

Abstract
Background
Left ventricular dysfunction and heart failure are strongly associated in humans with increased circulating levels of proinflammatory cytokines, T cells, and soluble intercellular cell adhesion molecule 1 (ICAM1). In mice, infiltration of T cells into the left ventricle contributes to pathological cardiac remodeling, but the mechanisms regulating their recruitment to the heart are unclear. We hypothesized that ICAM1 regulates cardiac inflammation and pathological cardiac remodeling by mediating left ventricular T‐cell recruitment and thus contributing to cardiac dysfunction and heart failure.
Methods and Results
In a mouse model of pressure overload–induced heart failure, intramyocardial endothelial ICAM1 increased within 48 hours in response to thoracic aortic constriction and remained upregulated as heart failure progressed. ICAM1‐deficient mice had decreased T‐cell and proinflammatory monocyte infiltration in the left ventricle in response to thoracic aortic constriction, despite having numbers of circulating T cells and activated T cells in the heart‐draining lymph nodes that were similar to those of wild‐type mice. ICAM1‐deficient mice did not develop cardiac fibrosis or systolic and diastolic dysfunction in response to thoracic aortic constriction. Exploration of the mechanisms regulating ICAM1 expression revealed that endothelial ICAM1 upregulation and T‐cell infiltration were not mediated by endothelial mineralocorticoid receptor signaling, as demonstrated in thoracic aortic constriction studies in mice with endothelial mineralocorticoid receptor deficiency, but rather were induced by the cardiac cytokines interleukin 1β and 6.
Conclusions
ICAM1 regulates pathological cardiac remodeling by mediating proinflammatory leukocyte infiltration in the left ventricle and cardiac fibrosis and dysfunction and thus represents a novel target for treatment of heart failure.
Keywords: adhesion molecules, heart failure, inflammation, remodeling
Subject Categories: Heart Failure, Remodeling, Inflammation
Introduction
The complex syndrome of heart failure (HF) is a leading cause of morbidity and mortality worldwide.1 Systemic serum markers of inflammation including the proinflammatory cytokines interleukin (IL) 1β and IL‐6 as well as tumor necrosis factor α and soluble intercellular cell adhesion molecule 1 (ICAM1)2 correlate with cardiac dysfunction and HF.3, 4, 5, 6 Nevertheless, clinical trials using tumor necrosis factor α inhibitors failed to improve HF outcomes,7, 8 emphasizing the need to better understand the mechanisms by which systemic inflammation influences the pathogenesis of HF. We and others showed high levels of circulating T cells in humans with HF, further supporting a role for systemic T‐cell–mediated inflammation in HF.9, 10, 11 Moreover, we recently demonstrated that T cells play a critical role in the pathogenesis of pressure overload–induced HF. Specifically, T cells were found to infiltrate the left ventricle (LV) of humans and mice with nonischemic HF, and the absence of T cells resulted in preserved cardiac systolic and diastolic function in a mouse model of pressure overload–induced HF. T‐cell recruitment to the LV was found to be essential to the pathogenesis of HF9; however, the mechanisms by which T cells are recruited to the LV in nonischemic HF remain poorly understood. ICAM1, an immunoglobulin‐like adhesion molecule that mediates leukocyte arrest and transendothelial migration from the blood stream to the site of inflammation,12, 13 is highly expressed in human intramyocardial vascular endothelium during myocardial infarction.14 Similarly, in experimental models of HF induced by suprarenal abdominal aortic constriction, ICAM1 expression is upregulated in the heart within the first week.15 We recently demonstrated increased ICAM1 expression in the vascular endothelium in mice after pressure overload–induced HF in association with LV systolic dysfunction and LV T‐cell infiltration.9 Consequently, we hypothesized that ICAM1 mediates T‐cell infiltration in the LV in response to pressure overload and contributes to cardiac remodeling and dysfunction during HF progression.
In the present study, we determined the dynamics of intramyocardial ICAM1 induction in mice subjected to pressure overload induced by transverse aortic constriction (TAC) and examined the pressure overload response of mice with genetic deletion of ICAM1. We showed, for the first time, that ICAM1‐deficient (ICAM1−/−) mice are protected from maladaptive cardiac remodeling and HF through mechanisms that include decreased cardiac fibrosis and CD3+ T‐cell and Ly6Chigh monocyte–mediated cardiac inflammation. We have also identified a mechanism by which ICAM1 is upregulated in the LV that is independent of endothelial cell–specific mineralocorticoid receptor (EC‐MR) signaling because mice deficient in EC‐MR (EC‐MR−/−) have increased LV endothelial ICAM1 expression and similar numbers of LV‐infiltrated T cells compared with EC‐MR+/+ mice.
Methods
Mice
Mice were bred and maintained under pathogen‐free conditions and treated in accordance with the guidelines of the National Institutes of Health for the care and use of laboratory animals. All protocols were approved by the Tufts Medical Center institutional animal care and use committee. Thirty male C57BL/6 wild‐type (WT) mice, 31 male ICAM1−/− mice16 (provided by Dan Bullard, University of Alabama, Birmingham), and 11 EC‐MR knockout EC‐MR−/− and EC‐MR+/+ male littermates17 were euthanized at age 10 to 14 weeks for tissue collection. The overall number of mice corresponding to each experiment is indicated in the related figure legend.
Mouse Model of TAC
Pressure overload was induced by constricting the transverse aorta of male mice aged 8 to 10 weeks, as described previously, to induce HF.18, 19 Sham‐operated mice underwent the same procedure but without constriction (4–8 per group unless otherwise indicated). At 48 hours and at 2 and 4 weeks after TAC, mice were euthanized and tissue was harvested for further analysis, as described.9
Quantitative Polymerase Chain Reaction of LV Samples
Total RNA was extracted from mouse heart LV tissues directly using Trizol (Invitrogen). RNA was then reverse‐transcribed to cDNA following Applied Biosystems’ protocol using MuLV reverse transcriptase and amplified by real‐time polymerase chain reaction (PCR) with SYBR green PCR mix (Applied Biosystems). Samples were quantified in triplicate using 40 cycles performed at 94°C for 30 seconds, 60°C for 45 seconds, and 72°C for 45 seconds using an ABI Prism 7900 Sequence Detection System (Applied Biosystems). The following primer sequences were used: Icam1, forward 5′‐GCT GTG CTT TGA GAA CTG TG ‐3′, reverse 5′ ‐GTG AGG TCC TTG CCT ACT TG ‐3′; Nppb, forward 5′ ‐CAC CGC TGG GAG GTC ACT ‐3′, reverse 5′ ‐GTG AGG CCT TGG TCC TTC AA ‐3′; Nppa, forward 5′ ‐GAG AGA CGG CAG TGC TTC TAG GC ‐3′, reverse 5′ ‐CGT GAC ACA CCA CAA GGG CTT AGG ‐3′; Col1A1, forward 5′ ‐AAG GGT CCC TCT GGA GAA CC ‐3′, reverse 5′ ‐TCT AGA GCC AGG GAG ACC CA ‐3′; Tgfb1, forward 5′ ‐CAC CGG AGA GCC CTG GAT A ‐3′, reverse 5′ ‐TGC CGC ACA CAG CAG TTC ‐3′; Acta2, forward 5′ ‐GCA TCC ACG AAA CCA CCT A ‐3′, reverse 5′ ‐CAC GAG TAA CAA ATC AAA GC ‐3′; Myh6, forward 5′‐CCT AGC CAA CTC CCC GTT CT ‐3′, reverse 5′ –GCC AAT GAG TAC CGC GTG A ‐3′; Myh7, forward 5′‐TGA GCC TTG GAT TCT CAA ACG T ‐3′, reverse 5′ –AGG TGG CTC CGA GAA AGG AA ‐3′; Il1b, forward 5′‐ ACT CCT TAG TCC TCG GCC A –3′, reverse 5′‐TGG TTT CTT GTG ACC CTG AGC ‐3′; Il6, forward 5′ –TGA TGG ATG CTA CCA AAC TGG‐3′, reverse 5′‐TTC ATG TAC TCC AGG TAG CTA TGG ‐3′; Gapdh, forward 5′‐ACC ACA GTC CAT GCC ATC AC ‐3′, reverse 5′ –TCC ACC ACC CTG TTG CTG TA – 3′.
Immunohistochemistry and Histological Analysis
Heart samples were excised, and the LV was separated from the right ventricle. Sections of the LV were immediately embedded in OCT or fixed in 10% formalin, embedded in paraffin, and cut into 5‐μm LV sections. Hematoxylin and eosin or picrosirius red staining was performed, as described previously.20, 21 Serial OCT frozen cryostat sections were stained with anti–mouse CD54 (YN1/1.7.4), anti–mouse CD31 (MEC13.3), or anti–mouse CD4 (GK1.5) purchased from BioLegend.
Quantitative Flow Cytometry
Flow cytometry was performed to analyze the immune profile present in response to TAC. The data were acquired on a FACSCanto analyzer (Becton Dickinson) and analyzed using FlowJo software. LV digested with collagenase type II (0.895 mg/mL) and protease XIV (0.5 mg/mL) and lymphoid organs were harvested from sham‐operated and TAC mice and stained with the following monoclonal antibodies: FITC–conjugated anti‐CD4 (GK1.5), APC‐Cy7–conjugated anti‐CD4 (GK1.5), FITC‐conjugated anti‐CD3e (145‐2C11), PE‐conjugated anti‐CD45.2 (104), APC‐Cy7–conjugated anti‐Ly‐6C (HK1.4), PerCP‐conjugated anti‐CD11b (M1/70), APC‐Cy7–conjugated anti‐CD44 (IM7), and PE‐conjugated anti‐CD62L (MEL‐14). All antibodies were purchased from BioLegend. Cells were surface stained by incubation with the relevant antibodies diluted in PBS plus 2% FBS for 20 minutes at room temperature, followed by 2 washes with PBS plus 2% FBS.
