

Abstract
Jak2 is a non-receptor tyrosine kinase that is involved in the control of cellular growth and proliferation. Due to its significant role in hematopoiesis, Jak2 is a frequent target for mutations in cancer, especially myeloid leukemia, lymphoid leukemia and the myeloproliferative neoplasms (MPN). These mutations are common amongst different populations all over the world and there is a great deal of effort to develop therapeutic drugs for the affected patients. Jak2 mutations, whether they are point, deletion, or gene fusion, most commonly result in constitutive kinase activation. Here, we explore the structure-function relation of various Jak2 mutations identified in cancer and understand how they disrupt Jak2 regulation. Current Jak2 inhibitors target the highly conserved active site in the kinase domain and therefore, these inhibitors may lack specificity. Based on our knowledge regarding structure-function correlations as they pertain to regulation of Jak2 kinase activity, an alternative approach for specific Jak2 targeting could be via allosteric inhibitor design. Successful reports of allosteric inhibitors developed against other kinases provide precedent for the development of Jak2 allosteric inhibitors. Here, we suggest plausible target sites in the Jak2 structure for allosteric inhibition. Such targets include the type II inhibitor pocket and substrate binding site in the kinase domain, the kinase-pseudokinase domain interface, SH2-JH2 linker region and the FERM domain. Thus, future Jak2 inhibitors that target these sites via allosteric mechanisms may provide alternative therapeutic strategies to existing ATP competitive inhibitors.
Keywords: allosteric inhibitors, Jak2 tyrosine kinase, mutations, MPN, structure-function
Introduction
Janus kinases (Jaks) are non-receptor tyrosine kinases, which play an important role in cytokine receptor signaling. The Jak family consists of four members; Jak1, Jak2, Jak3 and Tyk2. Jak1, Jak2 and Tyk2 are expressed ubiquitously, but Jak3 expression is restricted to myeloid and lymphoid tissues. Different cytokines activate different subsets of Jaks. One of the downstream substrates of the Jaks are the Signal Transducers and Activators of Transcription (STATs) and Jak-STAT signaling has been implied in the regulation of cellular growth and proliferation.
Jak-STAT signaling is highly regulated and any change in this controlled process can affect normal physiology. For example, Jak1 knockout mice die perinatally due to defects in signaling through a subset of cytokine receptors [1]. Jak2 has a non-redundant role in erythropoiesis, as the Jak2 knockout mice die embryonically at day 12.5 due to lack of definitive erythropoiesis [2, 3]. Jak3 knockout mice are viable, but have defects in lymphoid development and also present with Severe Combined ImmunoDeficiency (SCID) [4, 5, 6]. Mutations in Jak3 have also been seen in patients with autosomal SCID [7, 8]. Tyk2 knockout mice are viable, but exhibit defects in interferon and IL-12 signaling [9, 10]. Inhibition of Jak mediated signal transduction has been observed in diseases associated with the Human Papilloma Virus (HPV), Human cytomegalovirus (HCMV), Leishmania donovani and Ehrlichia chaffeensis. Jak inhibition has also been associated with tumor-related immunosuppression caused by the reduction of T-cell proliferation [11].
On the contrary, there has been mounting evidence for the occurrence of Jak mutations that cause constitutive activation in cancer. Specifically, Jak2 mutations have been identified in Acute Lymphoid Leukemia (ALL), Chronic Myelogenous Leukemia (CML), Myeloproliferative Neoplasia (MPN), lymphomas and myelomas. Constitutively active Jak2 has also been implicated in solid cancers of the breast, prostrate, head, and neck. While there are a wide variety of Jak2 mutations present in these disorders, it is important to understand the mechanism by which they lead to constitutive activation of the protein. As such, knowledge of the structure-function correlation for the various mutations will be useful in improvising the drug design strategies for better patient outcomes.
Current Jak2 inhibitors target the active site of the kinase domain and most of them are ATP-competitive. A major drawback in this design strategy has been in achieving specificity for Jak2 among the different Jak family members and also specificity for mutant Jak2 over the native form. However, such selectivity can be achieved by using allosteric inhibitors that are not ATP-competitive and bind to sites remote to the active site of the kinase. This approach has been successful in targeting kinases such as BCR-ABL, B-Raf, MEK, and Akt [12]. The focus of this review will be to analyze the structure-function aspects of the different Jak2 mutations and their implication in the relatively unexplored area of allosteric drug design for Jak2.
Jak2 Regulation
The protein structures of all four Jak family members share seven conserved Jak Homology (JH) domains encoding four major domains (Table 1). The C-terminal JH1 domain is a highly conserved kinase domain which contains the activation loop, primary phosphorylation sites (Y1007 and Y1008 in Jak2), and the ATP binding site (K882 in Jak2). JH2 is the pseudo kinase domain, which has similar structural properties to the kinase domain, but lacks catalytic activity. The pseudokinase domain is a unique structural feature of the Jak family members. Adjacent to the JH2 domain is the SH2 (Src Homology 2)-like domain, whose function is not yet fully understood. At the N-terminus of the protein is the FERM domain (F for 4.1 protein, E for ezrin, R for radixin and M for moesin), which facilitates interactions between Jak2 and a number of cell surface receptors [13].
