES or burst pacing protocols was significantly higher than that of WT mice (n=15; P<0.01); notably, DF value also showed a significant difference (P<0.05) between Rgs5 TG (n=10) and WT mice (n=15), suggesting the importance of Rgs5 in atrial tachyarrhythmia characteristics. In addition, the frequency spectrum showed a distribution containing multiple peaks, suggesting unstable periodic activity in Rgs5 KO mouse atria (Figure 4).
Figure 4.
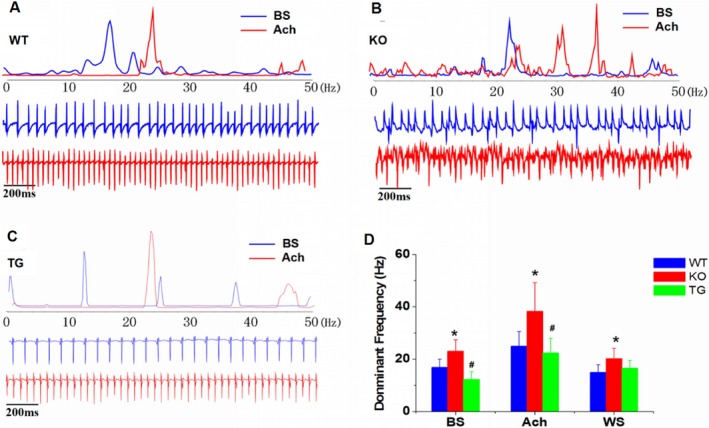
Frequency analysis determined with bipolar electrogram recordings in WT (n=15), Rgs5 KO (n=15), and Rgs5TG (n=10) mice. A through C, Dominant frequency (DF) of AT in the fast Fourier transform was analyzed at baseline and ACh state. D, Mean±SE DF values at baseline, ACh, and washout states were compared among the 3 groups. *P<0.05 vs WT and Rgs5 TG by ANOVA; # P<0.05 vs WT and Rgs5 KO by ANOVA. ACh indicates acetylcholine; AT, atrial tachyarrhythmia; BS, baseline; KO, knockout mice; TG, transgenic mice; WS, washout; WT, wild‐type mice.
Magnitude of the ACh‐Dependent K+ Current
Figure 5 shows the properties of the steady‐state IKAch in mouse atrial myocytes and describes the voltage clamp protocols employed. Rgs5 KO atrial myocytes had strong IKAch current when the test potential was set at −120 mV; the IKAch density was not significantly higher in Rgs5 KO than in WT atrial cells. However, atrial myocytes (n=8) from Rgs5 KO mice (n=5) exhibited a significant increase in the time course of activation and deactivation (P<0.01). In atrial myocytes (n=8) from Rgs5 TG mice (n=6), we found a smaller amplitude of IKAch than WT cells (n=10, from 6 mice), but this effect was not significant. Application of ACh also elicited rapid IKAch current in Rgs5 TG myocytes (n=8, from 6 mice), and time of activation and deactivation was shorter than in WT mice (P<0.05). In addition, mRNA expression levels of IKAch subunits Kir3.1 and Kir3.4 were similar between WT (n=5) and Rgs5 KO (n=5) or Rgs5 TG (n=5) mouse atria (P>0.05).
Figure 5.