Adult Mouse Cardiac Myocyte Isolation
Cardiac myocytes were isolated from WT and ICAM1−/− hearts 4 weeks after TAC, as described previously.22 Briefly, mice were administered heparin (200 U IP) and euthanized. The hearts were rapidly excised, cannulated via the aorta, and perfused in the Langendorff mode with perfusion buffer (113 mmol/L NaCl, 4.7 mmol/L KCl, 0.6 mmol/L NaH2PO4, 12 mmol/L NaHCO3, 0.6 mmol/L KH2PO4, 10 mmol/L HEPES, 1.2 mmol/L MgSO4·7H2O, 30 mmol/L taurine, 10 mmol/L 2,3‐butanedione monoxime, and 5.5 mmol/L glucose, pH 7.46, at 35°C). Hearts were then digested with perfusion buffer containing 773.78 μg/mL collagenase type 2 and 0.14 mg/mL trypsin. After 5‐minute digestion, the LV was separated, minced, and gently agitated in stopping buffer, which consists of perfusion buffer containing 10% FBS and 12.5 μmol/L CaCl2, allowing the myocytes to be dispersed. The cells were subsequently resuspended in stopping buffer with gradually increasing Ca2+ concentrations (0.062, 0.112, 0.212, 0.5, and 1.0 mmol/L). Cardiac myocytes were imaged with ×20 magnification, and width and length were determined using the Nikon Elements software.
Bone Marrow Monocyte Isolation and M1 or M2 Differentiation
WT and ICAM1−/− bone marrow (BM) cells were collected from femurs and tibias by flushing them with buffer (PBS with 0.5% BSA and 2 mmol/L EDTA) using a 25‐gauge needle. BM cells were disaggregated and passed through a 40‐μm cell strainer. From the resulting BM cell suspension, the Miltenyi Biotec isolation kit was used to isolate BM‐derived monocytes, which were incubated for 3 days with 1 μg/mL LPS (sc‐3535; Santa Cruz Biotechnology) or 20 ng/mL IL‐4 (214‐14; PeproTech) for differentiation to M1 or M2 monocytes, respectively. RNA was isolated using the Qiagen kit, and inducible nitric oxide synthase and arginase I expressions were determined through quantitative reverse transcription PCR for identifying the M1 and M2 monocyte populations. The following primer sequences were used: Nos2, forward 5′‐GCC ACC AAC AAT GGC AAC A ‐3′, reverse 5′ –CGT ACC GGA TGA GCT GTG AAT T ‐3′; Arg1, forward 5′‐TTG CGA GAC GTA GAC CCT GG ‐3′, reverse 5′ –CAA AGC TCA GGT GAA TCG GC ‐3′.
Cardiac Fibroblast Isolation and Differentiation to Myofibroblast
Perfused hearts from male WT and ICAM1−/− mice aged 8 to 10 weeks were digested with Liberase DL (Roche) for 25 minutes at 37°C. The resulting cell suspension was cultured in fibroblast growth media on 1% gelatin–coated plates. Purified cardiac fibroblast in passages 2 to 3 were treated with transforming growth factor β (100 ng/mL for 16 hours) to induce fibroblast transition to myofibroblasts, which was determined by measuring mRNA levels of α‐smooth muscle actin.
LV In Vivo Hemodynamic Studies
In vivo LV function was assessed by pressure–volume analysis in mice anesthetized with 2.5% isofluorane. A 1.4F pressure–volume catheter (SPR‐839; Millar Instruments) was advanced through the carotid artery and across the aortic valve into the LV. The absolute volume was calibrated, and pressure–volume data were assessed at steady state and during preload reduction. Hemodynamics were recorded and analyzed with IOX version 1.8.11 software (EMKA Instruments).23
In Vivo Transthoracic Echocardiography
M mode and 2‐dimensional images were obtained from the short‐axis view, as described previously.19, 23 LV end‐diastolic and end‐systolic diameters (shown as EDD and ESD, respectively, in the equation below) and heart rate were measured by averaging values obtained from 5 cardiac cycles. Fractional shortening (shown as FS) was calculated using the following standard equation: FS%=([EDD−ESD]/EDD)×100. The investigators were blinded to genotype during data acquisition and analysis.
Statistical Analysis
Data are expressed as the mean±SEM. Statistical analyses were performed with the unpaired Student t test or nonparametric Mann–Whitney test, as indicated, using GraphPad Prism software (GraphPad Software). When the unpaired t test was used, the outcome was confirmed to be normally distributed. When the nonparametric Mann–Whitney test was used, each condition's median and interquartile range was reported in the figure legend corresponding to each data set. Differences were considered statistically significant at P<0.05.
Results
Upregulation of ICAM1 in the LV in Response to TAC
To investigate the kinetics of endothelial cell activation during the progression of HF, we determined the expression of ICAM1 in the LV of WT mice at 48 hours and at 2 and 4 weeks after inducing LV pressure overload by TAC. We observed significant upregulation of ICAM1 mRNA in the LV of TAC mice compared with sham‐operated mice as early as 48 hours after TAC, which progressively increased by 2 and 4 weeks after TAC (Figure 1A). Endothelial ICAM1 protein expression followed kinetics similar to the LV mRNA, remaining significantly upregulated during the period of progressive systolic dysfunction and T‐cell infiltration, as shown previously.9 Platelet/endothelial cell adhesion molecule 1 was used to stain the intramyocardial endothelial vessels in parallel with ICAM1 in the same sections and was, as expected, constitutively expressed and not induced in response to TAC (Figure 1B). Taken together, our data demonstrate that endothelial ICAM1 is upregulated during the course of TAC as HF progresses, raising the question of whether ICAM1 mediates T‐cell infiltration of the LV later on as HF progresses and contributes to local cardiac inflammation and systolic dysfunction in response to LV pressure overload.
Figure 1.
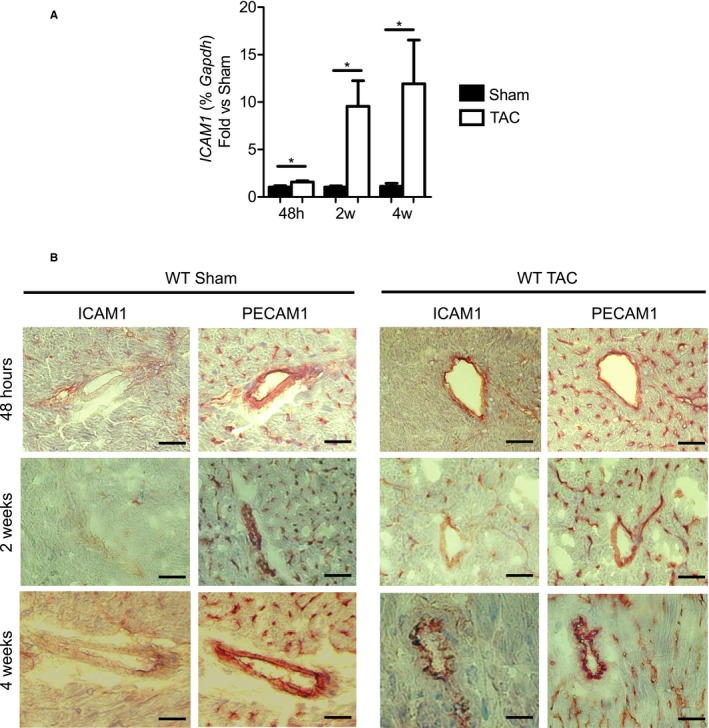
Upregulation of ICAM1 mRNA expression and ICAM1 protein in the LV, specifically in the vascular endothelium, in response to TAC. A, ICAM1 mRNA expression in the LV of WT mice determined by quantitative reverse transcription polymerase chain reaction represented as fold change for TAC vs sham at 48 hours and at 2 and 4 weeks after surgery. Statistical analysis used the nonparametric Mann–Whitney test. Data shown as median (interquartile range): at 48 hours: WT sham 1.044 (0.754–1.307) and WT TAC 1.483 (1.374–1.841); at 2 weeks: WT sham 1.115 (0.735–1.250) and WT TAC 6.916 (1.633–13.30); at 4 weeks: WT sham 1.035 (0.582–1.770) and WT TAC 3.957 (1.519–28.71). *P<0.05. B, Representative immunohistochemistry staining for ICAM1 and PECAM1 (used as positive control for endothelial cell staining in the same sections) in sham‐operated and TAC mice at 48 hours and at 2 and 4 weeks after surgery. Scale bar=50 μm. n=4 sham, n=6 to 9 TAC. h indicates hours; ICAM1, intercellular cell adhesion molecule 1; LV, left ventricle; PECAM1, platelet/endothelial cell adhesion molecule 1; TAC, transverse aortic constriction; w, weeks; WT, wild type.
ICAM1 Is Required for Leukocyte Infiltration Into the LV in Response to TAC
Given the well‐known role of ICAM1 as an endothelial cell adhesion molecule mediating leukocyte recruitment into tissues, the observed upregulation of ICAM1 in the intramyocardial vessels in response to TAC, and the recent findings that T cells infiltrate the LV and contribute to the pathogenesis of HF in response to TAC,9, 24 we evaluated whether ICAM1 was required for T‐cell infiltration of the pressure‐overloaded LV using WT and ICAM1−/− mice. In contrast to WT mice, in which CD45.2+ leukocytes infiltrated the LV in response to TAC, pressure overload did not induce a significant increase in LV leukocyte infiltration in ICAM1−/− mice (Figure 2A) as measured by flow cytometry. These leukocytes included CD3+ T cells and Ly6Chigh proinflammatory monocytes, both infiltrated in WT but not in ICAM1−/− mice in response to TAC (Figure 2B through 2D). The LV of ICAM1−/− mice also showed reduced infiltration of CD4+ T cells compared with WT mice in response to TAC (Figure 2E); CD4+ T cells have been shown to mediate cardiac dysfunction in response to TAC.9, 24 Taken together, our data indicate that ICAM1 regulates the infiltration of leukocytes, including CD3+ and CD4+ T cells and Ly6Chigh proinflammatory monocytes, into the LV in response to TAC.
Figure 2.