TABLE 1.
Jak2 Mutations that occur in various hematalogical disorders
| EXON LOCATION (Amino acids encoded) | MUTATION | DISEASE | REFERENCE |
|---|---|---|---|
| Kinase domain (849-1123) | |||
| Exon 20 (858–920) | R867Q | Ch-B-ALL | [110] |
| D873N | Ch-B-ALL | [110] | |
| T875N | AMKL cell line | [71] | |
| Exon 21 (921 – 962) | P933R | Ch-B-ALL | [110] |
| Pseudokinase domain (545-806) | |||
| Exon 16 (665 to 710) | I682F | [110] | |
| R683G | DS-B-ALL | [110,111, 112, 113] | |
| R683S | DS-B-ALL | [110,111, 112, 113] | |
| RQIns683 | [114] | ||
| I682-D686 (IREED) del | DS-B-ALL | [51] | |
| Exon 15 (623 to 664) | L624P | MPNs | [115] |
| E627E | MPNs | [51] | |
| I645V | MPNs | [51] | |
| Exon 14 (593–622) | H606Q | MPNs | [116] |
| K607N | AML | [51] | |
| H608Y | MPNs | [117] | |
| L611S | Ch-B-ALL | [20–24, 116, 118, 119–122] | |
| V617F | MPNs, CMML, JMML, RARS, AML, MDS | [51] | |
| V617I | MPNs | [115, 123] | |
| D620E+V617F | MPNs | [124] | |
| C616Y+V617F | MPNs | [51, 125] | |
| C618R+V617F | MPNs | [126] | |
| Exon 14 del | MPNs | [51] | |
| Exon 13 (548–592) | F557L | MPNs | [51] |
| V567A | MPNs | [51] | |
| L579F | MPNs | [51] | |
| H587N | MPNs | [51] | |
| S591L | MPNs | [51] | |
| R564L, R564Q | MPNs | [51] | |
| G571S, G571R | MPNs | [51] | |
| SH2-JH2 Linker (501-544) | |||
| Exon 12 (501–547) | T514M | MPNs | [51] |
| N533Y | MPNs | [51] | |
| F537I | MPNs | [127] | |
| K539L | MPNs | [47] | |
| L545V | MPNs | [51] | |
| F547V | MPNs | [128] | |
| F547L | MPNs | [51] | |
| H538QK539L | MPNs | [47, 128, 129] | |
| H538-K539del | MPNs | [51, 129] | |
| E543del | MPNs | [128] | |
| N542-E543del | MPNs | [47, 128, 129] | |
| E543-D544del | MPNs | [128, 129, 130] | |
| D544-L545del | MPNs | [129] | |
| F537-K539delinsL | MPNs | [47, 128, 129] | |
| H538-K539delinsL | MPNs | [128, 131] | |
| R541-E543delinsK | MPNs | [131, 132] | |
| F537-I546dup10+F547L | MPNs | [49, 128] | |
| 547insLI540-F547 | MPNs | [128] | |
| I540-E543delinsMK | MPNs | [128, 132] | |
| H538DK539LI540S | MPNs | [128, 129] | |
| I540-N542delinsS | MPNs | [128] | |
| F537-F547dup | MPNs | [128] | |
| V536-F547dup | MPNs | [128, 129] | |
| V536-I546dup11 | MPNs | [49, 128] | |
| I540-D544delinsMK | MPNs | [51] | |
| N542-D544delinsN | MPNs | [51] | |
| FERM domain (37-380) | |||
| Exon 8 (313–352) | R340Q | MPNs | [103] |
Abbreviations used: Ch-B-ALL - childhood B-cell precursor acute lymphoblastic leukemia, DS- down syndrome, AML - acute myeloid leukemia, AMKL - acute megakaryoblastic leukemia, CMML - chronic myelomonocytic leukemia, JMML - juvenile myelomonocytic leukemia, MPNs - myeloproliferative neoplasms, MDS - myelodysplastic syndrome, RARS - refractory anemia with ringed sideroblasts.
When there is no ligand bound to the receptor, Jak2 remains associated with the receptor in an inactive state. Once ligand binds, Jak2 is activated and it phosphorylates tyrosine residues on the intra-cellular tail of the receptor. Downstream effectors such as the STATs bind to the phosphorylated receptors and are subsequently phosphorylated by Jak2. Phosphorylated STATs dimerize and translocate into the nucleus where they activate the transcription of effector genes resulting in cell proliferation and differentiation [14]. While this paradigm of Jak2 activation is widely accepted, one key limitation of this model is the paucity of structural data in support of it.
