

Abstract
Background
Nitric oxide synthase uncoupling occurs under conditions of oxidative stress modifying the enzyme's function so it generates superoxide rather than nitric oxide. Nitric oxide synthase uncoupling occurs with chronic pressure overload, and both are ameliorated by exogenous tetrahydrobiopterin (BH4)—a cofactor required for normal nitric oxide synthase function—supporting a pathophysiological link. Genetically augmenting BH4 synthesis in endothelial cells fails to replicate this benefit, indicating that other cell types dominate the effects of exogenous BH4 administration. We tested whether the primary cellular target of BH4 is the cardiomyocyte or whether other novel mechanisms are invoked.
Methods and Results
Mice with cardiomyocyte‐specific overexpression of GTP cyclohydrolase 1 (mGCH1) and wild‐type littermates underwent transverse aortic constriction. The mGCH1 mice had markedly increased myocardial BH4 and, unlike wild type, maintained nitric oxide synthase coupling after transverse aortic constriction; however, the transverse aortic constriction–induced abnormalities in cardiac morphology and function were similar in both groups. In contrast, exogenous BH4 supplementation improved transverse aortic constricted hearts in both groups, suppressed multiple inflammatory cytokines, and attenuated infiltration of inflammatory macrophages into the heart early after transverse aortic constriction.
Conclusions
BH4 protection against adverse remodeling in hypertrophic cardiac disease is not driven by its prevention of myocardial nitric oxide synthase uncoupling, as presumed previously. Instead, benefits from exogenous BH4 are mediated by a protective effect coupled to suppression of inflammatory pathways and myocardial macrophage infiltration.
Keywords: hypertrophy, inflammation, myocardium, nitric oxide synthase, oxidative stress
Subject Categories: Oxidant Stress, Inflammation, Hypertrophy, Basic Science Research, Myocardial Biology
Introduction
Heart failure is a leading cause of morbidity and mortality that accounts for ≈35% of all cardiovascular deaths in the United States alone.1 Hypertension with cardiac hypertrophy and fibrosis are major risk factors for heart failure, and abnormal oxidative stress is thought to play a major role in the underlying pathophysiology.2 With many myocardial diseases, excess reactive oxygen species (ROS) are generated by multiple sources, including mitochondria; NADPH‐, xanthine‐, and monoamine oxidases; and the constitutive nitric oxide synthases (NOSs). In the latter case, NOS transitions from a nitric oxide to superoxide generator by processes involving oxidative modification,3 including S‐glutathionylation,4 elevated levels of the NOS inhibitor asymmetric dimethylarginine,5 and oxidation/depletion of the NOS cofactor tetrahydrobiopterin (BH4).6 The last mechanism is of particular interest because BH4 is available as an oral therapeutic for treating phenylketonuria.7 BH4 is required for electron transfer from the NOS reductase to oxidase domain,8, 9 but it also functions as an allosteric modulator of arginine binding10 and a stabilizer of the NOS homodimer.11 Reduced BH4 availability and NOS uncoupling are thought to play roles in a broad variety of cardiovascular diseases; in particular, oral supplementation with BH4 prevents both NOS uncoupling and associated cardiac hypertrophy and failure12, 13, 14 in models of pressure overload from transverse aortic constriction (TAC).3
Clinical efforts to translate BH4 as a cardiovascular therapy have met with mixed results (see Bendall et al15 for review). This may stem from inadequate cellular uptake or oxidation of BH4 following oral administration but equally from our lack of understanding of what BH4 targets to be effective. The conventional view is that orally supplemented BH4 becomes available in its reduced form and is transported intracellularly to restore NOS coupling and the nitric oxide/ROS balance. Most consumed BH4, however, is first systemically oxidized to 7,8‐dihydrobiopterin (BH2; which can provoke NOS uncoupling) and must be reduced to BH4 by dihydrofolate reductase (salvage pathway) to be biologically effective.16 This conversion may be limited by diseases with oxidative stress, reducing the benefit of BH4 therapy.17 The capacity for cellular uptake of BH4 may vary, and data suggest that intracellularly synthesized BH4 cannot be released to neighboring cells.18, 19 Mice with genetically induced endothelial‐selective enhancement of BH418 exhibit reduced atherosclerotic lesions.20 The same model displays reduced myocardial superoxide in hearts subjected to pressure overload, yet antihypertrophic and antifibrotic effects are absent, in contrast to mice receiving exogenous BH4.13
To further explore cell‐specific BH4 signaling in the heart, we developed mice in which BH4 synthesis is enhanced in cardiac myocytes by overexpressing the rate‐limiting enzyme of BH4 de novo synthesis, GTP cyclohydrolase 1 (GCH1),19 coupled to the α‐myosin heavy chain (α‐MHC) promoter.19 Prior studies reported marked elevation of myocardial BH4 and BH2 levels in the model, with NOS remaining coupled and neuronal NOS (NOS1) modestly enhanced at baseline.19, 21 We tested whether augmenting myocyte BH4 synthesis protects against adverse left ventricle (LV) remodeling from pressure overload, a condition in which both NOS1 and NOS3 (endothelial NOS) uncoupling contribute to ROS generation.12, 22 Surprisingly, we found minimal improvement in mGCH1 mice that underwent TAC, despite prevention of NOS uncoupling. In contrast, oral BH4 supplementation was very effective in both wild‐type (WT) mice and mice with cardiomyocyte‐specific overexpression of GCH1 (mGCH1) after TAC, although NOS was already effectively coupled and ROS generation was significantly reduced in the mGCH1 group. Instead, we found anti‐inflammatory effects only from exogenous BH4 treatment, including suppression of macrophage infiltration. These data show that NOS recoupling in stressed myocardium does not mediate the beneficial effects of BH4, thus revealing new cellular targets for cardiac BH4 therapy in pressure overload.
Methods
General Experimental Model
Heterozygous transgenic animals overexpressing the human GCH1 gene under control of the α‐MHC were bred to provide cardiomyocyte‐selective overexpressors (mGCH1) and WT littermate controls.19 Myocardial BH4 is ≈10× selectively higher in mGCH1 than WT, whereas plasma, skeletal muscle, and liver levels are similar.19 Mice (aged 2–3 months, matched for body size) underwent TAC or sham surgery12 and were followed serially by echocardiography for 3 weeks. At terminal study, the heart was rapidly excised, and the LV was dissected and placed in 10% formalin or liquid nitrogen for subsequent analysis. In a second experimental group, mice were fed a soft diet (BioServ) with or without added BH4 (200 mg/kg per day; Enzo Life Sciences or BioMarin Pharmaceuticals) and then subjected to TAC for 3 weeks. All animal protocols were approved by the animal care and use committee of Johns Hopkins University.
Echocardiography
Transthoracic echocardiography was performed in conscious mice using a 13‐MHz transducer (Acuson Sequoia c256; Siemens). M‐mode LV dimensions were averaged over 3 to 5 beats at physiological heart rates, and fractional shortening and LV mass were calculated. Echocardiography and image analyses were performed blinded to the experimental group.