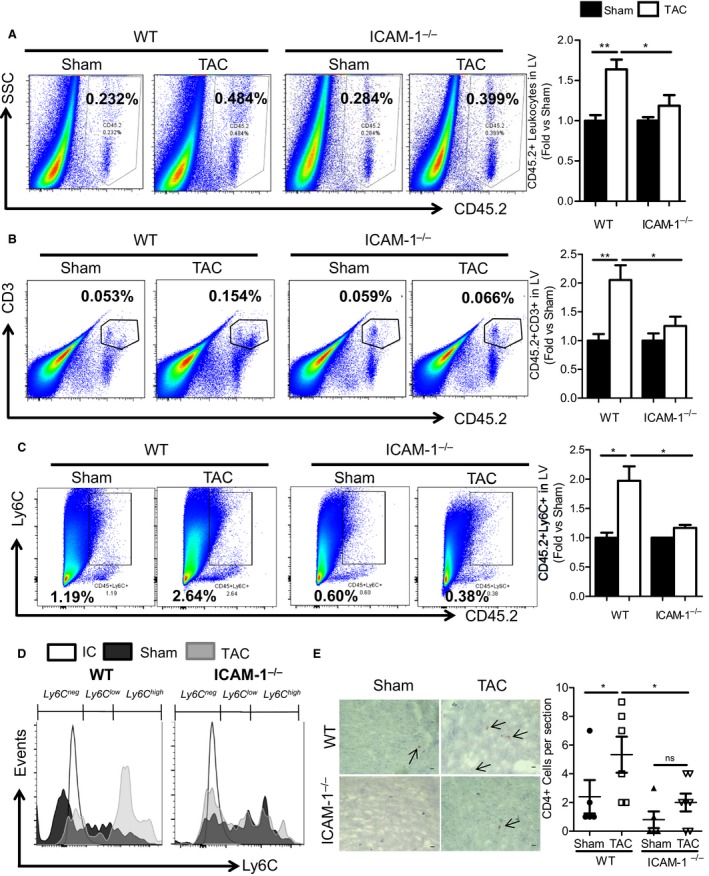
ICAM1‐mediated regulation of CD45.2+, CD3+, CD4+, and Ly6C+ leukocyte infiltration into the LV in response to TAC. A through C, Representative FACS plots of (A) LV CD45.2+ leukocyte, (B) CD3+ T‐cell, and (C) Ly6C+ monocyte infiltration in response to TAC or sham operation at 4 weeks in WT and ICAM1−/− mice. Bar graph on the right represents fold‐change quantification of the indicated leukocytes normalized to sham controls. D, Representative FACS histogram for Ly6C+ low and high subpopulations, gated on CD45.2+ CD11b+ myeloid cells infiltrated into the LV of sham‐ and TAC‐operated WT and ICAM1−/− mice. Ly6Cneg cells represent nonmonocyte myeloid cells, Ly6Clow represents patrolling monocytes, and Ly6Chigh represents proinflammatory monocytes. E, Representative pictures (×40 magnification) of immunohistochemistry staining for CD4+ T helper lymphocytes infiltrated into the LV in sham‐ and TAC‐operated WT and ICAM1−/− mice. Quantification of CD4+ cells per 6‐μm LV cross‐sections is represented. Scale bar=10 μm. n=5 to 7 sham, n=11 TAC. Statistical analysis used the unpaired t test. *P<0.05; **P<0.01. FACS indicates fluorescence‐activated cell sorting; ICAM1, intercellular cell adhesion molecule 1; LV, left ventricle; TAC, transverse aortic constriction; WT, wild type.
T Lymphocytes Are Equally Activated in the Heart‐Draining Lymph Nodes in Both WT and ICAM1−/− Mice at 4 Weeks After TAC
Given the role of ICAM1 expressed by the antigen‐presenting cells in the immune synapse during the presentation of some antigens,25 we next investigated whether the decreased LV T‐cell infiltration observed in ICAM1−/− mice was a result of endothelial ICAM1 mediating T‐cell recruitment or deficient T‐cell activation in the immune synapse of ICAM1−/− mice in response to TAC. We initially determined T‐cell activation in the mediastinal lymph nodes, the heart‐draining lymph nodes in which T cells are activated in response to TAC,9, 24 by evaluating the levels of CD62LlowCD44high–activated lymphocytes (Figure 3A). Both WT and ICAM1−/− mice had similar frequencies of CD62LlowCD44high–activated lymphocytes in the mediastinal lymph nodes in response to TAC at 4 weeks (Figure 3B). The percentage of circulating CD4+ T cells in blood was also similar between WT and ICAM1−/− mice in response to TAC (Figure 3C). Taken together, our data indicate that ICAM1 does not regulate T‐cell activation in the heart‐draining lymph nodes in response to TAC. This finding, together with the similar numbers of circulating T cells observed in WT and ICAM1−/− mice, suggests that the decreased T‐cell infiltration observed in the ICAM1−/− LV in response to TAC is likely related to reduced recruitment mediated by endothelial ICAM1, not to impaired T‐cell activation in the absence of ICAM1.
Figure 3.
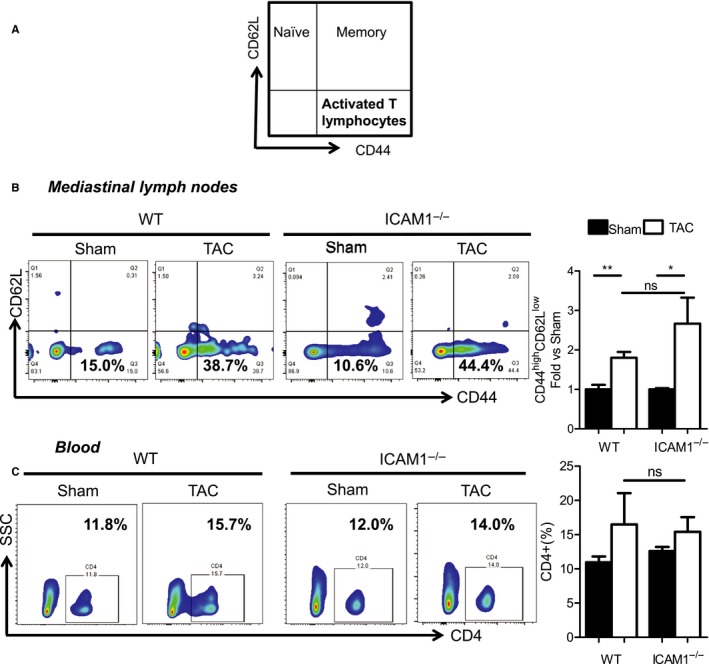
Lymphocyte activation in the heart‐draining mediastinal lymph nodes and circulating CD4+ T cells in WT and ICAM1−/− mice in response to TAC. A, Diagram indicating T‐cell subpopulations using T‐cell activation markers. B, Representative FACS plots showing naïve, memory, and activated T lymphocytes in the mediastinal lymph node lymphocyte gate at 4 weeks after TAC or sham surgery. Quantification of activated T lymphocytes (CD44high CD62Llow) is shown in the right bar graph with values normalized to sham control. C, Representative FACS plots showing blood‐circulating CD4+ T lymphocytes at 4 weeks after TAC or sham surgery. CD4+ cells are represented as percentages in the bar graph at right. n=5 sham, n=8 TAC. Statistical analysis used the unpaired t test. *P<0.05; **P<0.01. FACS indicates fluorescence‐activated cell sorting; ICAM1, intercellular cell adhesion molecule 1; TAC, transverse aortic constriction; WT, wild type.
Reduced LV Cardiac Myocyte Hypertrophy in ICAM1−/− Mice Compared With WT Mice in Response to TAC
We next evaluated cardiac hypertrophy after 4 weeks of pressure overload as a hallmark of pathological cardiac remodeling in response to TAC. LV mass increased to the same extent in WT and ICAM1−/− mice in response to TAC (Figure 4A). We further found that the length and width of cardiac myocytes isolated from both WT and ICAM1−/− mice at 4 weeks after undergoing TAC was increased compared with sham‐operated mice, but these values were significantly higher in WT mice compared with ICAM1−/− mice after TAC (Figure 4B). Interestingly, WT TAC LVs exhibited a pattern of increased mRNA levels of the atrial and brain natriuretic peptides (markers of pathological hypertrophy), but TAC LVs of ICAM1−/− mice did not show such increases (Figure 4C and 4D). Moreover, other hallmarks of pathological remodeling such as increased myosin heavy chain β and α isoform expression,26 although increased in WT TAC hearts, did not increase in ICAM1−/− mice (Figure 4E). Taken together, ICAM1−/− mice developed significantly less cardiomyocyte hypertrophy and exhibited decreased fetal gene reexpression and pathological hypertrophy in response to TAC.
Figure 4.