Strict regulatory control of Jak2 signaling is important under two circumstances. First, in the absence of ligand, Jak2 kinase activity must be maintained at a basal level, which is characterized by an absence of the activation loop phosphorylation and low phospho-transferase activity. Second, following activation, the Jak2-mediated signaling processes must be stopped via appropriate spatial and temporal negative feedback mechanisms. Collectively, these two levels of control are achieved through both cis and trans mechanisms. At the cis level, the regulation is achieved by the allosteric interaction between various Jak domains and by the phosphorylation/dephosphorylation of some of the 49 different tyrosine residues that are distributed throughout the Jak2 protein. Phosphorylation of Y1007 in the activation loop is the initiating and also an essential event for Jak2 activation [15]. Another level of cis regulation is achieved by the autoinhibition of the pseudokinase domain (JH2) over the kinase domain (JH1). As such, the JH2 domain suppresses the basal kinase activity of Jak2 in the absence of cytokine stimulation. The JH2 domain inhibits the kinase activity non-competitively by decreasing the maximum velocity (Vmax) of enzyme catalysis without changing its substrate affinity (Km) [16]. Ligand binding to the receptor causes conformational changes in the receptor/Jak2 complex, which relieves the autoinhibition and allows for subsequent Jak2 activation.
Post Jak2 activation, trans regulation occurs via negative feedback loops. The Jak-STAT signaling pathway stimulates the expression of proteins involved in the negative feedback regulation, thus terminating the proliferative signals induced by the ligand. Suppressor of Cytokine Signaling (SOCS) is a major regulator in this feedback loop. SOCS proteins that are expressed in response to Jak-STAT signaling, bind directly to active Jak2 via the SH2 domain and inhibit it. Alternately, SOCS binding also facilitates UE3 ligase mediated proteasomal degradation of Jak2. Concurrent with the role of SOCS in Jak2 negative regulation, mutations in the SOCS1 gene have been identified in the classical Hodgkin Lymphoma (cHL). Other trans regulators include phosphatases such as SHP1 and SHP2. They inactivate Jak2 through the dephosphorylation of Tyr 1007. Additionally, Lnk, an SH2 (B3) adaptor protein, was identified as an important negative regulator of Jak2 in hematopoietic cells [17]. Adipocyte fatty acid binding protein (AFABP/aP2), which serves as a fatty acid sensor for Jak2, was also recently identified as another negative regulator of Jak2 [18]. According to this report, when fatty acid levels are high in the cell as in the case of obesity, the AFABP/aP2 binds to and attenuates Jak2 kinase activity.
Jak2 mutations in Myeloproliferative Neoplasms (MPNs)
Deregulation of Jak2 kinase activity is a common event in various types of cancer, especially in hematological malignancies such as the BCR-ABL negative myeloproliferative neoplasms (MPNs). These are a class of stem cell derived hematological disorders include Polycythemia Vera (PV), Essential Thrombocythemia (ET) and Primary Myelofibrosis (PMF). They are clinically characterized by the presence of increased red blood cell, platelet and granulocyte counts along with bone marrow fibrosis, respectively [19]. MPN patients also bear a risk of leukemic transformation in the long term. William Dameshek first identified MPNs in 1951, but the molecular mechanism for the dysfunctional hematopoiesis in these patients remained unknown for over 50 years. In 2005, several research groups independently reported a Jak2 V to F substitution at amino acid 617 in a large percentage of MPN patients [20, 21, 22, 23, 24]. The V617F mutation occurs somatically in hematopoietic stem cells and leads to cytokine independent, constitutive Jak-STAT signaling. Following the identification of Jak2-V617F in MPN, there have been several reports of its occurrence in other hematological disorders including leukemia, lymphoma and myeloma (Table 1). Apart from the V617F mutation that occurs in exon 14, Jak2 mutations have also been found in exons 12 - 15 in patients presenting with various types of hematological disorders. These mutations deregulate the Jak-STAT signaling pathway and skew the proliferation and differentiation of hematopoietic stem cells during hematopoiesis. However, the mechanism(s) by which these different Jak2 mutations confer constitutive activation and understanding how any differences can be exploited for drug development have not been examined.
The Jak2-V617F Era
The identification of the Jak2-V617F mutation in 2005 stimulated research in various directions leading to approximately 700–800 publications solely regarding Jak2-V617F over the past 6 years. This mutation is found in at least 95% of PV patients and about 50% of all ET and PMF cases [19]. Investigations from a clinical point of view have focused on studying the occurrence of the Jak2-V617F mutation in other hematological disorders apart from MPNs, the development of diagnostic tools that can be used to screen for the V617F mutation in patients, and in the identification of novel Jak2 mutations in MPN patients. Microarray studies have also been conducted using patient samples in order to identify changes in the gene expression pattern induced by Jak2-V617F [25]. Furthermore, the gene expression pattern of Jak2-V617F-positive ET patients has been found to be different from that of Jak2-V617F-negative patients [26].