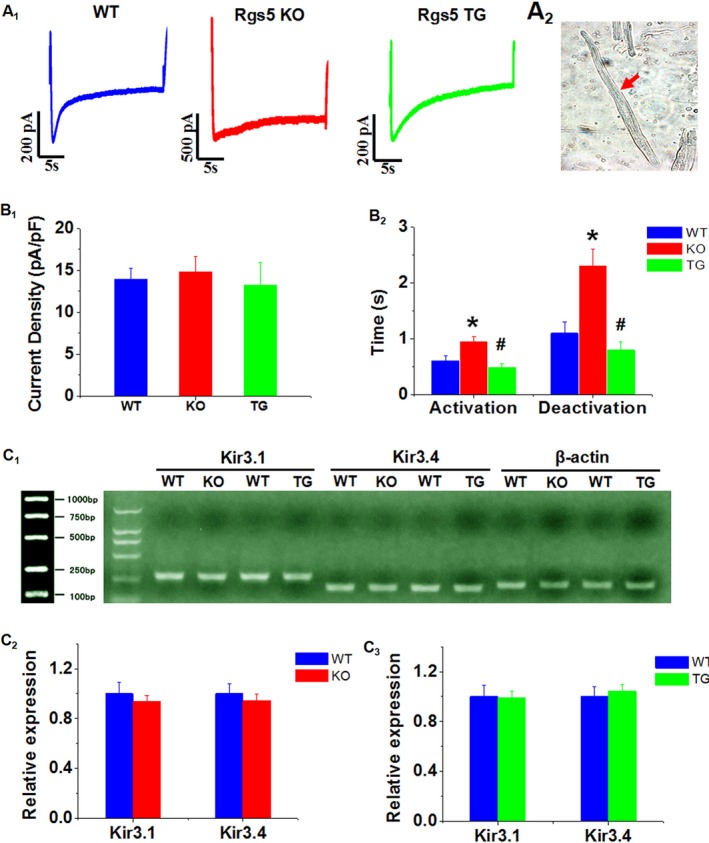
A, Representative IkAch recordings from WT (n=10), Rgs5 KO (n=8), and Rgs5 TG (n=8) atrial myocytes. B, Photograph of isolated cell from mouse atrium. C, Current density (peak current/cell capacitance) and (D) half‐maximal activation/deactivation time constant (t1/2) analysis of IKACh obtained from WT, Rgs5 KO, and Rgs5 TG mice. E through G, mRNA expression of Kir3.1 and Kir3.4 was determined in atria of mice. Relative abundance was calculated with value of WT as reference of 100%. *P<0.05 vs WT and Rgs5 TG by ANOVA; # P<0.05 vs WT and Rgs5 KO by ANOVA. KO indicates knockout mice; TG, transgenic mice; WT, wild‐type mice.
Discussion
The present study demonstrates that absence of Rgs5 facilitates bradycardia and AT during activation of muscarinic receptor by prolonged rapid deactivation of IKAch current. These novel findings suggest that Rgs5 is a critical factor that protects against vagal‐induced bradycardia and AT. To our knowledge, these data provide the first direct evidence demonstrating the crucial role of Rgs5 in regulation of ANS‐related arrhythmia.
Rgs5 in Controlling Heart Rate
Some GPCRs can couple to multiple isoforms of G proteins. For example, M2R and M3R interact with both Gαi2 and Gαq. The βγ subunits from Gα directly activate G‐protein‐coupled inwardly rectifying K+ (GIRK) currents and lead to negative chronotropic effects. This pathway is greatly regulated by RGS proteins. Coupling of G‐protein subtypes to muscarinic M2 receptor‐mediated HR slowing has been demonstrated in vitro using RGS‐insensitive Gαo and Gαi2 ES‐derived cardiomyocytes. RGS‐insensitive Gαo homozygous knock‐in cells demonstrated enhanced adenosine A1 and muscarinic M2 receptor‐mediated bradycardic responses.12 However, blocking GIRK channels largely abolished mutation‐induced enhancement of M2 receptor‐mediated response.
A previous study demonstrated that RGS5, RGS4, and RGS6 are highly expressed in the sinus node and surrounding right atrium.13, 14, 15 RGS4 and RGS6 null mice showed an increased bradycardic response to carbachol. Isolated hearts from mice deficient in these RGS proteins also display bradycardia, a sustained lower HR, and prolonged RR intervals, and isolated sinus node myocytes from these mice show impaired desensitization and slower deactivation of the GIRK current in response to agonist application. In the present study, Rgs5 protected against ACh‐induced bradycardia and prolonged SNRT. Furthermore, the predominant role of the parasympathetic effect in Rgs5 KO mice was also demonstrated in spectral measures of HRV. Such analysis has been widely used in mice, and there is agreement on the contribution of the different arms of the ANS to the frequency components. Taken together, the results indicate that RGS proteins have an important parasympathetic modulatory role in the SAN.