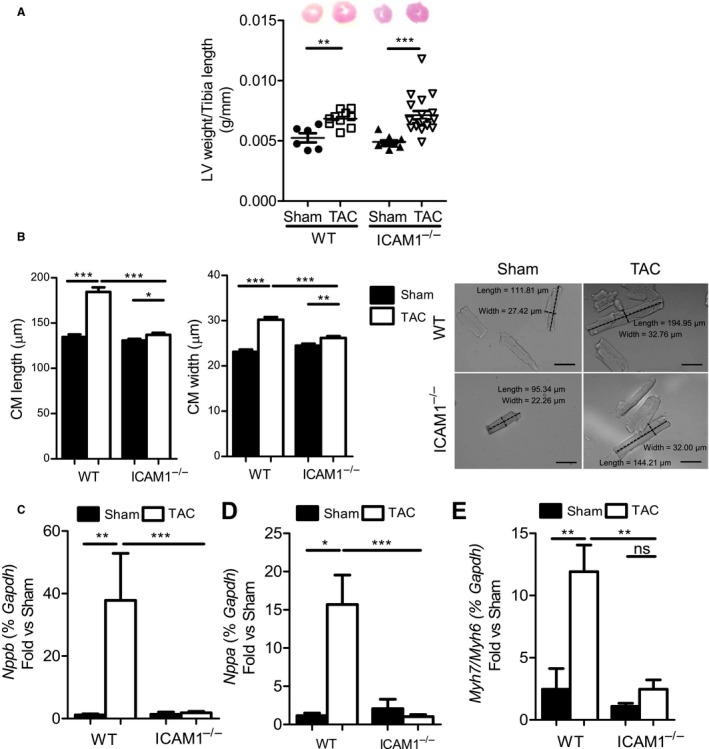
LV weight, CM dimensions, and expression of natriuretic peptides and MHC isoforms in response to TAC in ICAM1−/− mice. A, LV weight of WT and ICAM1−/− (n=6–9 sham, n=10–14 TAC) at 4 weeks after TAC, normalized to the tibia length. B, Dimensions of CMs isolated from WT and ICAM1−/− mice at 4 weeks after TAC or sham surgery. n=100 to 200 cells per group, from 2 independent CM isolations. Representative pictures are shown (magnification ×20). Scale bar=50 μm. Statistical analysis used the unpaired t test. C through E, Quantitative reverse transcription polymerase chain reaction of the hypertrophy markers (C) BNP and (D) ANP and the (E) ratio of MHCβ and MHCα. mRNA expression normalized to sham in the LV in response to TAC (n=7) or sham (n=5) surgery at 4 weeks. Statistical analysis used the nonparametric Mann–Whitney test. Data are shown as median (interquartile range): for BNP: WT sham 1.190 (0.508–1.876) and WT TAC 18.60 (6.985–91.82), ICAM1−/− sham 0.682 (0.502–2.921) and ICAM1−/− TAC 1.516 (1.168–2.236); for ANP: WT sham 1.268 (0.519–1.709) and WT TAC 14.33 (5.885–23.92), ICAM1−/− sham 1.273 (0.264–4.664) and ICAM1−/− TAC 0.6279 (0.509–1.797); for MHCβ/MHCα: WT sham 0.557 (0.416–2.395) and WT TAC 10.87 (8.446–14.01), ICAM1−/− sham 1.168 (0.600–1.521) and ICAM1−/− TAC 1.454 (1.045–3.935). *P<0.05, **P<0.01, ***P<0.005. ANP indicates atrial natriuretic peptide; BNP, brain natriuretic peptide; CM, cardiac myocyte; ICAM1, intercellular cell adhesion molecule 1; LV, left ventricle; ns, not significant; MHC, myosin heavy chain; TAC, transverse aortic constriction; w, weeks; WT, wild type.
ICAM1−/− Mice Do Not Develop Cardiac Fibrosis in Response to TAC
Cardiac fibrosis, another hallmark of maladaptive cardiac remodeling, likely promotes HF in response to pressure overload induced by TAC. Based on our findings that ICAM1−/− mice had decreased T‐cell infiltration in the LV in response to TAC, and given the role of T cells in cardiac fibrosis in this model,9 we hypothesized that ICAM1−/− mice would not develop cardiac fibrosis in response to TAC. TAC induced an expected increase in both LV interstitial (Figure 5A) and perivascular (Figure 5B) fibrosis in WT mice. In contrast, LV fibrosis was completely absent in ICAM1−/− mice in response to TAC (Figure 5A and 5B). Markers of fibrosis such as transforming growth factor β, collagen type I, and α‐smooth muscle actin were also elevated in WT mice in response to TAC but not in ICAM1−/− mice (Figure 6A through 6C). To investigate the cellular mechanism by which ICAM1 deletion inhibits LV fibrosis in pressure overload, we next evaluated whether ICAM1 expressed in BM‐derived monocytes or cardiac fibroblasts could contribute to the observed profibrotic response in WT mice by measuring M2 macrophage and myofibroblast phenotypes from the ICAM1−/− mice. Differentiation of BM monocytes from ICAM1−/− mice toward the M2 profibrotic macrophage phenotype expressing arginase 1 or toward the M1 proinflammatory macrophage phenotype expressing inducible nitric oxide synthase was not different from WT monocytes (Figure 6D and 6E). Moreover, WT and ICAM1−/− adult cardiac fibroblasts differentiated similarly toward profibrotic myofibroblasts in vitro in response to transforming growth factor β (Figure 6F). Taken together, these data indicate that ICAM1 expression in BM‐derived monocytes or in cardiac fibroblast does not contribute to their transition toward a profibrotic phenotype in vitro and support the concept that ICAM1 contributes to cardiac fibrosis by mediating LV T‐cell infiltration in response to pressure overload in vivo.
Figure 5.
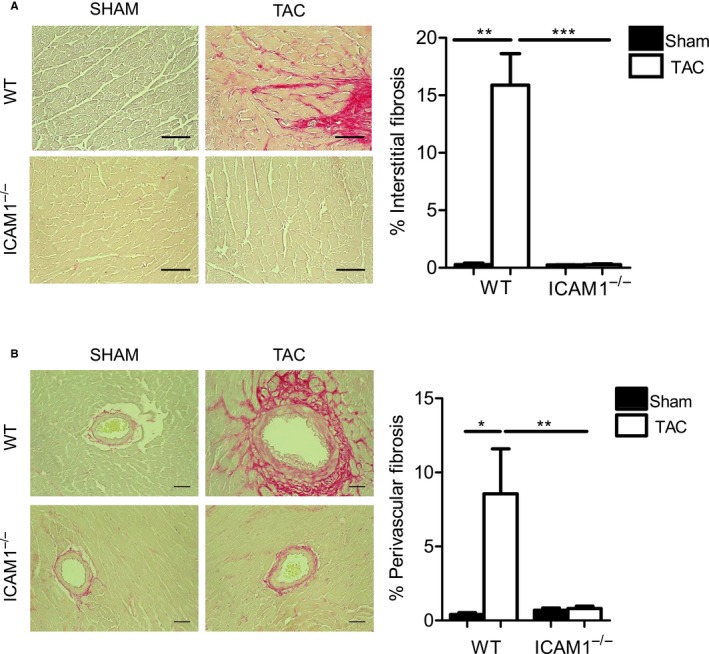
Absence of fibrosis in ICAM1−/− in response to TAC. A, Representative photomicrographs (×40 magnification) and quantification (bar graph on the right) of myocardial interstitial fibrosis evaluated by picrosirius red staining of left ventricle sections from sham and TAC mice at 4 weeks. B, Representative pictograms (×20 magnification) shown on the left and percentage of perivascular fibrosis on the right. Scale bar=50 μm. n=3 to 5 sham, n=3 to 8 TAC. Statistical analysis used the unpaired t test. *P<0.05, **P<0.01, ***P<0.005. ICAM1 indicates intercellular cell adhesion molecule 1; TAC, transverse aortic constriction; WT, wild type.
Figure 6.
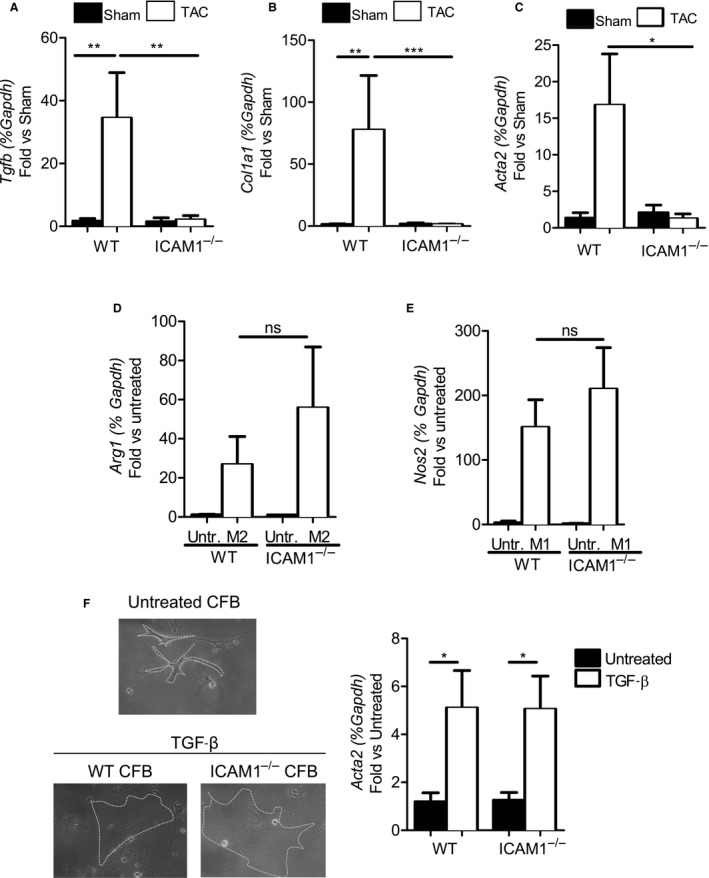
Absence of fibrosis mRNA marker upregulation in ICAM1−/− in response to TAC despite equal capacity for M2 profibrotic and myofibroblast differentiation in vitro. A through C, Quantitative reverse transcription polymerase chain reaction in the left ventricle for the fibrosis markers (A) TGF‐β, (B) collagen I, and (C) α‐SMA. mRNA expression normalized to sham. n=3 to 4 sham, n=6 to 9 TAC. Statistical analysis used the nonparametric Mann–Whitney test. Data shown as median (interquartile range): for TGF‐β: WT sham 1.594 (0.297–3.180) and WT TAC 7.209 (5.212–80.73), ICAM1−/− sham 0.2096 (0.186–3.964) and ICAM1−/− TAC 0.472 (0.263–1.768); for collagen I: WT sham 0.758 (0.615–2.431) and WT TAC 12.51 (3.596–109.6), ICAM1−/− sham 1.318 (0.321–3.388) and ICAM1−/− TAC 1.048 (0.840–1.610); for α‐SMA: WT sham 0.642 (0.552–2.824) and WT TAC 7.838 (0.441–37.80), ICAM1−/− sham 2.008 (0.240–3.953) and ICAM1−/− TAC 0.601 (0.233–1.704). D and E, WT and ICAM1−/− bone marrow–derived monocytes polarized to M2 and M1 macrophages in the presence of 20 ng/mL IL‐4 or 1 μg/mL LPS, respectively, for 3 days. Polarization was determined by mRNA expression of (D) arginase 1 for M2 and (E) inducible nitric oxide synthase for M1 macrophages. n=3 untreated, n=3 to 4 treated with IL‐4 or LPS. Statistical analysis used the unpaired t test. F, WT and ICAM1−/− cardiac fibroblast polarized to myofibroblast in the presence of 100 ng/mL TGF‐β for 16 hours. Polarization determined by mRNA expression of α‐SMA, Magnification ×20 representative pictures of each condition are shown on the left. n=4 to 6 untreated, n=6 to 7 TGF‐β treated. Statistical analysis used the unpaired t test.*P<0.05, **P<0.01, ***P<0.005. α‐SMA, α‐smooth muscle actin; ICAM1 indicates intercellular cell adhesion molecule 1; IL, interleukin; TAC, transverse aortic constriction; TGF‐β, transforming growth factor β; Untr., untreated; WT, wild type.