One challenging question that arose as a consequence of the discovery of V617F was, how can one mutation, that is, the Jak2-V617F lead to three different disorders; namely, PV, ET, and PMF? To address this issue, several cases have been presented [27]. Since the Jak2 V617F is found to be homozygous in PV patients and heterozygous in ET patients, it is argued that gene dosage may be a determining factor. Mouse models developed for MPN also support this case [28]. Another possibility is the presence of additional mutations in genes apart from Jak2, which in combination with V617F may determine the specific pathology. Further, germline genetic variations may also be responsible for the similar, yet distinct phenotypes observed clinically among the Jak2-V617F positive patients. For example, in 2009, three independent groups reported a predisposition allele called the “46/1” haplotype which explained the higher occurrence of the Jak2-V617F mutation among first-degree relatives of MPN patients [29, 30, 31].
Another line of current research includes the understanding of the molecular mechanism for the constitutive activation of Jak2-V617F. The point mutation at Val 617 occurs in the auto-inhibitory pseudokinase domain (JH2) of Jak2 and therefore allows the kinase to evade negative cis regulation (Figure 1). Kinetic studies of Jak2-V617F have shown that although its Vmax is similar to that of Jak2-WT, it has a lower Km and hence an increased affinity towards substrates leading to hyperactivation [32]. Interestingly, an earlier report had shown there was no difference in the enzyme kinetics of Jak2-V617F when compared to Jak2-WT [33]. Nevertheless, in vitro assays reported by several groups have demonstrated that Jak2-V617F is hyperkinetic [23, 34, 35] and that the FERM and SH2 domains are required for its function [36, 37]. These reports have shown that the FERM domain is important for the association of Jak2-V617F with the erythropoietin receptor (EpoR), while the SH2-like domain is required for cross-phosphorylation and aggregation in the receptor complexes at the membrane. This in turn implies that the constitutive activation of Jak2-V617F is mainly at the level of relieving the autoinhibition of the pseudokinase domain over the kinase domain. The V617F mutation by itself does not confer independence from the functions of other Jak2 domains. However, in order to obtain an understanding as to how a point mutation causes constitutive activation, more information at the level of protein structure is needed. Since a full length Jak2 crystal structure does not exist, homology models and molecular dynamic simulations have been useful in predicting the mechanism of these mutations [34, 38, 39]. In particular, the creation of full-length homology models and the initial descriptions of novel structure-function relationships of the different domains by Kroemer and colleagues has been the starting point in this area of research [40, 41].
FIGURE 1. The Jak2 Protein Structure and Hotspots for Jak2 Mutations.

Cartoon representation of the full length Jak2 homology model prepared using VMD 1.8.6. showing the kinase domain (tan), pseudokinase domain (blue), SH2-like domain (red) and the FERM domain (green). Also shown are the regions where most of the known Jak2 mutations occur; exon 14 (purple spheres) and exon 12 (yellow spheres).
The homology model of Jak2 JH1 and JH2 domains indicated that the activation loop in the kinase domain and V617 in the pseudokinase domain could be involved in an autoinhibitory interaction [40]. Recently, using molecular dynamic simulations and mutagenesis studies in vitro, our group along with that of Lee et al. and Dusa et al., reported that the V617F mutation in the pseudokinase domain (JH2) of Jak2 interacts with the neighboring F595, which in turn reduces the JH1-JH2 autoinhibitory interaction at the JH1/JH2 interface and leads to constitutive activation [34, 35, 38]. However, at the trans level of regulation, several negative feedback regulators such as the SOCS and Lnk are still activated. Thus, one wonders how does the Jak2-V617F mutant protein escape the inhibitory effect of these trans regulators? Jak2-V617F seems to evade this negative regulation by hyper methylating the SOCS3 promoter thereby making it insensitive to STAT binding or hyper phosphorylating the SOCS3 protein thus making it more sensitive to degradation [42, 43]. Additionally, an HDAC inhibitor, in combination with a Jak2 inhibitor, has been shown to reduce Jak2-V617F signaling in vitro [44]. Thus, gene suppression through epigenetic modifications could also be a possible explanation for the resistance of Jak2 V617F against the cellular regulators of signaling.
Identification of Jak2-V617F and availability of the crystal structure for Jak2 kinase domain has also opened the doors to the development of small molecule inhibitors for Jak2. Several mouse models for MPNs that have been created as either transgenic or knock-in models using the V617F allele [28, 45, 46] have been very useful in testing the broad range of Jak2 inhibitors that are being developed for use in the MPN patients. Apart from V617F, additional mutations in the exon 14 region that cause constitutive activation of Jak2 in human diseases include C618R and D620E in PV, E627E and C616Y in unclassified MPN, L611S in ALL, ΔIREED in Down syndrome, K607N in AML, and R683G in Down Syndrome-ALL (Table 1).