Histology and Immunostaining
Myocardium was fixed in 10% formalin and stained with Masson's trichrome to assess interstitial fibrosis. Quantification was performed using ImageJ software (National Institutes of Health), examining 5 to 6 independent regions in each heart. For enzyme immunohistochemistry, deparaffinized tissue sections were heated in 10 mmol/L citrate buffer (pH 6.0) in the microwave oven for antigen retrieval. After blocking with 20% goat serum, sections were incubated with primary antibody at 4°C overnight. Following quenching endogenous peroxidase with 3% hydrogen peroxide, sections were incubated with biotinylated secondary antibody and, subsequently, streptavidin–horseradish peroxidase conjugate. Last, sections were incubated in 3,3′‐diaminobenzidine solution and counterstained with hematoxylin.
Superoxide Determination
Superoxide production was determined by lucigenin‐enhanced chemiluminescence23 or detection of 2‐hydroxyethidium (2‐OH‐E) using high‐performance liquid chromatography (HPLC), as described previously. For chemiluminescence, lucigenin (5 μmol/L) was added to LV homogenates, and the proportion of ROS generated by NOS was determined by subsequent addition of L‐NAME (1 mmol/L; the inactive isomer D‐NAME (1 mmol/L) was a negative control). Finally, the superoxide scavenger 4,5‐dihydroxy‐1,3‐benzene disulphonic acid (10 mmol/L; Tiron; Sigma‐Aldrich) was added, and recording was resumed for 4 minutes. The relative chemiluminescence levels were evaluated over the last 120 seconds of each period and expressed as the Tiron‐inhibitable fraction. Data are presented in relative light units (RLU) per milligram of protein per second. For HPLC detection of dihydroethidium conversion to 2‐OH‐E, LV homogenates were incubated with dihydroethidium (50 μmol/L) for 15 minutes at 37°C and lysed with methanol. Hydrochloric acid (100 mmol/L) was added, and the samples were loaded into the autosampler for detection. Subsets of supernatants were preincubated with L‐NAME (1 mmol/L). The separation of dihydroethidium, 2‐OH‐E, and ethidium was performed using the gradient HPLC system with a reverse‐phase column and quantified using fluorescence detection set at 510 nm (excitation) and 595 nm (emission). All results were expressed as the Tiron‐inhibitable fraction of 2‐OH‐E.
Gene Expression Analysis
Total cardiac RNA was extracted using Trizol reagent (Invitrogen), according to the manufacturer's instructions, and reverse transcribed. Messenger RNA was analyzed by quantitative real‐time polymerase chain reaction using TaqMan (Applied Biosystems) or custom SYBR Green primers. Polymerase chain reaction primers were the same as in a recent study.24 Data were normalized to GAPDH gene expression.
Myocardial BH2/BH4 Analysis
BH4, BH2, and fully oxidized biopterin levels were determined by chromatography, as described previously.19 Briefly, LVs were snap‐frozen in liquid nitrogen and stored at −80°C until further use. Samples were homogenized in PBS (50 mmol/L, pH 7.4), containing dithioerythritol (1 mmol/L) and EDTA (100 μmol/L) and deproteinated using trichloroacetic acid (2 mol/L) and dithioerythritol (1 mmol/L). After centrifugation for 15 minutes at 16 000 × g at 4°C, supernatants were injected into an HPLC system in which biopterins were separated using a carbon‐18 column (Hichrom) and quantified using sequential fluorescence (Jasco) and electrochemical (Coulochem III; ESA Inc) detection. Standard curves were calculated by injected known concentrations of each biopterin, and results were normalized to the sample's protein content (bicinchoninic acid assay).
Statistical Analysis
Data are presented as mean±SEM or median with interquartile range. Within‐group comparisons (eg, same genotype or same treatment group) involving the remaining 2 parameters (time course and genotype or treatment) were performed using a 2‐way repeated‐measures ANOVA, with post hoc multiple comparisons to baseline using a Dunnett test, and between‐group comparisons were done at the same time point by the Tukey test. For a given genotype and treatment (drug or TAC), data were also analyzed using 1‐way repeated‐measures ANOVA. ANOVA without repeated measures was used as appropriate and is noted. Other tests including nonpaired 2‐tailed Student t tests and nonparametric tests (Kruskal–Wallis or Mann–Whitney U) were performed as appropriate and are indicated in the figure legends. All statistical analysis was performed using GraphPad Prism version 6 (GraphPad Software).
Results
Pressure Overload Induces Similar LV Dilation and Dysfunction in WT and mGCH1 Mice
Both WT and mGCH1 mice exhibited similar myocardial dysfunction and remodeling following 3 weeks of TAC. Figure 1A (left) displays representative M‐mode echocardiograms at baseline and after 3 weeks of sustained TAC, and Figure 1B shows summary data for fractional shortening. LV dilation and reduced ejection fraction after TAC were also similar between groups (Figure S1A through S1C). Echo‐derived estimates of LV hypertrophy rose similarly (Figure 1B, upper), although postmortem ratios of heart weight/body weight (Figure 1B, lower) and heart weight/tibia length (Figure S1D) revealed less hypertrophy in mGCH1 mice. Nevertheless, TAC‐stimulated myocardial fibrosis was similar between groups (Figure 1C).
Figure 1.
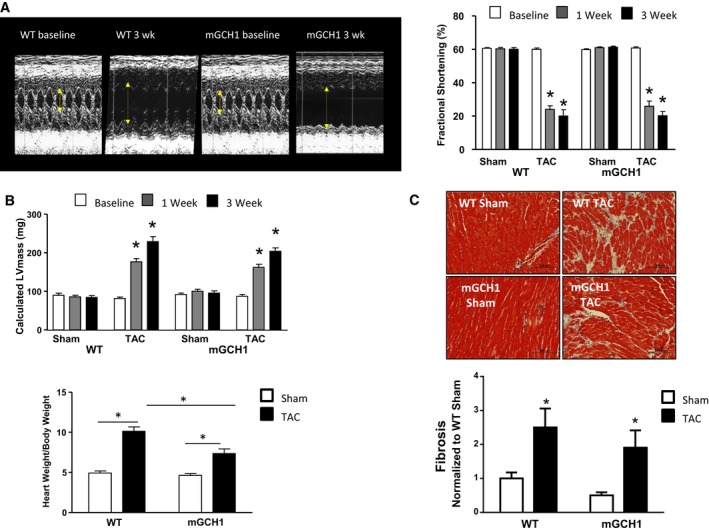
Mice overexpressing mGCH1 in cardiomyocytes develop progressive LV hypertrophy and fibrosis with TAC similar to littermate (WT) controls. A, Sample M‐mode echocardiograms show increased LV dilation and reduced fractional shortening after 3 weeks of TAC. Average LV fractional shortening 3 weeks after TAC is shown on the right. Results of repeated‐measures 2‐way ANOVA: P<0.0001 for TAC effect; P=0.7 for genotype group and interaction. Results of repeated measures 1‐way ANOVA within genotype groups: P<0.0001 for TAC; *P<0.0001 vs baseline in same group by the Dunnett multiple comparisons test, n=8–10 per bar). B, (Upper) Echo‐derived LV mass increased similarly in both groups. Results of repeated‐measures 2‐way ANOVA: P<0.0001 for TAC effect; P=0.2 for genotype, 0.09 for interaction. *P<0.0001 vs baseline by the Dunnett test, n=8–10 per bar). (Lower) Heart weight/body weight ratio at 3 weeks after TAC or sham surgery confirmed LV hypertrophy in both groups (*P<0.01), which was less marked in mGCH1 hearts (*P<0.05, n=8–10 per group). C, (Upper) Sample histopathology shows increased LV fibrosis (Masson's trichrome) in both groups after TAC. (Lower) Average data (*P<0.05 vs sham‐operated controls; n=5–6 per group, each with 5–6 regions in each heart separately quantified). LV indicates left ventricle; mGCH1, cardiomyocyte‐specific GTP cyclohydrolase overexpression; TAC, transverse aortic constriction; wk, weeks; WT, wild type.