ICAM1−/− Mice Do Not Develop Cardiac Dysfunction and Heart Failure in Response to TAC
We next sought to determine the effect of ICAM1 deletion in cardiac function in the TAC model of HF, using invasive hemodynamics and echocardiography analysis. LV end‐diastolic pressure, which is elevated in HF, increased 3‐fold in WT mice in response to TAC. Despite equal increases in LV pressure overload, end‐diastolic pressure was significantly reduced in ICAM1−/− mice compared with WT in response to TAC (Figure 7A and 7B). Cardiac systolic and diastolic function determined by the indexes for maximum rate of ventricular pressure increase and decrease obtained from the hemodynamics studies remained completely preserved in ICAM1−/− mice after TAC (Figure 7C and 7D). Finally, although ICAM1−/− mice showed a significant reduction in fractional shortening by echocardiography in response to TAC (27.15±4.89 TAC versus 35.32±4.02 sham), this reduction was not as severe as in WT mice after TAC (19.96±2.864 TAC versus 44.37±3.405 sham) (Figure 7E, Table). These studies demonstrated that ICAM1 contributes to cardiac dysfunction in response to pressure overload induced by TAC.
Figure 7.
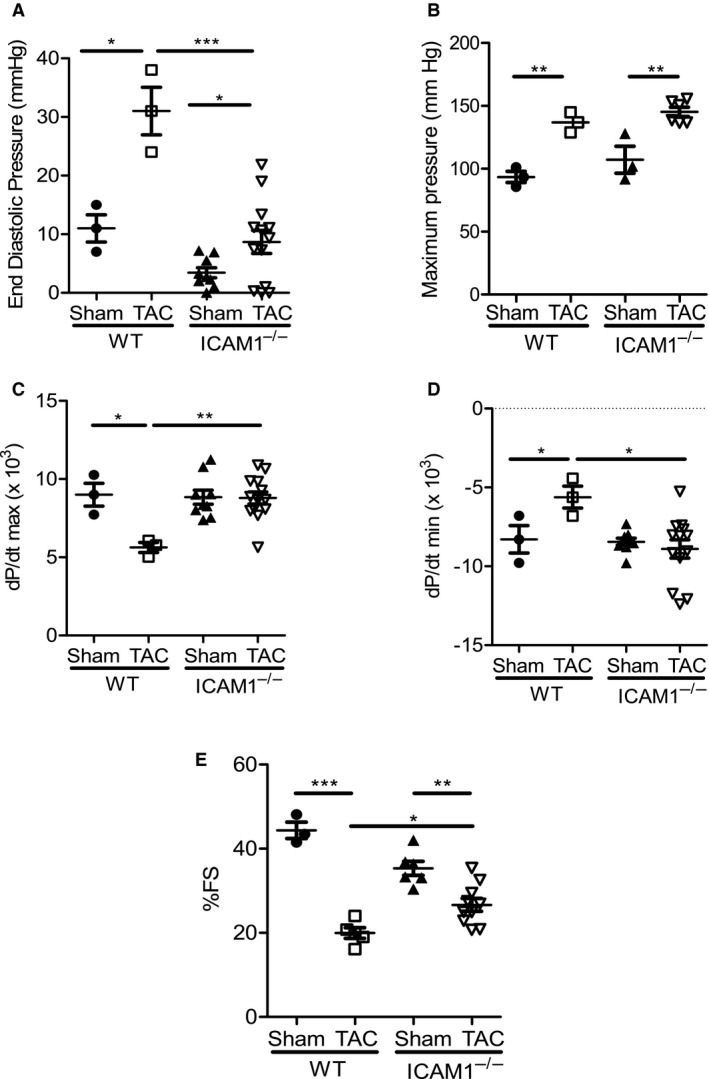
Improved cardiac function in response to TAC‐induced heart failure in ICAM1−/− mice. A through C, Invasive hemodynamic measurements in WT and ICAM1−/− mice measured at 4 weeks after TAC and sham surgery (n=3 WT, n=6–12 ICAM1−/−), (A) end‐diastolic pressure, (B) maximum pressure (C) contractile function (dP/dtmax), and (D) relaxation function (dP/dtmin). E, Fractional shortening at 4 weeks after sham or TAC surgery in WT and ICAM1−/− mice (n=3–6 sham, n=5–10 TAC). Statistical analysis used the unpaired t test. *P<0.05, **P<0.01, ***P<0.005. FS indicates fractional shortening; ICAM1 indicates intercellular cell adhesion molecule 1; TAC, transverse aortic constriction; WT, wild type.
Table 1.
WT and ICAM1−/− Mice Cardiac Function Phenotype Characterization 4 Weeks After TAC
| WT | ICAM1−/− | |||
|---|---|---|---|---|
| Sham (n=3) | TAC (n=5) | Sham (n=6) | TAC (n=10) | |
| EDD, mm | 3.375±0.074 | 3.983±0.575 | 3.802±0.362 | 4.137±0.279 |
| ESD, mm | 1.880±0.135 | 3.191±0.552a | 2.464±0.343 | 3.044±0.389a |
| AWT, mm | 0.904±0.081 | 1.113±0.241 | 0.9319±0.111 | 0.9688±0.167 |
| PWT, mm | 1.005±0.079 | 1.011±0.258 | 0.8504±0.071 | 0.9241±0.115 |
| HR, bpm | 470±75.498 | 444±53.665 | 445.0±44.16 | 454.5±53.87 |
Statistical analysis used the unpaired t test. AWT indicates anterior wall thickness; bpm, beats per minute; EDD, end‐diastolic dimension; ESD, end‐systolic dimension; HR, heart rate; ICAM1 indicates intercellular cell adhesion molecule 1; PWT, posterior wall thickness; TAC, transverse aortic constriction; WT, wild type.
P<0.01.
ICAM1 Upregulation in the Pressure‐Overloaded LV is Independent of EC‐MR Signaling and Associated With Early Upregulation of Cardiac IL‐1β and IL‐6
We explored potential mechanisms that may be driving the increase in ICAM1 expression in cardiac endothelial cells in response to pressure overload. Mineralocorticoid receptor signaling is known to be activated in HF, and prior studies demonstrate that EC‐MR activation upregulates the expression of ICAM1 in vitro in human coronary endothelial cells27 and in the heart in vivo in response to constriction of the ascending aorta.28 Based on these data, we hypothesized that EC‐MR signaling drives LV endothelial ICAM1 upregulation in response to TAC. To test this hypothesis, we subjected EC‐MR−/− mice to TAC.17 After 4 weeks of pressure overload, EC ICAM1 was upregulated in EC‐MR−/− mice to the same extent as in EC‐MR+/+ littermates (Figure 8A), and this correlated with a similar increase in CD4+ T‐cell recruitment independent of the presence of EC‐MR (Figure 8B). Moreover, the expression of the proinflammatory cytokines IL‐6 and IL‐1β, known to be endothelial activators leading to endothelial adhesion molecule upregulation,12, 29 were significantly increased to the same extent in the LVs of both EC‐MR−/− and EC‐MR+/+ mice (Figure 8C). Furthermore, the time course of IL‐1β and IL‐6 upregulation in the LV of WT mice correlated with the upregulation of ICAM1 over the course of TAC (Figure 1A), being evident as early as 48 hours after TAC and remaining upregulated as adverse cardiac remodeling progressed (Figure 8D). Altogether, our studies demonstrated that EC‐MR does not play a role in endothelial LV ICAM1 upregulation and suggested a mechanism by which IL‐1β and IL‐6 produced by resident LV cells as early as 48 hours after TAC induce endothelial ICAM1, which drives LV leukocyte infiltration, further cardiac inflammation, and adverse cardiac remodeling in an EC‐MR–independent manner.
Figure 8.