Jak2 Exon 12 mutations
In 2007, Scott et al. reported the occurrence of exon 12 mutations in Jak2-V617F negative MPN patients [47]. There are at least 20 different exon 12 mutations that have been reported to date, which include single or multiple point, insertion, deletion and duplication mutations (Table 1). These mutations are found almost exclusively in PV.
The exon 12 mutations are located structurally in the linker region located between the SH2 domain and pseudokinase domain (Figure 1). Functionally, this linker has been implicated in relaying the engagement of cytokines to the extracellular portion of the receptor with the relief of JH2 mediated autoinhibition. Therefore, it is thought that exon 12 mutations lead to constitutive activation of Jak2 by altering the interaction between JH1 and JH2 domains similar to that of Jak2-V617F. Molecular dynamics simulation studies conducted for the various Jak2 exon 12-15 mutations show that in most of the clinically occurring mutations, the JH1-JH2 interface is open indicating disruption of JH2-mediated autoinhibition and subsequent constitutive activation [39].
The 46/1 Jak2 haplotype has been found to predispose patients to both Jak2-V617F and exon 12 mutation positive MPNs [29, 48]. On the other hand, Jak2-V617F positive PV patients have a distinct clinical phenotype from that of PV patients carrying the exon 12 mutations. While patients with exon 12 mutations have isolated erythrocytosis, Jak2-V617F positive patients present a trilineage pattern of erythrocytosis, leukocytosis and thrombocytosis. PV patients with Jak2 exon 12 mutations have higher hemoglobin levels, lower erythropoietin levels, lower platelet counts, and lower leukocyte counts than Jak2-V617F positive PV patients [49]. Additionally, mitotic recombination leading to homozygosity is more likely to occur in Jak2-V617F-positive PV patients than those that carry the exon 12 mutations. This indicates that even though both Jak2-V617F and exon 12 mutations are constitutively active, their downstream signaling pathways are different, which eventually leads to variation in their pathophysiology and clinical phenotypes. Accordingly, Zou et al. reported that the catalytic activity, downstream signaling, ability to bind the erythropoietin receptor and the transforming properties of Jak2-V617F are significantly higher when compared to the exon 12 mutant, Jak2-K539L [50]. Contrary to this report however, Scott et al. reported that exon 12 mutants including Jak2-K539L have increased Jak2 signaling when compared to Jak2-V617F [47]. Interestingly, there are no apparent differences in the assay conditions used by these two groups.
While Jak2-V617F and exon 12 mutations are predominant in MPN patients, Ma et al. reported that Jak2 mutations also occur in exons 13 and 15 [51] (Table 1). This indicates that the pseudokinase domain serves as a hotspot for mutations that lead to constitutive Jak2 activation. Based on the antisymmetrical interface between the kinase and pseudokinase domains, most of the Jak2 mutations identified to date occur in the N-lobe of the pseudokinase domain, which participates in the autoinhibition of the C-lobe of the kinase domain.
Jak2 chromosomal translocations
The first evidence for Jak2 constitutive activation in human cancers came in 1997 when the TEL-Jak2 fusion gene was identified among leukemia patients [52, 53]. The chromosomal translocation occurred between ETV6/TEL located on 12p13 chromosome and Jak2 on 9p24, resulting in a fusion gene that contains the N-terminal dimerization domain of TEL and the C-terminal kinase domain of Jak2. The chimeric protein exhibits constitutive kinase activation mediated by increased homodimerization. Since then, at least 14 different chromosomal translocations involving the Jak2 gene have been identified in different hematological malignancies, including both myeloid and lymphoid disorders (Table 2).
TABLE 2.
Jak2 Chromosomal Translocations
| TRANSLOCATION | FUSION PARTNER | DISEASE | REFERENCE |
|---|---|---|---|
| t(9;12)(p24;p13) | TEL-Jak2 | T-ALL, precursor B-ALL, atypical CML, MDS | [52,53,66] |
| t(9;22)(p24;q11.2), t(9;22)(p24;q11) | BCR-Jak2 | atypical CML, AML | [54,55] |
| t(8;9)(p22;p24) | PCM1-Jak2 | atypical CML, precursor B-ALL, CEL, AML, T-cell lymphoma, PMF, MDS/MPN | [56–59] |
| del(9)(p13:p24) | PAX5b | ALL | [60–61] |
| t(5;9)(p14.1;p24.1) | SSBP2 | precursor B-ALL | [64] |
| t(4;9)(q21;p24) | SEC31A-Jak2 | classical Hodgkin Lymphoma (cHL) | [65] |
| t(9;12)(p24;q13) | NFE2c | MDS | [66] |
| add (9) (p24) and del (21)(q11.2) | AML1 (RUNX1)c | MDS | [66] |
| t(3;9)(q21;p24) | RPN1 c | CIMF | [67] |
| t(9;22)(p24;q11.2) | Unknown | B-ALL | [68] |
| t(2;9)(p21;p24) | Unknown | PMF | [69] |
| t(8;9)(q22;p24) | Unknown | PMF | [69] |
| t(9;17)(p24;q23) | Unknown | PV | [69] |
| t(4;9)(q25;p24) | Unknown | PMF | [69] |
| t(8;9)(q13;p24) | Unknown | DLBCL | [69] |
Abbreviations: AML1 - acute myeloid leukemia 1, BCR - B-cell receptor, CML - chronic myeloid leukemia, CEL - chronic eosinophilic leukemia, cHL - classic hodgkin’s lymphoma, CIMF - chronic idiopathic myelofibrosis, DLBCL - diffuse large B-cell lymphoma, ETV6 - Ets variant gene 6, NFE2 - nuclear factor erythroid-derived 2, PAX5 - paired box gene 5, PCM1 - Human autoantigen pericentriolar material 1, PMF - primary myelofibrosis, PV - polycythemia vera, RPN1 - ribophorin 1, SSBP2 - single stranded DNA binding protein 2, T-ALL - T cell ALL.