Effect of Pressure Overload on BH4 Synthesis, Oxidation, and NOS‐Derived Superoxide in mGCH1 Mice
A potential explanation for the lack of overall protection against TAC in mGCH1 mice was BH4 oxidation, which would reduce its capacity to support NOS function. Accordingly, we assayed BH4, BH2, and total biopterins by HPLC. Concentrations of all 3 were 1 to 2 orders of magnitude higher in sham control mGCH1 myocardium (Figure 2A). These levels remained high after 3 weeks of TAC, with a trend toward slightly further increases in BH4 in mGCH1 mice (P=0.06). The ratio of BH4 to its oxidized products rose in both groups after TAC (P<0.05 each). BH4 levels did not decline significantly after TAC in the littermate controls (unlike in some earlier studies12, 13). Total myocardial superoxide trended toward increase in WT mice (123.7±17.5 versus 189.3±28.0 RLU/mg per second, P=0.09) but remained low in mGCH1 transgenic mice (121.1±14.8 versus 135.6±19.1 RLU/mg per second, P=0.55). Notably, ROS generation attributable to NOS more than doubled in WT mice with TAC, whereas this was low and remained so in mGCH1 mice with TAC (Figure 2B) (P=0.021 for the interaction of TAC and genotype by 2‐way ANOVA, P<0.005 for WT versus mGCH1 mice with TAC by the Tukey multiple comparisons test, n=6–10). Both groups had similar values for sham controls. We tested whether differences in NOS uncoupling might relate to altered expression of NOS1 or NOS3 (Figure 2C) but found no changes from TAC in either group. NOS2 was not detectable in this model, consistent with prior data.12 These results showed that despite preventing myocardial NOS uncoupling, myocyte‐specific enhancement of BH4 generally failed to ameliorate cardiac pathological remodeling from TAC.
Figure 2.
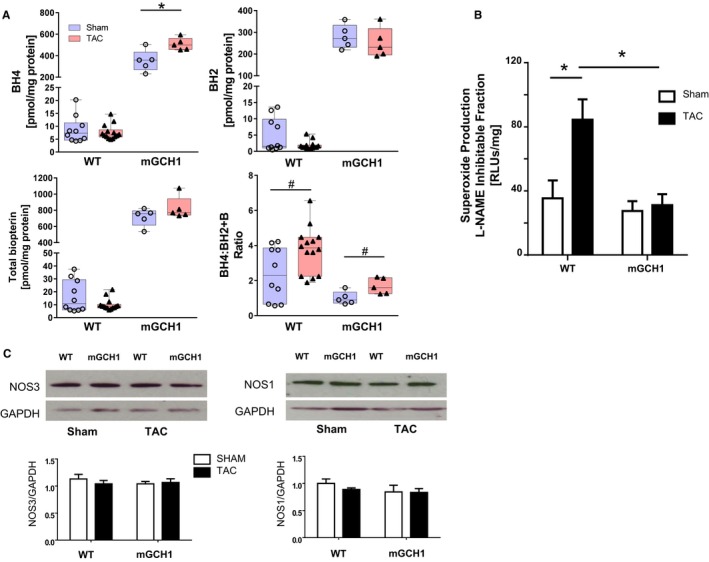
Increased LV biopterin content in mGCH1 mice decreases oxidant stress. A, BH4, BH2, and total biopterin are markedly increased in mGCH1 mice (all P<0.0001, n=5 per group). With the exception of the tendency of a small rise in BH4 in mGCH1 LV (*P=0.06 by Mann–Whitney test), TAC did not alter biopterins significantly in either genotype; however, the ratio of BH4/BH2 plus biopterin was higher at 3 weeks after TAC in both genotypes (# P<0.05 by Mann–Whitney test; WT, n=10–14; mGCH1, n=5). B, NOS‐derived superoxide increased in WT but not in mGCH1 hearts after TAC. P=0.021 for the interaction of TAC and genotype by 2‐way ANOVA, *P<0.01 by the Tukey multiple comparisons test, (n=6–10). Baseline levels were similar in the 2 groups. C, NOS1 (n=4–5) and NOS3 (n=7–9) protein expression was similar in both genotypes. BH4 indicates tetrahydrobiopterin; LV, left ventricle; mGCH1, cardiomyocyte‐specific GTP cyclohydrolase overexpression; NOS, nitric oxide synthase; RLU, relative light units; TAC, transverse aortic constriction; WT, wild type.
Exogenous BH4 Rescues Both WT and mGCH1 Mice From LV Dysfunction Caused by Pressure Overload
To test whether BH4 supplementation could rescue mGCH1 hearts, which already had coupled myocardial NOS after TAC and myocyte BH4 at 50× above normal, we repeated the study but also provided exogenous BH4 to the animals. BH4 ameliorated function, hypertrophy, and fibrosis in WT hearts with TAC, as expected, but it also induced the same improvements in mGCH1 hearts (Figure 3A through 3C, Figure S2). Although repeated‐measures 2‐way ANOVA revealed an interaction (P=0.012) between TAC time course and genotype in mice receiving exogenous BH4, individual between‐group comparisons at weeks 1 and 3 were not significantly different (P>0.8). Consistent with these changes, mRNA expression of hypertrophy‐responsive genes including B‐type natriuretic peptide, the ratio of β‐MHC/α‐MHC, and thrombospondin‐425, 26, 27—all similarly elevated by TAC in control and mGCH1 hearts—each declined with oral BH4 treatment (Figure 3D).
Figure 3.