Rgs5 and Vagal‐Related AT
Myocardial refractoriness, which is markedly affected by ACh, is determined by sodium current (INa) and GIRK currents. Activation of GIRK current results in membrane hyperpolarization as well as shortening of action potential duration and effective refractory period (ERP). The change in atrial ERP may predispose to induction and maintenance of atrial fibrillation (AF). GIRK4 KO mice, which lack IKAch channels, appear to be completely resistant to carbachol‐induced atrial fibrillation.16 Thus, any increased activity in inhibitory G‐protein signaling may predispose to AF. Delivery of cell‐penetrating peptides to the posterior left atrium to disrupt M2‐Gαi coupling prolonged refractoriness in the left atrium and suppressed vagally induced AF.17
RGS5, a member of the regulator of G‐protein‐signaling protein superfamily, acts as a GTPase‐activating factor for a number of Gαi/q‐coupled receptor‐mediated pathways and negatively regulates Gαi/q‐coupled receptor‐mediated signaling. Toumi et al.18 reported that expression of RGS2 is >150‐fold higher than that of RGS4 in the mouse atrium. Notably, Rgs2−/− mice are more susceptible to PES‐induced AT/AF and had greater percentage of sustained AT/AF. This observation was associated with abbreviated AERP and alteration of muscarinic receptor‐gated K+ flux in Rgs2−/− mice.18 Thus, as a member of the same subfamily, Rgs5 may affect atrial electrophysiology by disruption of the muscarinic receptor‐mediated signaling pathway. In the current study, delayed deactivation of IKAch in RGS5‐deficient atrial myocytes slowed channel closing, prolonging membrane hyperpolarization. This effect would be expected to produce the dramatic increase in ACh‐induced shortening of AERP, together with a significantly increased DF after ACh stimulation; these effects resulted in increased inducibility and duration of atrial arrhythmias.
Limitation
First, in a mouse model, it is difficult to distinguish AF from AT, particularly with an arrhythmia of short duration. Second, although these results indicate a role for Rgs5 in regulating vagal‐related atrial tachyarrhythmia, alteration of intrinsic and extrinsic cardiac ANS cannot be identified in mouse heart. Additional studies will need to directly record activity of vagus nervous trunk and ganglia plexus in a large animal model.
Conclusion
In conclusion, the present study demonstrated that, as a critical regulator of parasympathetic activation in the heart, Rgs5 prevents vagal‐related bradycardia and AT through negatively regulating the IKAch current. Involvement of additional mechanisms requires further exploration.
Sources of Funding
This research was supported by a National Natural Science Foundation of China grant (Grant No.: 81400246) and a China Postdoctoral Science Foundation grant (Grant No.: 2015M571570). The funder has designed this study and decided to publish the manuscript.
Disclosures
None.
(J Am Heart Assoc. 2016;5:e002783 doi: 10.1161/JAHA.115.002783)
References
- 1. Shen MJ, Zipes DP. Role of the autonomic nervous system in modulating cardiac arrhythmias. Circ Res. 2014;114:1004–1021. [DOI] [PubMed] [Google Scholar]
- 2. Schauerte P, Scherlag BJ, Pitha J, Scherlag MA, Reynolds D, Lazzara R, Jackman WM. Catheter ablation of cardiac autonomic nerves for prevention of vagal atrial fibrillation. Circulation. 2000;102:2774–2780. [DOI] [PubMed] [Google Scholar]
- 3. Ehrlich JR, Cha TJ, Zhang L, Chartier D, Villeneuve L, Hebert TE, Nattel S. Characterization of a hyperpolarization‐activated time‐dependent potassium current in canine cardiomyocytes from pulmonary vein myocardial sleeves and left atrium. J Physiol. 2004;557:583–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cha TJ, Ehrlich JR, Chartier D, Qi XY, Xiao L, Nattel S. Kir3‐based inward rectifier potassium current: potential role in atrial tachycardia remodeling effects on atrial repolarization and arrhythmias. Circulation. 2006;113:1730–1737. [DOI] [PubMed] [Google Scholar]