PAX5-Jak2 is not constitutively active.
Not fully characterized
Chromosomal translocations between the B-cell Receptor (BCR) and Jak2 genes have been reported in atypical Chronic Myeloid Leukemia (aCML) and Acute Myeloid Leukemia (AML) [54, 55]. The human autoantigen pericentriolar material 1 (PCM1) gene has also been implicated as a fusion partner of Jak2 in atypical CML, AML, Chronic Eosinophilic Leukemia (CEL), PMF, unclassifiable Myelodysplastic/Myeloproliferative neoplasm (MDS/MPN), precursor B- Acute Lymphoblastic Leukemia (B-ALL) and T-cell lymphoma [56, 57, 58, 59]. Nebral et al. reported an ALL fusion of Jak2 with the N-terminus of the Paired box protein 5 (PAX5), a master regulator of B-cell development [60, 61]. The DNA binding domains along with the nuclear localization signal of PAX5 are fused with the Jak2 kinase domain. Therefore, the chimeric PAX5-Jak2 is located in the nucleus and is capable of binding to the wild type PAX5 targets. Interestingly, the DNA binding domain of PAX5 has not been shown to mediate dimerization and the kinase activity of the PAX5-Jak2 has not been examined. Therefore, the effect of the fusion partner on Jak2 activity cannot be predicted. Based on the recent findings of histone phosphorylation by Jak2 in the nucleus [62], it is possible that the PAX5-Jak2 affects gene expression in ALL by altering the status of histone phosphorylation. This would be interesting to determine as other PAX5 fusions such as PAX5-TEL and PAX5-ELN have been shown to act as transcriptional repressors via a dominant negative mechanism on wild type PAX5 [63]. Fusion of the Single strand DNA binding protein 2 (SSBP2) with Jak2 has been reported in precursor B-ALL [64]. Recently, a novel SEC31A-Jak2 fusion was identified in classical Hodgkin Lymphoma (cHL) and shown to constitutively activate the Jak-STAT signaling pathway [65]. Interestingly, in this case the constitutive activation of Jak2 did not depend on the WD40 repeats or the proline rich domain in the SEC31A fusion partner, which facilitates protein-protein interactions. The role of the required domain of the SEC31A partner has not yet been identified. Apart from the Jak2 fusion partners described above, NF-E2, RUNX1 (AML1) and RPN1 have also been identified as putative partners involved in chromosomal translocation with Jak2 [66, 67]. Six additional chromosomal translocations involving Jak2 have been reported, but the fusion partners have not been identified [68, 69]. The functional and molecular characterization of these chimeric proteins may reveal their role in the tumorigenesis of the respective cancers.
The occurrence of Jak2 fusion genes in hematological malignancies is relatively rare when compared to that of the point mutations. While point mutations disrupt auto regulation and lead to ligand hypersensitivity in MPNs, they are generally not sufficient to cause a more deleterious phenotype such as leukemia. However, the clinical phenotypes of Jak2 fusion genes are very aggressive and have rapid progression to blast phase [70]. Further, in most cases of Jak2 fusion, there is a common pattern of an N-terminal dimerization domain being fused with the C-terminal Jak2 kinase domain. Yet, the different fusion genes confer different clinical phenotypes. This may be ascribed to the variability in the site of Jak2 fusion (Figure 2). Given the role of the different JH domains on Jak2 kinase activity, the variation in the clinical phenotypes observed might correlate with the Jak2 domains included in the chromosomal translocation. For example, in the case of the ETV6/TEL-Jak2 fusion found in ALL and T-cell lymphoblastic leukemia, a partial JH2 domain is present along with the entire JH1 domain. However, in the case of ETV6/TEL-Jak2 fusion present in AML, complete JH1 and JH2 domains are present. Further, the N-terminus domains from the different partner genes may influence Jak2 kinase activity by variant mechanisms of regulation. Also, the fusion mutations may occur in different types of somatic cells. Hence, the observed phenotype may vary based on the level of kinase activity of the chimeric protein and also the nature of the cells in which they occur.