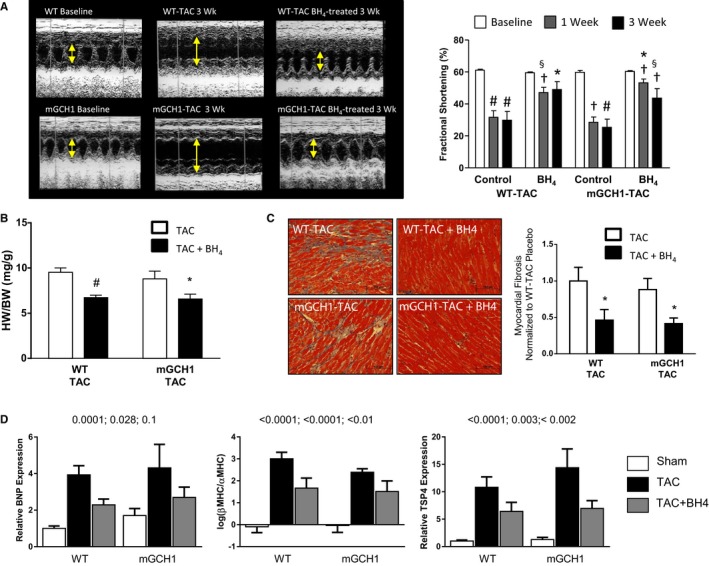
BH4 treatment rescues LV dysfunction, hypertrophy, and fibrosis induced by TAC in WT and mGCH1 mice. A, Sample M‐mode echocardiograms show increased LV dilation and fractional shortening after 3 weeks of TAC. Both were improved in mice treated with BH4. Summary data are shown in the right panel; Within‐genotype 2‐way repeated‐measures ANOVA: P<0.0001 for time, P<0.03 for the time–treatment interaction. Within‐treatment 2‐way repeated‐measures ANOVA: P<0.0001 for time, P>0.5 for time–genotype in placebo‐treated, P=0.014 for interaction in BH4‐treated groups. P<0.05 by 1‐way repeated‐measures ANOVA in all individual groups. Post hoc multiple comparisons: # P<0.005, † P<0.04 vs baseline within same group (Dunnett test), *P<0.01, § P<0.05 for TAC plus BH4 vs TAC at each time point (Tukey test, n=6–9). B, HW/BW ratio was lower in BH4‐treated mice of either genotype; (n=6–9, # P<0.01, *P<0.05). C, BH4 treatment reduced myocardial fibrosis in both groups (n=6–9, *P<0.05 vs TAC control by unpaired t test). D, BNP, β‐MHC/α‐MHC ratio, and TSP4 mRNA expression all increased with TAC and were attenuated by BH4 treatment. Because both genotypes showed the same response, 1‐way ANOVA was performed on the combined data. P values are shown for TAC vs sham, TAC vs TAC plus BH4, and sham vs TAC plus BH4 based on the Tukey post hoc multiple comparisons test (n=6–9). BH4 indicates tetrahydrobiopterin; BNP, B‐type natriuretic peptide; BW, body weight; HW, heart weight; LV, left ventricle; mGCH1, cardiomyocyte‐specific GTP cyclohydrolase overexpression; MHC, myosin heavy chain; NOS, nitric oxide synthase; RLU, relative light units; TAC, transverse aortic constriction; TSP4, thrombospondin 4; WT, wild type.
Oral BH4 supplementation significantly elevated myocardial BH4, BH2, and total biopterin measured 3 weeks after TAC in WT mice (Figure 4A) (results for the 2 commercial BH4 sources tested were combined because they were similar). In mGCH1 mice, levels were much higher at baseline and actually declined slightly with exogenous BH4 added. Consistent with prior studies, myocardial O2 − declined in WT mice when treated with exogenous BH4 (area under the curve of 920±60 versus 757±25 for 2‐OH‐E; P<0.03; control value 773±38), principally related to a decline in NOS‐derived O2 − (Figure 4B); however, mGCH1 mice showed no increase in O2 − or change with the addition of oral BH4 (Figure 4C). These results show that exogenous BH4 therapy improves TAC‐induced cardiac remodeling and dysfunction but without myocardial NOS uncoupling occurring or being reversed in mGCH1 mice.
Figure 4.
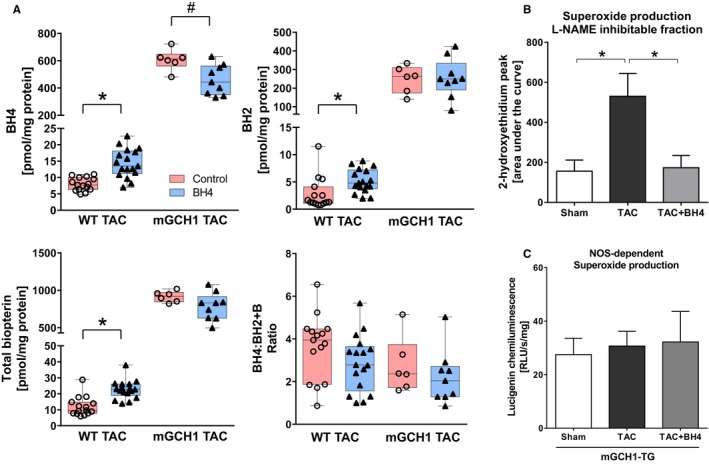
Modulation of tissue BH4 and myocardial NOS uncoupling by exogenous BH4 treatment. A, Myocardial BH4, BH2, and total biopterin significantly rose in WT mice with TAC treated with oral BH4 (*P<0.005 by Mann–Whitney test, n=15–17). In mGCH1 mice, there was a slight decline in myocardial BH4 (# P<0.05 by Mann–Whitney test, n=6–9), although it remained markedly elevated over WT‐TAC controls. The ratio of BH4/BH2 plus biopterin was unaltered with exogenous therapy. B, High‐performance liquid chromatography analysis of 2‐hydroxyethidium revealed NOS‐derived superoxide production rose in WT‐TAC mice, which was attenuated in exogenous BH4‐supplemented mice (*P<0.05, n=4–8). C, Lucigenin chemiluminescence assay showed that NOS‐derived superoxide production remained as low as sham in mGCH1‐TAC mice. Exogenous BH4 supplementation did not affect superoxide production (n=6–10). B indicates biopterin; BH2, 7,8‐dihydrobiopterin; BH4, tetrahydrobiopterin; LV, left ventricle; mGCH1, cardiomyocyte‐specific GTP cyclohydrolase overexpression; NOS, nitric oxide synthase; RLU, relative light units; TAC, transverse aortic constriction; WT, wild type.
Exogenous BH4 Blocks Inflammatory Signaling and Macrophage Infiltration From TAC
The capacity of exogenous BH4 to reverse TAC pathophysiology even in mGCH1 hearts, in which NOS uncoupling was not observed, suggests that other cellular targets or signals are involved. Given recent evidence of BH4 involvement in inflammatory cell signaling,28 we hypothesized that exogenous but not myocyte‐selective BH4 augmentation could block these pathways. Figure 5 shows the results of gene expression analysis for inflammatory cell–specific surface markers and proinflammatory cytokines. Both neutrophil chemokine receptor CXCR2 and macrophage surface antigen galectin 3 showed increased expression after TAC in both genotypes. Both were significantly reduced back to control levels by BH4 treatment. Similarly, the expression of interleukins 6 and 1β, both proinflammatory cytokines, were augmented by TAC and reduced by BH4.
Figure 5.