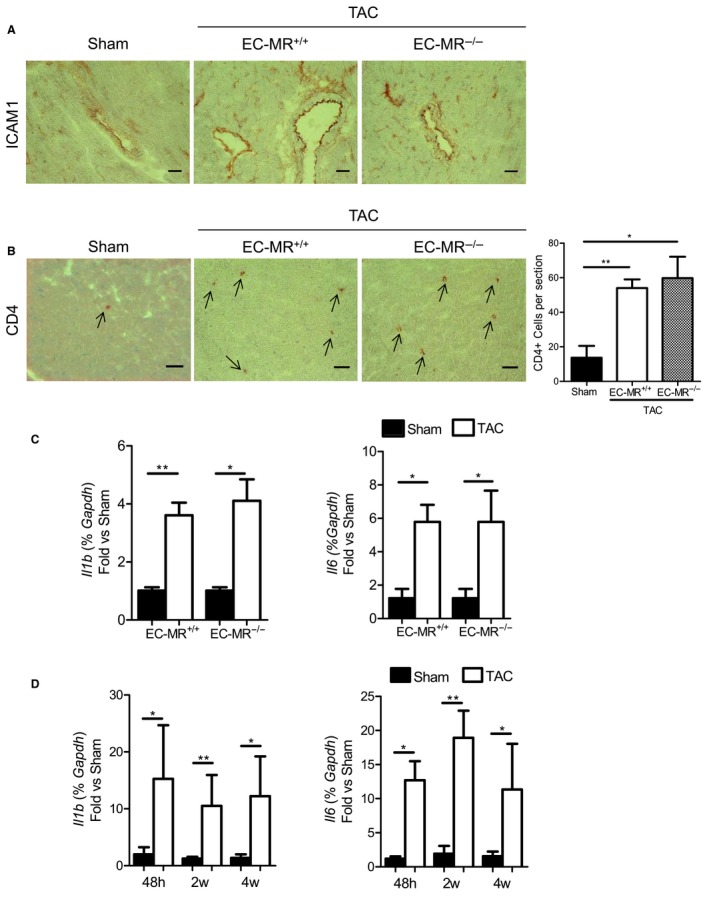
ICAM1 expression, LV T‐cell recruitment, and LV cytokine expression at 4 weeks after TAC are independent of EC‐MR signaling. A and B, Representative immunohistochemistry staining (×20 magnification) for ICAM1 (A) and CD4 (B) in sham, EC‐MR +/+ and EC‐MR −/− TAC mice at 4 weeks after surgery. On the right, quantification of CD4+ T cells per LV section after TAC (B). Scale bar=50 μm. C, mRNA expression of the cytokines IL‐1β and IL‐6 on the LV of EC‐MR −/− and EC‐MR +/+ littermates in response to TAC at 4 weeks normalized to sham. n=3 sham, n=4 TAC. Statistic analysis used the unpaired t test. D, mRNA expression of the cytokines IL‐1β and IL‐6 on the LV of WT mice in response to TAC at 48 hours and at 2 and 4 weeks, normalized to sham. n=4 to 7 sham, n=4 to 9 TAC. Statistic analysis used the nonparametric Mann–Whitney test. Data shown as median (interquartile range): for IL‐1β: at 48 hours, WT sham 1.045 (0.369–4.534) and WT TAC 7.105 (4.022–34.63); at 2 weeks, WT sham 0.898 (0.580–2.110) and WT TAC 3.537 (1.807–10.40); at 4 weeks, WT sham 0.702 (0.592–2.465) and WT TAC 3.960 (1.240–18.51); for IL‐6: at 48 hours, WT sham 1.249 (0.544–1.802) and WT TAC 11.92 (8.317–17.87); at 2 weeks, WT sham 0.952 (0.538–2.777) and WT TAC 19.16 (6.427–31.26); at 4 weeks, WT sham 0.962 (0.365–3.022) and WT TAC 3.808 (2.429–9.796). *P<0.05, **P<0.01. EC‐MR indicates endothelial cell–specific mineralocorticoid receptor; h, hours; ICAM1 indicates intercellular cell adhesion molecule 1; IL, interleukin; LV, left ventricle; TAC, transverse aortic constriction; w, weeks; WT, wild type.
Discussion
This study demonstrates that ICAM1 functions as a critical regulator of pathological cardiac remodeling in pressure overload–induced HF by mediating the recruitment of proinflammatory leukocytes in the LV, leading to cardiac inflammation, cardiac fibrosis, and cardiac dysfunction. We report for the first time that ICAM1−/− mice are protected from cardiac inflammation and fibrosis and do not develop HF in the setting of LV pressure overload. We provide a mechanism by which T cells and proinflammatory monocytes are recruited to the LV via intramyocardial endothelial ICAM1, which is upregulated independent of EC‐MR signaling. Our data suggest that ICAM1 upregulation in the heart may be temporally related to pressure overload induction of cardiac IL‐1β and IL‐6, known activators of ICAM1 expression. Our findings also suggest that ICAM1 expression in antigen‐presenting cells is not necessary for T‐cell activation in the heart‐draining lymph nodes at the onset of HF induced by LV pressure overload. Our data support a model in which LV pressure overload initially results in cardiac cytokine release that induces endothelial ICAM1 expression, in turn promoting later leukocyte recruitment to the LV in the onset of HF. Once infiltrated, LV leukocytes further release cytokines and promote cardiac inflammation, cardiac fibrosis, and cardiac dysfunction resulting in HF. In the absence of ICAM1, leukocyte recruitment, cardiac inflammation, and fibrosis are significantly reduced and result in improved cardiac function. Our data positions ICAM1 as a central player in pressure overload–induced HF.
Multiple studies over the years have identified ICAM1 as a critical regulator of leukocyte arrest and transendothelial migration to allow leukocyte tissue infiltration in cardiovascular disease.12, 30, 31 In the human heart, endothelial ICAM1 is upregulated after myocardial infarction, correlating with high infiltration of inflammatory leukocytes.14 We and others have shown that endothelial ICAM1 is upregulated at the onset of pressure overload induced by either TAC or suprarenal abdominal aortic constriction in rodents, concomitantly with cardiac inflammation and T‐cell infiltration of the LV.9, 15, 32 These data led us to hypothesize that ICAM1 mediates T‐cell recruitment to the LV during pathological cardiac remodeling in HF. Our data confirm that expression of endothelial ICAM1 at 2 and 4 weeks after TAC correlates with T‐cell infiltration of the LV and with a highly proinflammatory cytokine environment that can result in intramyocardial endothelial cell activation. Moreover, we demonstrated LV endothelial ICAM1 as early as 48 hours after TAC, preceding LV leukocyte infiltration.9
Our studies using ICAM1−/− mice and quantitative flow cytometry demonstrate for the first time that Ly‐6Chigh monocytes, known to play a role in cardiac remodeling after myocardial infarction,33, 34 are also recruited to the LV together with T cells in response to TAC at 4 weeks. Our results are in line with a recent report that suggests Ly6Chigh monocytes play a role in maladaptive cardiac remodeling at 3 weeks after TAC, whereas Ly6Clow monocytes participate in early adaptive cardiac remodeling to TAC.35 We also demonstrated that this process is ICAM1 dependent, positioning ICAM1 as a major contributor to cardiac inflammation, which likely contributes to the pathogenesis of HF in this model of pressure overload–induced HF.
Although ICAM1 was originally cloned from endothelial cells, it is now known to be expressed on essentially all leukocyte subsets in addition to vascular endothelial cells.36 These leukocytes include several antigen‐presenting cells in which ICAM1 functions as an accessory molecule in the immune synapse in response to some specific antigens.36 Consequently, ICAM1 expressed in nonendothelial cells also likely contributes to the phenotype of the ICAM1−/− mice. Nevertheless, our findings suggest that ICAM1 expressed by antigen‐presenting cells does not play a role in T‐cell activation in response to TAC because T cells were activated similarly in the heart‐draining lymph nodes of WT and ICAM1−/− mice in response to TAC. Although the antigens activating the T‐cell immune response in TAC remain unknown,9, 24 we interpret our findings as suggesting that the antigens involved in this response do not require ICAM1 in the immune synapse for optimal presentation to achieve T‐cell activation. In addition, the similar frequencies of activated T cells and blood‐circulating CD4+ T cells in sham‐operated WT and ICAM1−/− mice are in line with the initial description of ICAM1−/− mice, indicating no differences in circulating T cells compared with WT mice.16 Moreover, the frequency of circulating T cells was also similar between WT and ICAM1−/− mice in response to TAC, suggesting that the reduced LV T‐cell infiltration observed in ICAM1−/− mice is not due to a defect in T‐cell activation promoted by antigen‐presenting cell–expressed ICAM1 but rather to a defect in LV T‐cell infiltration mediated by endothelial ICAM1.
We interpret our findings indicating that ICAM1−/− mice develop LV hypertrophy but not fibrosis and systolic and diastolic dysfunction as supporting the intriguing possibility that ICAM1 normally promotes a pathological hypertrophic program, whereas deletion of ICAM1 is sufficient to retain a compensatory hypertrophy phenotype during pressure overload. The lack of transition to the fetal or pathological gene expression pattern, including atrial and brain natriuretic peptides and myosin heavy chain β observed in ICAM1−/− mice after TAC,37, 38 also supports this model. Notably, cardiac myocytes isolated from mice lacking ICAM1 in response to TAC are smaller in length and width than WT cardiac myocytes, suggesting a previously unappreciated role for ICAM1 in cardiac hypertrophy that could directly imply LV‐infiltrated T cells. We and others have shown previously that T‐cell–deficient mice do not develop cardiac hypertrophy in response to TAC9, 24; however, we also demonstrated that pharmacological depletion of T cells did not prevent cardiac hypertrophy in response to TAC, despite preventing cardiac dysfunction,9 similar to our results in ICAM1−/− mice and to a previous report that neutralized ICAM1 pharmacologically in rats during pressure overload.15 We speculate that in pharmacologically T‐cell–depleted mice, in ICAM1−/− mice, and in ICAM1‐inhibited rats, the remaining T cells that infiltrate the LV can still contribute to hypertrophy by secreting prohypertrophic mediators such as angiotensin II.39 Interestingly, in all of these settings in mice, we observed blunted LV fibrosis in response to TAC. Our in vitro data demonstrated that ICAM1 expressed in monocytes and fibroblasts is not required for their transition to profibrotic macrophages or myofibroblasts in vitro. These data suggest that the lack of fibrosis in ICAM1−/− mice in vivo is related to a lack of endothelial ICAM1 that prevents T‐cell infiltration, which is necessary for fibrosis. We cannot rule out a role for nonendothelial ICAM1 in vivo, and this possibility will require further investigation in tissue‐specific ICAM1‐deficient mice. Alternatively, soluble ICAM1 resulting from matrix metalloprotease and elastase cleavage of membrane‐bound ICAM136 could also contribute to cardiac inflammation by acting on β2‐integrins in infiltrated leukocytes and inducing their proinflammatory activation, as described in a model of lung injury.40 Soluble ICAM1 has also been shown to engage unknown receptors in the central nervous system and to trigger Src activation,41 and Src, p38 MAPK, and ERK1/2 are involved in cardiac hypertrophy and fibrosis.42, 43, 44 It could be possible that soluble ICAM1 functions through similar mechanisms in cardiac macrophages (by engaging β2‐integrins) and in cardiac resident cells (by engaging an unknown receptor) at the onset of pressure overload and hemodynamic stress and contributes to signaling cascades that result in adverse cardiac remodeling, in addition to membrane‐bound ICAM1 engagement by infiltrated LV leukocytes. These actions of soluble ICAM1 merit further investigation because soluble ICAM1 is significantly increased in plasma of patients with HF.2
Our echocardiography results demonstrate that despite ICAM1−/− mice having a slight increase in end‐diastolic diameter in response to TAC, and thus a decrease in fractional shortening compared with sham, these values are within the healthy‐heart range and are significantly different from those of WT mice. Our results are in line with a recent study in which treatment of WT mice with an ICAM1 function‐blocking antibody resulted in increased fractional shortening compared with mice treated with control antibody in response to TAC at 2 weeks.32 Moreover, our detailed hemodynamics analyses demonstrated that both systolic and diastolic function was preserved in ICAM1−/− mice at the onset of TAC. A mechanism for preserved cardiac function in ICAM1−/− mice is explained by the lack of fibrosis, possibly mediated by decreased LV leukocyte infiltration. In addition, ICAM1 has been found to be expressed in human, dog, and rat cardiac myocytes14, 30, 45 and shown to adhere to neutrophils via Mac1 in in vitro experiments resulting in impaired cardiac myocyte contractility and cardiac myocyte oxidative injury.46, 47 Given the striking preservation of contractility and relaxation in ICAM1−/− mice in response to TAC, we suggest the intriguing possibility that cardiac myocyte ICAM1 may adhere to LV‐infiltrated leukocytes and trigger signals that result in impaired cardiac myocyte function. This hypothesized mechanism requires further investigation.