FIGURE 2. Jak2 Chromosomal Translocations.

N-terminus domains from the different fusion partners are fused with the C-terminus of Jak2 including as a minimum the kinase domain (tan). OP-Octapeptide, WD40 - beta transducin repeat motifs terminating in W-D dipeptide, LisH - LIS1 homology domain, TK - Tyrosine kinase, PK - pseudo kinase, SH2 - Src Homology 2, HLH - Helix Loop Helix.
Lack of Mutations in the Jak2 Kinase domain
In the case of kinases such as B-Raf, RET, FLT3, KIT and EGFR, the kinase domain serves to be a hotspot for somatic mutations that cause constitutive activation in cancers. Such mutations generally occur in the ATP binding region, catalytic region, activation loop and P-loop of the kinase domain. These mutations cause constitutive activation by destabilizing the inactive conformation of the kinase. However, in Jak2 only a single substitution mutation, T875N, in the kinase domain has been shown to cause MPN in murine models [71]. The Jak2-T875N mutation occurs in the N-terminal lobe of the kinase domain and is predicted to cause constitutive activation by altering the surface properties of the domain and by disrupting the autoinhibition of the JH2 domain over the JH1 domain. Further, a few other kinase domain mutations have been identified in ch-B-ALL (Table 1). This overall rare incidence of kinase domain mutations indicates a rather tight control of Jak2 kinase activity by other cis acting JAK homology domains, which are difficult to disrupt by mutations in the kinase domain itself. Keeping this in mind, it is important to understand that current Jak2 small inhibitors target the Jak2 kinase domain, which does not carry these gain-of-function mutations. Therefore, inhibiting Jak2 kinase activity via the selective targeting of JAK regulatory domains may serve to improve the therapeutic index and specificity of the drugs.
Need for Alternative Approaches to ATP mimetic Jak2 Inhibitors
Current Jak2 kinase inhibitors are categorized as either class I or class II drugs. Class I drugs were developed specifically for Jak2 based on available protein structural information. Class II drugs are those that were developed against other kinases, but were found to have beneficial off-target effects on Jak2. Based on the currently available results from Jak2 inhibitor clinical trials, the benefits of these drugs include improvement of splenomegaly, pruritus, erythrocytosis, leukocytosis and thrombocytosis [72, 73]. Though these drugs have positive effects in terms of reducing symptomologies associated with the MPN phenotype, they are unable to hinder the deleterious fibrosis or significantly reduce the mutant-allelic burden in the bone marrow. Also, toxicity, in the forms of anemia, nausea, vomiting and diarrhea, has been observed in these patients.
The current design of Jak2 selective inhibitors is based on structural information available from homology models or crystal structures for the wild type Jak2 kinase domain. Most of these inhibitors target the ATP-binding pocket in the kinase domain and hence are ATP-competitive. ATP-competitive inhibitors bind tightly to the kinase domain in its active conformation and prevent ATP binding or hydrolysis, thereby reducing kinase activity. The kinase domains of proteins from different kinase families have distinct conformations in their inactive state. However, upon activation, they assume a very similar active state conformation [74]. Targeting the active kinase conformation may provide increased sensitivity towards constitutively active mutant Jak2. On the other hand, this strategy may also result in non-specific effects through action on Jak2-WT and other important kinases that are active in the cell. Accordingly, results from the clinical trials reveal that treatment with current generation ATP-competitive Jak2 inhibitors results in myelosuppression and anemia. This could be a possible effect of these drugs on wild type Jak2, which plays an important role in normal hematopoiesis. Although it would be premature to judge the general success or failure of existing ATP-competitive drugs, the results clearly indicate the need for specific targeting of mutant forms of Jak2. Such specific ablation of Jak2 mutant activity would, in theory, reduce the mutant allelic burden and hence improve the therapeutic window. Apart from the issue of selectivity, development of drug resistance is also higher with the use of ATP competitive inhibitors. Using in vitro assays, it was shown that mutation of residues Y931, Y958 and P960 in the hinge region of the Jak2 kinase domain confer constitutive activation along with resistance to ATP-competitive Jak2 inhibitors, including INCB018424 [75]. These findings indicate the possibility of such secondary mutations occurring in patients during prolonged treatment with ATP-competitive inhibitors. Therefore, in order to gain specificity and avoid drug resistance, alternative strategies such as allosteric inhibitor design, should be considered.
Current Allosteric Inhibitors for Kinases in Cancer
Since the role of kinases in cancer has become more obvious in the recent years, kinases have become attractive targets for inhibition. An important part of drug design against kinases includes the selection of the drug binding site that would achieve high target specificity. Additionally, the nature of interactions between the inhibitor and the target atoms at the binding site would determine its binding affinity and potency. Therefore, the active site, where the phosphate from ATP is catalytically transferred to the substrate, would be an ideal targeting site for kinases. However, the catalytic activity of kinases can also be moderated allosterically from sites that are remote to the active site. Binding of a drug to the allosteric site can therefore moderate the catalytic activity indirectly through a network of residues that connects to the active site, without directly competing with the ATP.