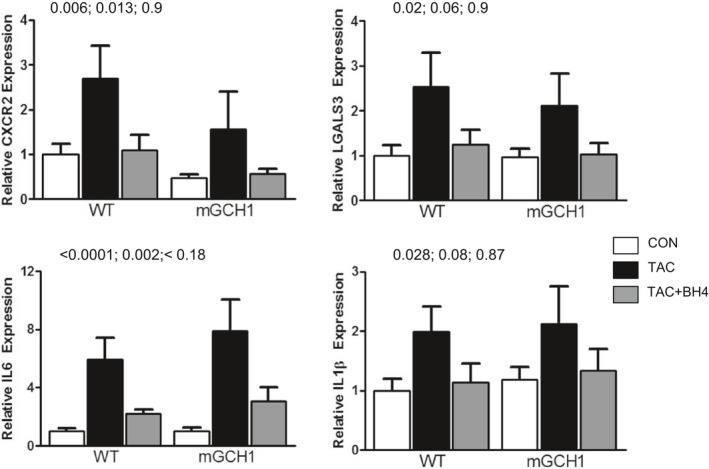
BH4 treatment downregulates mRNA expression of inflammatory marker genes CXCR2, LGALS3 (surface markers for neutrophils and macrophages, respectively), and cytokines IL‐6 and IL‐1β. P values are shown for TAC vs sham surgery, TAC vs TAC plus BH4, and sham vs TAC plus BH4, using a 1‐way ANOVA, followed by Tukey multiple comparisons test (n=6–9). BH4 indicates tetrahydrobiopterin; IL, interleukin; mGCH1, cardiomyocyte‐specific GTP cyclohydrolase overexpression; TAC, transverse aortic constriction; WT, wild type.
To determine whether inflammatory cells were indeed suppressed in hearts treated with BH4, we performed immunohistochemical staining of myocardium for macrophages. Inflammation peaks early (within 3 days) after TAC and is composed mostly of macrophages.29, 30 Exogenous BH4 supplementation also augmented myocardial BH4 at this time (in pmol/mg protein: sham 4.5 [interquartile range 3.9–6.1], TAC 7.8 [interquartile range 6.7–10.2], TAC plus BH4 15.3 [interquartile range 11.6–16.6], P<0.05 for TAC versus TAC plus BH4 by Kruskal–Wallis test). After 3 days, TAC had induced marked LV macrophage accumulation, but this was suppressed in BH4‐supplemented mice (Figure 6A and 6B). Echocardiography at this same time point showed significantly lower LV end‐systolic dimension and LV mass but improved fractional shortening and ejection fraction in BH4‐recipient mice (Figure 6C). These results show that exogenous BH4 supplementation attenuates macrophage infiltration and improves early maladaptive responses of the heart to TAC.
Figure 6.
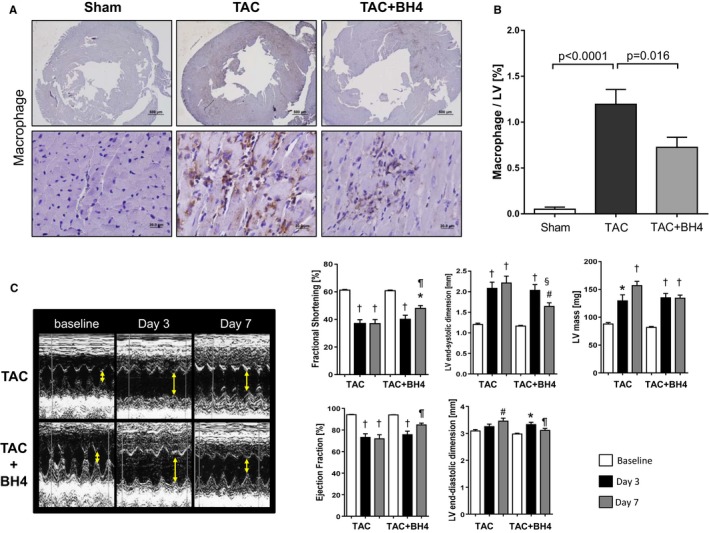
BH4 supplementation reduces early macrophage infiltration and subsequent LV dysfunction in TAC‐treated mice. A, Macrophage infiltration in hearts 3 days after TAC surgery. Sections were stained with anti–Mac‐3 antibody. Representative microphotographs (×20, ×400) are shown. B, Quantification of macrophage infiltration in the myocardium. Macrophage‐positive area per entire LV was determined in the low power fields (sham, n=10; TAC, n=20; TAC plus BH4, n=29). C, M‐mode echocardiograms show dilated LV dimension and decreased fractional shortening 3 days after TAC surgery in both groups with and without BH4 supplementation. Fractional shortening and LV dimension restored 7 days after TAC in the BH4‐treated group compared with TAC mice (n=10–20). Two‐way ANOVA: P<0.05 for time–treatment interaction for all parameters except LV mass (P=0.11); P<0.0001 for time effect for all variables, P<0.05 for treatment effect for all but LV end‐systolic dimension (P=0.06) and LV mass (P=0.17). Post hoc comparisons (Tukey test): # P<0.05, *P<0.01, † P<0.001 vs baseline. ¶ P<0.05, § P<0.01 for BH4 vs TAC at each time point. BH4 indicates tetrahydrobiopterin; LV, left ventricle; TAC, transverse aortic constriction.
Discussion
This study addresses a critical question for understanding the benefits of exogenous BH4 supplementation in hypertrophic heart disease by finding that the effects of BH4 are not mediated by myocardial NOS recoupling. This result is based on 2 primary observations. First, despite the biochemical rescue of NOS uncoupling in mGCH1 mice that underwent TAC, hearts responded with similar dysfunction, fibrosis, and induction of pathological gene programs as in WT mice, in which NOS uncoupling was induced. Second, exogenous BH4 supplementation improved the TAC response in both WT and mGCH1 mice despite having no further impact on NOS coupling in mGCH1 mice. The WT response replicated prior data by showing concomitant recoupling of NOS with improved pathophysiology with oral BH4. These observations indicate that BH4 exerts effects beyond reducing myocardial NOS‐derived superoxide production and suggest a new mechanism for BH4 in limiting the response to LV pressure overload.
The failure of myocyte‐specific BH4 augmentation to counter TAC pathobiology is similar to previous observations in mice in which GCH1 was overexpressed in cells expressing the Tie‐2 promoter (mostly endothelial), in which endothelial GCH1 overexpression provided no protection. It is conceivable that both cell sources are required, although this would mean that each compartment could fully offset any pathophysiology benefits from higher BH4 synthesis in the other, which seems unlikely. Both models are consistent with data showing that BH4 is not exported outside its cell of origin18, 19 but rather functions as an intracellular modulator. Consequently, evidence of NOS uncoupling in tissue with mixed cell types that each express NOS may not reveal the relevant pool and/or target of externally supplemented BH4.
Another potential explanation for failure of the mGCH1 model is that high levels of BH4 in myocytes were oxidized to BH2, which would counter benefits against TAC‐induced pathobiology. Earlier work had shown that although lower doses of exogenous BH4 were protective against TAC, higher doses were not,31 and this correlated to a decline in ratios of BH4/BH2 plus biopterin at the higher doses. With TAC, there was a somewhat lower ratio of BH4/BH2 plus biopterin in the mGCH1 transgenic mice versus WT in the present study. Despite this, the phenotype was almost the same after TAC and certainly not worse in the mGCH1 overexpressors. Furthermore, NOS‐derived ROS was reduced in the TAC mGCH1 myocardium. Importantly, adding even more exogenous BH4 did not significantly affect this ratio, BH2 and biopterin levels were still high in mGCH1 transgenic mice with TAC, yet TAC‐induced LV dysfunction, hypertrophy, and fibrosis were attenuated. Consequently, failure of mGCH1 overexpression to protect against TAC cannot be attributed to an inadequate ratio of BH4/BH2 plus biopterin.