- 5. Mark MD, Herlitze S. G‐protein mediated gating of inward‐rectifier K+ channels. Eur J Biochem. 2000;267:5830–5836. [DOI] [PubMed] [Google Scholar]
- 6. Ross EM, Wilkie TM. GTPase‐activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS‐like proteins. Annu Rev Biochem. 2000;69:795–827. [DOI] [PubMed] [Google Scholar]
- 7. Xiao B, Zhang Y, Niu WQ, Gao PJ, Zhu DL. Haplotype‐based association of regulator of G‐protein signaling 5 gene polymorphisms with essential hypertension and metabolic parameters. Clin Chem Lab Med. 2009;47:1483–1488. [DOI] [PubMed] [Google Scholar]
- 8. Li HL, He CW, Feng JH, Zhang Y, Tang QZ, Bian ZY, Bai X, Zhou H, Jiang H, Scott PH, Qin M, Huang H, Liu PP, Huang CX. Regulator of G protein signaling 5 protects against cardiac hypertrophy and fibrosis during biomechanical stress of pressure overload. Proc Natl Acad Sci USA. 2010;107:13818–13823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Qin M, Huang H, Wang T, Hu H, Liu Y, Cao H, Li H, Huang C. Absence of Rgs5 prolongs cardiac repolarization and predisposes to ventricular tachyarrhythmia in mice. J Mol Cell Cardiol. 2012;53:880–890. [DOI] [PubMed] [Google Scholar]
- 10. Griffioen KJ, Wan R, Okun E, Wang X, Lovett‐Barr MR, Li Y, Mughal MR, Mendelowitz D, Mattson MP. GLP‐1 receptor stimulation depresses heart rate variability and inhibits neurotransmission to cardiac vagal neurons. Cardiovasc Res. 2011;89:72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Qin M, Huang H, Wang T, Hu H, Liu Y, Gu YW, Cao H, Li HL, Huang CX. Atrial tachyarrhythmia in Rgs5‐null mice. PLoS One. 2012;7:e46856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fu Y, Huang X, Zhong H, Mortensen RM, D'Alecy LG. Neubig RR endogenous RGS proteins and G alpha subtypes differentially control muscarinic and adenosine‐mediated chronotropic effects. Circ Res. 2006;98:65–66. [DOI] [PubMed] [Google Scholar]
- 13. Cifelli C, Rose RA, Zhang HJ, Bolz JV, Bolz SS, Backx PH, Heximer SP. RGS4 regulates parasympathetic signaling and heart rate control in the sinoatrial node. Circ Res. 2008;103:527–535. [DOI] [PubMed] [Google Scholar]
- 14. Fu Y, Huang XY, Piao L, Lopatin AN, Neubig RN. Endogenous RGS proteins modulate SA and AV nodal functions in isolated heart: implications for sick sinus syndrome and AV block. Am J Physiol Heart Circ Physiol. 2007;292:H2532–H2539. [DOI] [PubMed] [Google Scholar]
- 15. Posokhova E, Wydeven N, Allen KL, Wickman K, Martemyanov KA. RGS6/Gβ5 complex accelerates IKAch gating kinetics in atrial myocytes and modulates parasympathetic regulation of heart rate. Circ Res. 2010;107:1350–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kovoor P, Wickman K, Maguire CT, Pu W, Gehrmann J, Berul CI, Clapham DE. Evaluation of the role of I(KACh) in atrial fibrillation using a mouse knockout model. J Am Coll Cardiol. 2001;37:2136–2143. [DOI] [PubMed] [Google Scholar]
- 17. Aistrup GL, Villuendas R, Ng J, Gilchrist A, Lynch TW, Gordon D, Cokic I, Mottl S, Zhou R, Dean DA, Wasserstrom JA, Goldberger JJ, Kadish AH, Arora R. Targeted G‐protein inhibition as a novel approach to decrease vagal atrial fibrillation by selective parasympathetic attenuation. Cardiovasc Res. 2009;83:481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tuomi JM, Chidiac P, Jones DL. Evidence for enhanced M3 muscarinic receptor function and sensitivity to atrial arrhythmia in the RGS2‐deficient mouse. Am J Physiol Heart Circ Physiol. 2010;298:H554–H561. [DOI] [PubMed] [Google Scholar]