Elucidation of the mechanisms that drive the upregulation of endothelial ICAM1 represent an important avenue in understanding of the contribution of ICAM1 and proinflammatory leukocyte recruitment to cardiac dysfunction and failure. The mineralocorticoid receptor is activated by the hormone aldosterone, which is elevated in serum in humans with HF and in animal models of HF induced by aortic constriction.48, 49, 50 Aldosterone induces ICAM1 expression in human coronary artery endothelial cells in vitro through its actions via mineralocorticoid receptor signaling.27 Our results demonstrating that ICAM1, IL‐1β, and IL‐6 levels in the LV and LV T‐cell infiltration are similarly induced in EC‐MR−/− and EC‐MR+/+ mice in response to TAC suggest that ICAM1 is upregulated independently of EC‐MR signaling. Consequently, our data suggest that ICAM1 may instead be upregulated in response to the cytokines IL‐1β and IL‐6, which have been shown previously to be expressed by cardiac‐resident cells under stress51, 52 and are known to mediate endothelial cell activation.29 Moreover, our results suggest that the beneficial effect of aldosterone antagonists in HF patients may occur independently of EC‐MRs that involve ICAM1‐mediated LV leukocyte infiltration and may be due to mineralocorticoid receptor inhibition in other cells including macrophages and cardiomyocytes, as demonstrated previously in mouse models of hypertension‐mediated cardiomyophathy using tissue‐specific deletion of mineralocorticoid receptor.53, 54
Collectively, our findings demonstrate for the first time that ICAM1 is necessary for pressure overload–induced cardiac inflammation, fibrosis, and resulting cardiac dysfunction and HF. Our data provide an ICAM1‐dependent mechanism through which effector T cells are recruited into the LV in response to pressure overload, perhaps due to cardiac expression of cytokines that activate endothelial ICAM1 expression. Future studies using tissue‐specific ICAM1‐deficient mice will explore whether ICAM1 expressed in other cell compartments in the heart in addition to the endothelium may contribute to maladaptive cardiac remodeling in HF. Our data provide a mechanism through which effector T cells are recruited into the LV in response to pressure overload. This understanding supports the potential to target ICAM1‐mediated T‐cell recruitment into the LV to improve the function of the failing heart.
Sources of Funding
These studies were supported by National Institutes of Health (NIH) K99‐HL097406, NIH RO1 HL123658 (Dr Alcaide), NIH T32 HL 69770 (Dr Karas, Dr Nevers), NIH 1KL2TR001063‐01 (Dr Blanton), NIH RO1 HL095590 (Dr Jaffe), by American Heart Association Grant in Aid‐ AHA GIA 13GRNT 14560068 (Dr Alcaide) and AHA EIA 18290005 (Dr Jaffe), and by the Instituto de Salud Carlos III MCT PI 10/01096 (Dr Abadía Molina).
Disclosures
None.
Acknowledgments
We would like to acknowledge Dr Luscinskas from the Brigham and Women's hospital, Boston, MA, for providing scientific advice in this manuscript, Dr Bullard, from the University of Alabama, Birmingham, AL for providing the ICAM1−/− mice, Dr Noujaim, Dr Galper, and Dr Paruchuri, from Tufts Medical Center (TMC), for their support in cardiac myocyte isolation from adult mice and experimental advice, respectively. Nathan Li from the Histology core at Tufts University, and the members of the Molecular Cardiology Research Institute (TMC), Boston, MA, for helpful suggestions and discussions of this article.
(J Am Heart Assoc. 2016;5:e003126 doi: 10.1161/JAHA.115.003126)
References
- 1. Braunwald E. Heart failure. JACC Heart Fail. 2013;1:1–20. [DOI] [PubMed] [Google Scholar]
- 2. Yin WH, Chen JW, Jen HL, Chiang MC, Huang WP, Feng AN, Lin SJ, Young MS. The prognostic value of circulating soluble cell adhesion molecules in patients with chronic congestive heart failure. Eur J Heart Fail. 2003;5:507–516. [DOI] [PubMed] [Google Scholar]
- 3. Kalogeropoulos A, Georgiopoulou V, Psaty BM, Rodondi N, Smith AL, Harrison DG, Liu Y, Hoffmann U, Bauer DC, Newman AB, Kritchevsky SB, Harris TB, Butler J. Inflammatory markers and incident heart failure risk in older adults: the Health ABC (Health, Aging, and Body Composition) study. J Am Coll Cardiol. 2010;55:2129–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moe GW, Marin‐Garcia J, Konig A, Goldenthal M, Lu X, Feng Q. In vivo TNF‐alpha inhibition ameliorates cardiac mitochondrial dysfunction, oxidative stress, and apoptosis in experimental heart failure. Am J Physiol Heart Circ Physiol. 2004;287:H1813–H1820. [DOI] [PubMed] [Google Scholar]
- 5. Vasan RS, Sullivan LM, Roubenoff R, Dinarello CA, Harris T, Benjamin EJ, Sawyer DB, Levy D, Wilson PW, D'Agostino RB. Inflammatory markers and risk of heart failure in elderly subjects without prior myocardial infarction: the Framingham Heart Study. Circulation. 2003;107:1486–1491. [DOI] [PubMed] [Google Scholar]
- 6. Deswal A, Petersen NJ, Feldman AM, Young JB, White BG, Mann DL. Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation. 2001;103:2055–2059. [DOI] [PubMed] [Google Scholar]
- 7. Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT. Randomized, double‐blind, placebo‐controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor‐alpha, in patients with moderate‐to‐severe heart failure: results of the anti‐TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation. 2003;107:3133–3140. [DOI] [PubMed] [Google Scholar]
- 8. Mann DL, McMurray JJ, Packer M, Swedberg K, Borer JS, Colucci WS, Djian J, Drexler H, Feldman A, Kober L, Krum H, Liu P, Nieminen M, Tavazzi L, van Veldhuisen DJ, Waldenstrom A, Warren M, Westheim A, Zannad F, Fleming T. Targeted anticytokine therapy in patients with chronic heart failure: results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation. 2004;109:1594–1602. [DOI] [PubMed] [Google Scholar]
- 9. Nevers T, Salvador AM, Grodecki‐Pena A, Knapp A, Velazquez F, Aronovitz M, Kapur NK, Karas RH, Blanton RM, Alcaide P. Left ventricular T cell recruitment contributes to the pathogenesis of heart failure. Circ Heart Fail. 2015;8:776–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yu Q, Watson RR, Marchalonis JJ, Larson DF. A role for T lymphocytes in mediating cardiac diastolic function. Am J Physiol Heart Circ Physiol. 2005;289:H643–H651. [DOI] [PubMed] [Google Scholar]
- 11. Li N, Bian H, Zhang J, Li X, Ji X, Zhang Y. The Th17/Treg imbalance exists in patients with heart failure with normal ejection fraction and heart failure with reduced ejection fraction. Clin Chim Acta. 2010;411:1963–1968. [DOI] [PubMed] [Google Scholar]
- 12. Alcaide P, Auerbach S, Luscinskas FW. Neutrophil recruitment under shear flow: it's all about endothelial cell rings and gaps. Microcirculation. 2009;16:43–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gerhardt T, Ley K. Monocyte trafficking across the vessel wall. Cardiovasc Res. 2015;107:321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Niessen HW, Lagrand WK, Visser CA, Meijer CJ, Hack CE. Upregulation of ICAM‐1 on cardiomyocytes in jeopardized human myocardium during infarction. Cardiovasc Res. 1999;41:603–610. [DOI] [PubMed] [Google Scholar]
- 15. Kuwahara F, Kai H, Tokuda K, Niiyama H, Tahara N, Kusaba K, Takemiya K, Jalalidin A, Koga M, Nagata T, Shibata R, Imaizumi T. Roles of intercellular adhesion molecule‐1 in hypertensive cardiac remodeling. Hypertension. 2003;41(3 Pt 2):819–823. [DOI] [PubMed] [Google Scholar]
- 16. Sligh JE Jr, Ballantyne CM, Rich SS, Hawkins HK, Smith CW, Bradley A, Beaudet AL. Inflammatory and immune responses are impaired in mice deficient in intercellular adhesion molecule 1. Proc Natl Acad Sci USA. 1993;90:8529–8533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barrett MK, Bender SB, Hong K, Yang Y, Aronovitz M, Jaisser F, Hill MA, Jaffe IZ. Endothelial mineralocorticoid receptors differentially contribute to coronary and mesenteric vascular function without modulating blood pressure. Hypertension. 2015;66:988–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chintalgattu V, Ai D, Langley RR, Zhang J, Bankson JA, Shih TL, Reddy AK, Coombes KR, Daher IN, Pati S, Patel SS, Pocius JS, Taffet GE, Buja LM, Entman ML, Khakoo AY. Cardiomyocyte PDGFR‐beta signaling is an essential component of the mouse cardiac response to load‐induced stress. J Clin Invest. 2010;120:472–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Patten RD, Pourati I, Aronovitz MJ, Alsheikh‐Ali A, Eder S, Force T, Mendelsohn ME, Karas RH. 17 Beta‐estradiol differentially affects left ventricular and cardiomyocyte hypertrophy following myocardial infarction and pressure overload. J Card Fail. 2008;14:245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bozkurt B, Kribbs SB, Clubb FJ Jr, Michael LH, Didenko VV, Hornsby PJ, Seta Y, Oral H, Spinale FG, Mann DL. Pathophysiologically relevant concentrations of tumor necrosis factor‐alpha promote progressive left ventricular dysfunction and remodeling in rats. Circulation. 1998;97:1382–1391. [DOI] [PubMed] [Google Scholar]