If neither myocyte nor endothelial cell BH4 and NOS recoupling can explain the capacity of exogenous BH4 to ameliorate TAC pathobiology, what else could be involved? Based on several recent studies linking BH4 and NOS uncoupling with inflammation,32 we tested whether this might be ameliorated only by exogenous BH4 treatment. The results confirmed this, showing that oral BH4 suppressed myocardial macrophage accumulation early after TAC, when inflammatory infiltration is most prominent,29 and suppressed multiple markers of inflammation even after several weeks. Early macrophage infiltration after pressure overload is an important contributor to subsequent maladaptive LV remodeling because its suppression is protective against pressure overload coupled to transforming growth factor β30 and phosphoinositide 3 kinase γ–dependent mechanisms.29
Inflammation reflected by circulating cytokines such as interleukin 6 and C‐reactive protein augments biopterin synthesis by stimulating GCH1, and this is thought to be protective because humans with a genetic haploinsuficiency for this gene develop worse cytokine‐induced endothelial dysfunction.32 Kossmann et al33 later showed that angiotensin II stimulated NOS uncoupling and that nitro‐oxidative stress is linked to inflammatory monocytes. Using a model of diphtheria toxin‐driven lysosome M‐specific cell linage ablation, they showed that loss of these cells ameliorated defective NOS function and enhanced BH4 synthesis. Interestingly, this was also achieved by gene or pharmacological inhibition of inducible NOS (NOS2), indicating circular feedback between inflammatory cells and NOS‐derived superoxide. Macrophages are among lysozyme M‐positive cells,34 so this finding is compatible with our results. Macrophages that lack GCH1 develop more ROS and less nitrite when stimulated with cytokines, and both are rescued by BH4 supplementation.28 This may work in part through the activation of nuclear factor erythroid 2–related factor 2 (Nrf2), a key transcription factor regulating antioxidant genes, because Nrf2‐activated gene expression is suppressed in GCH1‐deficient macrophages and antioxidant transcriptional factor Nrf2 is known to inhibit inflammatory processes.35, 36, 37 Prior studies have shown that exogenous BH4 reduces myocardial oxidant stress in the same pressure‐overload model12, 13; however, although we previously presumed this was due to reversal of NOS coupling, the present data indicate that this may happen even if NOS is already coupled.
It is worth considering whether exogenous BH4 may have affected other proteins that require it as a cofactor. BH4 modulates alkyl glycerol mono‐oxygenase, but to date there is no evidence about its involvement in cardiovascular disease. BH4 regulation of aromatic amino acid hydroxylases (phenylalanine, tyrosine, and tryptophan) is not known to modify cardiac stress remodeling. If anything, enhancing tyrosine hydroxylase might stimulate sympathetic signaling, which would not be expected to suppress hypertrophy. Tryptophan hydroxylase catalyzes synthesis of 5‐hydroxytryptophan, a precursor of serotonin. Blocking, not stimulating, serotonin receptors can counter cardiac hypertrophy,38 so this too is an unlikely explanation.
Our study has several limitations. We cannot determine the cell‐specific role of NOS uncoupling in a complex tissue like myocardium, so proof of exactly which cells are targeted by exogenous BH4 remains lacking. In addition, inflammatory cells can generate superoxide by multiple NOS‐ and BH4‐independent pathways, and BH4 has potent antioxidant properties itself. We have not yet developed a macrophage‐targeted GCH1 overexpression model; therefore, whether this mimics the effects of exogenous BH4 remains unclear. Depleting macrophages could test whether these cells are required for BH4 efficacy, although this alone countered pressure‐overload maladaptation.39 Generating a chimera model lacking GCH1 in macrophages by bone marrow transplantation from Tie2‐driven GCH1 knockout mice might detect intrinsic macrophage–BH4 interaction but would not necessarily clarify exogenous BH4 effects. Still, our study directly confronts the generally held notion that supplemental BH4 improves the stressed heart via NOS recoupling. Although this still may have occurred, it would have to be in cells, in which this change is generally invisible to tissue‐level analysis of NOS function and superoxide generation.
We used the lucigenin chemiluminescence assay for O2 − measurement, although lucigenin can itself uncouple NOS; however, the relative increase in NOS‐derived superoxide after TAC, as measured by this technique, was very similar to 2‐OH‐E detection by HPLC (Figures 2B and 4B) and does not suggest this as a potential source of error. Consequently, we think that the lucigenin concentrations used did not themselves stimulate ROS. Last, we tested 2 different BH4 sources, both were pure (6R)‐stereoisomer formulations and neither of which contained inactive ingredients for drug stabilization; because both raised myocardial BH4 levels similarly, we think they were essentially interchangeable.
In conclusion, the present findings show that NOS‐derived ROS measured at the cardiac tissue level is not a predictor of pressure overload–induced myopathic remodeling and that its absence does not preclude benefits from exogenously applied BH4. Although myocyte dysfunction is a prominent feature of the disease, high levels of BH4 expression in these cells are insufficient to ameliorate it, whereas exogenously provided BH4 does. If these data translate to humans, then the presence of inflammatory cascades may be a better predictor than NOS‐coupling status in assessing the efficacy of exogenous BH4 therapy.
Sources of Funding
This work was supported by the Fondation Leducq Transatlantic Network of Excellence, National Institutes of Health RO1HL119012, HL‐07227, and the British Heart Foundation (RG/11/15/29375).
Disclosures
None.
Supporting information
Figure S1. Echocardiographic measurements of chamber (A and B) end‐diastolic and end‐systolic dimensions and calculated total ejection fraction (C) at each time point in WT and mGHC1 mice in both sham‐ and TAC‐operated groups. Volumes increased and ejection fraction declined similarly in both groups with TAC. Within‐group 1‐way ANOVA post hoc comparison (Tukey) results: *P<0.005 vs baseline; † P<0.0001 vs baseline. The responses in WT and mGCH1 groups were not significantly different. D, Heart weight/tibia length ratio for both WT and mGCH1 groups in sham and TAC groups. *P<0.001 vs sham. These data analyzed by Kruskal–Wallis test due their nonnormal distribution. mGCH1 indicates mouse GTP cyclohydrolase 1; TAC, transverse aortic constriction; WT, wild type.
Figure S2. Echocardiographic measurements of chamber end‐systolic volume and calculated ejection fraction at each time point in wild‐type mice and mice with cardiomyocyte‐specific overexpression of GTP cyclohydrolase 1 in TAC groups administered control soft diet or diet supplemented with BH4. BH4 treatment suppressed chamber remodeling (less rise in end‐systolic dimension) and increased ejection fraction. Within‐group 1‐way ANOVA post hoc comparison (Tukey) results: *P<0.005 vs baseline; † P<0.05 vs baseline, # P=0.06 vs baseline. § P<0.05, ¶ P<0.01 vs control within same genotype in the same week (n=6–9).
(J Am Heart Assoc. 2016;5:e003208 doi: 10.1161/JAHA.116.003208)
Accompanying Figures S1 and S2 are available at http://jaha.ahajournals.org/content/5/3/e003208/suppl/DC1
References
- 1. Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Judd SE, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Mackey RH, Magid DJ, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER III, Moy CS, Mussolino ME, Neumar RW, Nichol G, Pandey DK, Paynter NP, Reeves MJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Wong ND, Woo D, Turner MB; American Heart Association Statistics C and Stroke Statistics S . Heart disease and stroke statistics–2014 update: a report from the American Heart Association. Circulation. 2014;129:e28–e292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sag CM, Santos CX, Shah AM. Redox regulation of cardiac hypertrophy. J Mol Cell Cardiol. 2014;73:103–111. [DOI] [PubMed] [Google Scholar]
- 3. Karbach S, Wenzel P, Waisman A, Munzel T, Daiber A. eNOS uncoupling in cardiovascular diseases—the role of oxidative stress and inflammation. Curr Pharm Des. 2014;20:3579–3594. [DOI] [PubMed] [Google Scholar]
- 4. Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MA, Chen YR, Druhan LJ, Zweier JL. S‐glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature. 2010;468:1115–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sharma S, Smith A, Kumar S, Aggarwal S, Rehmani I, Snead C, Harmon C, Fineman J, Fulton D, Catravas JD, Black SM. Mechanisms of nitric oxide synthase uncoupling in endotoxin‐induced acute lung injury: role of asymmetric dimethylarginine. Vascul Pharmacol. 2010;52:182–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vasquez‐Vivar J, Martasek P, Whitsett J, Joseph J, Kalyanaraman B. The ratio between tetrahydrobiopterin and oxidized tetrahydrobiopterin analogues controls superoxide release from endothelial nitric oxide synthase: an EPR spin trapping study. Biochem J. 2002;362:733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Levy HL, Milanowski A, Chakrapani A, Cleary M, Lee P, Trefz FK, Whitley CB, Feillet F, Feigenbaum AS, Bebchuk JD, Christ‐Schmidt H, Dorenbaum A. Efficacy of sapropterin dihydrochloride (tetrahydrobiopterin, 6R‐BH4) for reduction of phenylalanine concentration in patients with phenylketonuria: a phase III randomised placebo‐controlled study. Lancet. 2007;370:504–510. [DOI] [PubMed] [Google Scholar]
- 8. Crabtree MJ, Channon KM. Synthesis and recycling of tetrahydrobiopterin in endothelial function and vascular disease. Nitric Oxide. 2011;25:81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bec N, Gorren AC, Voelker C, Mayer B, Lange R. Reaction of neuronal nitric‐oxide synthase with oxygen at low temperature. Evidence for reductive activation of the oxy‐ferrous complex by tetrahydrobiopterin. J Biol Chem. 1998;273:13502–13508. [DOI] [PubMed] [Google Scholar]
- 10. Liu Q, Gross SS. Binding sites of nitric oxide synthases. Methods Enzymol. 1996;268:311–324. [DOI] [PubMed] [Google Scholar]
- 11. Klatt P, Schmidt K, Lehner D, Glatter O, Bachinger HP, Mayer B. Structural analysis of porcine brain nitric oxide synthase reveals a role for tetrahydrobiopterin and L‐arginine in the formation of an SDS‐resistant dimer. EMBO J. 1995;14:3687–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B, Lazzarino G, Paolocci N, Gabrielson KL, Wang Y, Kass DA. Oxidant stress from nitric oxide synthase‐3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest. 2005;115:1221–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moens AL, Takimoto E, Tocchetti CG, Chakir K, Bedja D, Cormaci G, Ketner EA, Majmudar M, Gabrielson K, Halushka MK, Mitchell JB, Biswal S, Channon KM, Wolin MS, Alp NJ, Paolocci N, Champion HC, Kass DA. Reversal of cardiac hypertrophy and fibrosis from pressure overload by tetrahydrobiopterin: efficacy of recoupling nitric oxide synthase as a therapeutic strategy. Circulation. 2008;117:2626–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Silberman GA, Fan TH, Liu H, Jiao Z, Xiao HD, Lovelock JD, Boulden BM, Widder J, Fredd S, Bernstein KE, Wolska BM, Dikalov S, Harrison DG, Dudley SC Jr. Uncoupled cardiac nitric oxide synthase mediates diastolic dysfunction. Circulation. 2010;121:519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bendall JK, Douglas G, McNeill E, Channon KM, Crabtree MJ. Tetrahydrobiopterin in cardiovascular health and disease. Antioxid Redox Signal. 2014;20:3040–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hasegawa H, Sawabe K, Nakanishi N, Wakasugi OK. Delivery of exogenous tetrahydrobiopterin (BH4) to cells of target organs: role of salvage pathway and uptake of its precursor in effective elevation of tissue BH4. Mol Genet Metab. 2005;86(suppl 1):S2–S10. [DOI] [PubMed] [Google Scholar]
- 17. Cunnington C, Van Assche T, Shirodaria C, Kylintireas I, Lindsay AC, Lee JM, Antoniades C, Margaritis M, Lee R, Cerrato R, Crabtree MJ, Francis JM, Sayeed R, Ratnatunga C, Pillai R, Choudhury RP, Neubauer S, Channon KM. Systemic and vascular oxidation limits the efficacy of oral tetrahydrobiopterin treatment in patients with coronary artery disease. Circulation. 2012;125:1356–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alp NJ, Mussa S, Khoo J, Cai S, Guzik T, Jefferson A, Goh N, Rockett KA, Channon KM. Tetrahydrobiopterin‐dependent preservation of nitric oxide‐mediated endothelial function in diabetes by targeted transgenic GTP‐cyclohydrolase I overexpression. J Clin Invest. 2003;112:725–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carnicer R, Hale AB, Suffredini S, Liu X, Reilly S, Zhang MH, Surdo NC, Bendall JK, Crabtree MJ, Lim GB, Alp NJ, Channon KM, Casadei B. Cardiomyocyte GTP cyclohydrolase 1 and tetrahydrobiopterin increase NOS1 activity and accelerate myocardial relaxation. Circ Res. 2012;111:718–727. [DOI] [PubMed] [Google Scholar]
- 20. Takaya T, Hirata K, Yamashita T, Shinohara M, Sasaki N, Inoue N, Yada T, Goto M, Fukatsu A, Hayashi T, Alp NJ, Channon KM, Yokoyama M, Kawashima S. A specific role for eNOS‐derived reactive oxygen species in atherosclerosis progression. Arterioscler Thromb Vasc Biol. 2007;27:1632–1637. [DOI] [PubMed] [Google Scholar]
- 21. Sears CE, Bryant SM, Ashley EA, Lygate CA, Rakovic S, Wallis HL, Neubauer S, Terrar DA, Casadei B. Cardiac neuronal nitric oxide synthase isoform regulates myocardial contraction and calcium handling. Circ Res. 2003;92:e52–e59. [DOI] [PubMed] [Google Scholar]
- 22. Niu X, Watts VL, Cingolani OH, Sivakumaran V, Leyton‐Mange JS, Ellis CL, Miller KL, Vandegaer K, Bedja D, Gabrielson KL, Paolocci N, Kass DA, Barouch LA. Cardioprotective effect of beta‐3 adrenergic receptor agonism: role of neuronal nitric oxide synthase. J Am Coll Cardiol. 2012;59:1979–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim YM, Guzik TJ, Zhang YH, Zhang MH, Kattach H, Ratnatunga C, Pillai R, Channon KM, Casadei B. A myocardial Nox2 containing NAD(P)H oxidase contributes to oxidative stress in human atrial fibrillation. Circ Res. 2005;97:629–636. [DOI] [PubMed] [Google Scholar]