- 21. Georgescu SP, Aronovitz MJ, Iovanna JL, Patten RD, Kyriakis JM, Goruppi S. Decreased metalloprotease 9 induction, cardiac fibrosis, and higher autophagy after pressure overload in mice lacking the transcriptional regulator p8. Am J Physiol Cell Physiol. 2011;301:C1046–C1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lim CC, Apstein CS, Colucci WS, Liao R. Impaired cell shortening and relengthening with increased pacing frequency are intrinsic to the senescent mouse cardiomyocyte. J Mol Cell Cardiol. 2000;32:2075–2082. [DOI] [PubMed] [Google Scholar]
- 23. Blanton RM, Takimoto E, Lane AM, Aronovitz M, Piotrowski R, Karas RH, Mendelsohn ME. Protein kinase G Ialpha inhibits pressure overload‐induced cardiac remodeling and is required for the cardioprotective effect of sildenafil in vivo. J Am Heart Assoc. 2012;1:e003731 doi: 10.1161/JAHA.112.003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Laroumanie F, Douin‐Echinard V, Pozzo J, Lairez O, Tortosa F, Vinel C, Delage C, Calise D, Dutaur M, Parini A, Pizzinat N. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation. 2014;129:2111–2124. [DOI] [PubMed] [Google Scholar]
- 25. Huppa JB, Davis MM. T‐cell‐antigen recognition and the immunological synapse. Nat Rev Immunol. 2003;3:973–983. [DOI] [PubMed] [Google Scholar]
- 26. Miyata S, Minobe W, Bristow MR, Leinwand LA. Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ Res. 2000;86:386–390. [DOI] [PubMed] [Google Scholar]
- 27. Caprio M, Newfell BG, la Sala A, Baur W, Fabbri A, Rosano G, Mendelsohn ME, Jaffe IZ. Functional mineralocorticoid receptors in human vascular endothelial cells regulate intercellular adhesion molecule‐1 expression and promote leukocyte adhesion. Circ Res. 2008;102:1359–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuster GM, Kotlyar E, Rude MK, Siwik DA, Liao R, Colucci WS, Sam F. Mineralocorticoid receptor inhibition ameliorates the transition to myocardial failure and decreases oxidative stress and inflammation in mice with chronic pressure overload. Circulation. 2005;111:420–427. [DOI] [PubMed] [Google Scholar]
- 29. Luscinskas FW, Cybulsky MI, Kiely JM, Peckins CS, Davis VM, Gimbrone MA Jr. Cytokine‐activated human endothelial monolayers support enhanced neutrophil transmigration via a mechanism involving both endothelial‐leukocyte adhesion molecule‐1 and intercellular adhesion molecule‐1. J Immunol. 1991;146:1617–1625. [PubMed] [Google Scholar]
- 30. Mann DL. Inflammatory mediators and the failing heart: past, present, and the foreseeable future. Circ Res. 2002;91:988–998. [DOI] [PubMed] [Google Scholar]
- 31. de Gaetano M, Dempsey E, Marcone S, James WG, Belton O. Conjugated linoleic acid targets beta2 integrin expression to suppress monocyte adhesion. J Immunol. 2013;191:4326–4336. [DOI] [PubMed] [Google Scholar]
- 32. Yoshida Y, Shimizu I, Katsuumi G, Jiao S, Suda M, Hayashi Y, Minamino T. p53‐Induced inflammation exacerbates cardiac dysfunction during pressure overload. J Mol Cell Cardiol. 2015;85:183–198. [DOI] [PubMed] [Google Scholar]
- 33. Hilgendorf I, Gerhardt LM, Tan TC, Winter C, Holderried TA, Chousterman BG, Iwamoto Y, Liao R, Zirlik A, Scherer‐Crosbie M, Hedrick CC, Libby P, Nahrendorf M, Weissleder R, Swirski FK. Ly‐6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res. 2014;114:1611–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dutta P, Nahrendorf M. Monocytes in myocardial infarction. Arterioscler Thromb Vasc Biol. 2015;35:1066–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Weisheit C, Zhang Y, Faron A, Kopke O, Weisheit G, Steinstrasser A, Frede S, Meyer R, Boehm O, Hoeft A, Kurts C, Baumgarten G. Ly6C(low) and not Ly6C(high) macrophages accumulate first in the heart in a model of murine pressure‐overload. PLoS One. 2014;9:e112710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ramos TN, Bullard DC, Barnum SR. ICAM‐1: isoforms and phenotypes. J Immunol. 2014;192:4469–4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Krenz M, Robbins J. Impact of beta‐myosin heavy chain expression on cardiac function during stress. J Am Coll Cardiol. 2004;44:2390–2397. [DOI] [PubMed] [Google Scholar]
- 38. McMullen JR, Jennings GL. Differences between pathological and physiological cardiac hypertrophy: novel therapeutic strategies to treat heart failure. Clin Exp Pharmacol Physiol. 2007;34:255–262. [DOI] [PubMed] [Google Scholar]
- 39. Hoch NE, Guzik TJ, Chen W, Deans T, Maalouf SA, Gratze P, Weyand C, Harrison DG. Regulation of T‐cell function by endogenously produced angiotensin II. Am J Physiol Regul Integr Comp Physiol. 2009;296:R208–R216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schmal H, Czermak BJ, Lentsch AB, Bless NM, Beck‐Schimmer B, Friedl HP, Ward PA. Soluble ICAM‐1 activates lung macrophages and enhances lung injury. J Immunol. 1998;161:3685–3693. [PubMed] [Google Scholar]
- 41. Otto VI, Gloor SM, Frentzel S, Gilli U, Ammann E, Hein AE, Folkers G, Trentz O, Kossmann T, Morganti‐Kossmann MC. The production of macrophage inflammatory protein‐2 induced by soluble intercellular adhesion molecule‐1 in mouse astrocytes is mediated by src tyrosine kinases and p42/44 mitogen‐activated protein kinase. J Neurochem. 2002;80:824–834. [DOI] [PubMed] [Google Scholar]
- 42. Zhu X, Fang J, Jiang DS, Zhang P, Zhao GN, Zhu X, Yang L, Wei X, Li H. Exacerbating pressure overload‐induced cardiac hypertrophy: novel role of adaptor molecule Src homology 2‐B3. Hypertension. 2015;66:571–581. [DOI] [PubMed] [Google Scholar]
- 43. Zhang S, Weinheimer C, Courtois M, Kovacs A, Zhang CE, Cheng AM, Wang Y, Muslin AJ. The role of the Grb2‐p38 MAPK signaling pathway in cardiac hypertrophy and fibrosis. J Clin Invest. 2003;111:833–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang Y. Mitogen‐activated protein kinases in heart development and diseases. Circulation. 2007;116:1413–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gwechenberger M, Mendoza LH, Youker KA, Frangogiannis NG, Smith CW, Michael LH, Entman ML. Cardiac myocytes produce interleukin‐6 in culture and in viable border zone of reperfused infarctions. Circulation. 1999;99:546–551. [DOI] [PubMed] [Google Scholar]
- 46. Davani EY, Dorscheid DR, Lee CH, van Breemen C, Walley KR. Novel regulatory mechanism of cardiomyocyte contractility involving ICAM‐1 and the cytoskeleton. Am J Physiol Heart Circ Physiol. 2004;287:H1013–H1022. [DOI] [PubMed] [Google Scholar]
- 47. Entman ML, Youker K, Shoji T, Kukielka G, Shappell SB, Taylor AA, Smith CW. Neutrophil induced oxidative injury of cardiac myocytes. A compartmented system requiring CD11b/CD18‐ICAM‐1 adherence. J Clin Invest. 1992;90:1335–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Morris BJ, Davis JO, Zatzman ML, Williams GM. The renin‐angiotensin‐aldosterone system in rabbits with congestive heart failure produced by aortic constriction. Circ Res. 1977;40:275–282. [DOI] [PubMed] [Google Scholar]
- 49. Young M, Head G, Funder J. Determinants of cardiac fibrosis in experimental hypermineralocorticoid states. Am J Physiol. 1995;1:E657–E662. [DOI] [PubMed] [Google Scholar]
- 50. Somanna NK, Yariswamy M, Garagliano JM, Siebenlist U, Mummidi S, Valente AJ, Chandrasekar B. Aldosterone‐induced cardiomyocyte growth, and fibroblast migration and proliferation are mediated by TRAF3IP2. Cell Signal. 2015;27:1928–1938. [DOI] [PubMed] [Google Scholar]
- 51. Suzuki S, Shishido T, Funayama A, Netsu S, Ishino M, Kitahara T, Sasaki T, Katoh S, Otaki Y, Watanabe T, Shibata Y, Mantovani A, Takeishi Y, Kubota I. Long pentraxin PTX3 exacerbates pressure overload‐induced left ventricular dysfunction. PLoS One. 2013;8:e53133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Saxena A, Chen W, Su Y, Rai V, Uche OU, Li N, Frangogiannis NG. IL‐1 induces proinflammatory leukocyte infiltration and regulates fibroblast phenotype in the infarcted myocardium. J Immunol. 2013;191:4838–4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rickard AJ, Morgan J, Bienvenu LA, Fletcher EK, Cranston GA, Shen JZ, Reichelt ME, Delbridge LM, Young MJ. Cardiomyocyte mineralocorticoid receptors are essential for deoxycorticosterone/salt‐mediated inflammation and cardiac fibrosis. Hypertension. 2012;60:1443–1450. [DOI] [PubMed] [Google Scholar]
- 54. Rickard AJ, Morgan J, Tesch G, Funder JW, Fuller PJ, Young MJ. Deletion of mineralocorticoid receptors from macrophages protects against deoxycorticosterone/salt‐induced cardiac fibrosis and increased blood pressure. Hypertension. 2009;54:537–543. [DOI] [PubMed] [Google Scholar]