- 24. Rainer PP, Hao S, Vanhoutte D, Lee DI, Koitabashi N, Molkentin JD, Kass DA. Cardiomyocyte‐specific transforming growth factor beta suppression blocks neutrophil infiltration, augments multiple cytoprotective cascades, and reduces early mortality after myocardial infarction. Circ Res. 2014;114:1246–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Frolova EG, Sopko N, Blech L, Popović ZB, Li J, Vasanji A, Drumm C, Krukovets I, Jain MK, Penn MS, Plow EF, Stenina OI. Thrombospondin‐4 regulates fibrosis and remodeling of the myocardium in response to pressure overload. FASEB J. 2012;26:2363–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mustonen E, Aro J, Puhakka J, Ilves M, Soini Y, Leskinen H, Ruskoaho H, Rysa J. Thrombospondin‐4 expression is rapidly upregulated by cardiac overload. Biochem Biophys Res Commun. 2008;373:186–191. [DOI] [PubMed] [Google Scholar]
- 27. Narouz‐Ott L, Maurer P, Nitsche DP, Smyth N, Paulsson M. Thrombospondin‐4 binds specifically to both collagenous and non‐collagenous extracellular matrix proteins via its C‐terminal domains. J Biol Chem. 2000;275:37110–37117. [DOI] [PubMed] [Google Scholar]
- 28. McNeill E, Crabtree MJ, Sahgal N, Patel JR, Chuaiphichai S, Iqbal AJ, Hale AB, Greaves DR, Channon KM. Regulation of iNOS function and cellular redox state by macrophage Gch1 reveals specific requirements for tetrahydrobiopterin in NRF2 activation. Free Radic Biol Med. 2015;79:206–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Damilano F, Franco I, Perrino C, Schaefer K, Azzolino O, Carnevale D, Cifelli G, Carullo P, Ragona R, Ghigo A, Perino A, Lembo G, Hirsch E. Distinct effects of leukocyte and cardiac phosphoinositide 3‐kinase gamma activity in pressure overload‐induced cardiac failure. Circulation. 2011;123:391–399. [DOI] [PubMed] [Google Scholar]
- 30. Kuwahara F, Kai H, Tokuda K, Takeya M, Takeshita A, Egashira K, Imaizumi T. Hypertensive myocardial fibrosis and diastolic dysfunction: another model of inflammation? Hypertension. 2004;43:739–745. [DOI] [PubMed] [Google Scholar]
- 31. Moens AL, Ketner EA, Takimoto E, Schmidt TS, O'Neill CA, Wolin MS, Alp NJ, Channon KM, Kass DA. Bi‐modal dose‐dependent cardiac response to tetrahydrobiopterin in pressure‐overload induced hypertrophy and heart failure. J Mol Cell Cardiol. 2011;51:564–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Antoniades C, Cunnington C, Antonopoulos A, Neville M, Margaritis M, Demosthenous M, Bendall J, Hale A, Cerrato R, Tousoulis D, Bakogiannis C, Marinou K, Toutouza M, Vlachopoulos C, Leeson P, Stefanadis C, Karpe F, Channon KM. Induction of vascular GTP‐cyclohydrolase I and endogenous tetrahydrobiopterin synthesis protect against inflammation‐induced endothelial dysfunction in human atherosclerosis. Circulation. 2011;124:1860–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kossmann S, Hu H, Steven S, Schonfelder T, Fraccarollo D, Mikhed Y, Brahler M, Knorr M, Brandt M, Karbach SH, Becker C, Oelze M, Bauersachs J, Widder J, Munzel T, Daiber A, Wenzel P. Inflammatory monocytes determine endothelial nitric‐oxide synthase uncoupling and nitro‐oxidative stress induced by angiotensin II. J Biol Chem. 2014;289:27540–27550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Goren I, Allmann N, Yogev N, Schurmann C, Linke A, Holdener M, Waisman A, Pfeilschifter J, Frank S. A transgenic mouse model of inducible macrophage depletion: effects of diphtheria toxin‐driven lysozyme M‐specific cell lineage ablation on wound inflammatory, angiogenic, and contractive processes. Am J Pathol. 2009;175:132–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brune B, Dehne N, Grossmann N, Jung M, Namgaladze D, Schmid T, von Knethen A, Weigert A. Redox control of inflammation in macrophages. Antioxid Redox Signal. 2013;19:595–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kong X, Thimmulappa R, Craciun F, Harvey C, Singh A, Kombairaju P, Reddy SP, Remick D, Biswal S. Enhancing Nrf2 pathway by disruption of Keap1 in myeloid leukocytes protects against sepsis. Am J Respir Crit Care Med. 2011;184:928–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kong X, Thimmulappa R, Kombairaju P, Biswal S. NADPH oxidase‐dependent reactive oxygen species mediate amplified TLR4 signaling and sepsis‐induced mortality in Nrf2‐deficient mice. J Immunol. 2010;185:569–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ayme‐Dietrich E, Marzak H, Lawson R, Mokni W, Wendling O, Combe R, Becker J, El Fertak L, Champy MF, Matz R, Andriantsitohaina R, Doly S, Boutourlinsky K, Maroteaux L, Monassier L. Contribution of serotonin to cardiac remodeling associated with hypertensive diastolic ventricular dysfunction in rats. J Hypertens. 2015;33:2310–2321. [DOI] [PubMed] [Google Scholar]
- 39. Kain D, Amit U, Yagil C, Landa N, Naftali‐Shani N, Molotski N, Aviv V, Feinberg MS, Goitein O, Kushnir T, Konen E, Epstein FH, Yagil Y, Leor J. Macrophages dictate the progression and manifestation of hypertensive heart disease. Int J Cardiol. 2016;203:381–395. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Echocardiographic measurements of chamber (A and B) end‐diastolic and end‐systolic dimensions and calculated total ejection fraction (C) at each time point in WT and mGHC1 mice in both sham‐ and TAC‐operated groups. Volumes increased and ejection fraction declined similarly in both groups with TAC. Within‐group 1‐way ANOVA post hoc comparison (Tukey) results: *P<0.005 vs baseline; † P<0.0001 vs baseline. The responses in WT and mGCH1 groups were not significantly different. D, Heart weight/tibia length ratio for both WT and mGCH1 groups in sham and TAC groups. *P<0.001 vs sham. These data analyzed by Kruskal–Wallis test due their nonnormal distribution. mGCH1 indicates mouse GTP cyclohydrolase 1; TAC, transverse aortic constriction; WT, wild type.
Figure S2. Echocardiographic measurements of chamber end‐systolic volume and calculated ejection fraction at each time point in wild‐type mice and mice with cardiomyocyte‐specific overexpression of GTP cyclohydrolase 1 in TAC groups administered control soft diet or diet supplemented with BH4. BH4 treatment suppressed chamber remodeling (less rise in end‐systolic dimension) and increased ejection fraction. Within‐group 1‐way ANOVA post hoc comparison (Tukey) results: *P<0.005 vs baseline; † P<0.05 vs baseline, # P=0.06 vs baseline. § P<0.05, ¶ P<0.01 vs control within same genotype in the same week (n=6–9).