There are four general types of kinase inhibitors based on their binding site and mode of inhibition: ATP-competitive inhibitors (type I), inhibitors that bind to the inactive kinase conformation (type II), allosteric inhibitors, and covalent inhibitors [76]. Of these, type II and allosteric inhibitors provide high kinase selectivity. Selective targeting of Jak2 is important because of its critical role in hematopoiesis, for which the allosteric inhibitors may be suitable. The term “allostery” means “different shape”. Allosteric inhibitors are non-ATP competitive and inhibit kinase activity by binding to a site that is remote to the active site. This approach can help achieve target specificity and increased drug efficacy. There are several examples in the literature for such non-ATP competitive inhibitors that are being currently tested for treatment in various cancers. A review of these drugs may provide perspective for a similar design against Jak2 in hematological malignancies.
BCR-ABL
Despite having good efficacy, the first generation inhibitors for BCR-ABL, such as imatinib, induced secondary mutations in the kinase that resulted in drug resistance. These mutations mostly occur in the P-loop and alter the inactive conformation of the BCR-ABL kinase domain, to which the imatinib binds preferentially. Consequently, more potent second-generation inhibitors, such as dasatinib, nilotinib and bosutinib, were developed [77]. Both the first and second generation of BCR-ABL inhibitors bind to the active site of the kinase domain. While imatinib has been shown to be non-ATP competitive, it still binds to the inactive kinase near the active site. A BCR-ABL T315I mutation, which disrupts the gatekeeper residue of the kinase domain, is resistant to both first and second-generation inhibitors. Henceforth, several alternative approaches, such as the development of non-ATP competitive inhibitors, are being pursued to combat this resistance mutation. Current allosteric inhibitors for BCR-ABL target three regions that are remote to the active site:
Myristoyl binding cleft: Since myristoylation has been known to stabilize ABL in its inactive conformation, the binding of inhibitors to this pocket in the ABL kinase domain are predicted to have the same effect. GNF-2 is a lead compound that binds to this cleft and allosterically inhibits BCR-ABL kinase activity (Table 3). GNF-2 has a 4,6-disubstituted pyrimidine core structure, which is essential for its activity and it binds to the cylindrical myristate-binding cavity of the Abl kinase domain in a trans conformation. The 4-trifluoromethoxyaniline group interacts at the base of the pocket and is essential for activity. However, substitutions at the pyrimidine 6-positions can be tolerated since they are exposed to the solvent [78]. The selectivity of these compounds for BCR-ABL is exceptional and they also have significant in vivo efficacy when used in combination with imatinib or nilotinib [79]. Therefore, GNF-2 may have good clinical efficacy, reduced drug resistance, and decreased toxicity due to its high selectivity.
Switch pocket inhibitors: DCC-2036 is an inhibitor that binds to a switch control pocket in the BCR-ABL kinase domain and locks it in the inactive conformation [80]. During the active state, R386 in the Abl1 kinase domain stabilizes the phosphorylated Y393 in the activation loop. However, in the inactive state R386 forms a salt bridge with E282 under the αC-helix. Therefore, DCC-2036 was designed with a tetrahydro-isoquinoline ring that hydrogen bonds to both E282 and R386 in order to target the inactive Abl1. Further, DCC-2036 also has a carboxamide-substituted pyridine ring, which hydrogen bonds with the backbone of the ATP hinge residue M318 as a second docking site in order to have increased binding energy and potency. In preclinical studies, this drug has shown good oral bioavailability, limited off-target effects, and excellent safety. It is currently being evaluated in phase I/II clinical trials for the treatment of imatinib-refractory CML.
Substrate-competitive inhibitors: ON012380 is an inhibitor that binds to the substrate-binding site of the ABL kinase domain and inhibits it at least ten-fold more potently than imatinib [81]. ON012380 was designed to have a structure that is unrelated to ATP or any other purine or pyrmidine nucleosides such that it blocks the substrate binding site rather than the ATP binding site. This inhibitor has good efficacy and reduced toxicity in murine models. However, the activity of ON012380 in clinical trials has not been tested.
TABLE 3.
Current allosteric inhibitors for BCR-ABL and Jak2 that target sites other than the ATP-binding site
| NAME | STRUCTURE | TARGET KINASES | IC50 (nM) | PREDICTED MECHANISM OF INHIBITION | REF. |
|---|---|---|---|---|---|
| GNF-2 |
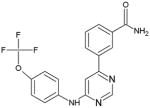
|
BCR-ABL | 138 | Targets the myristoyl binding cleft | [79] |
| DCC-2036 |
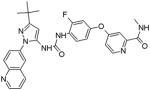
|
BCR-ABL | 5.8 | Targets the switch control pocket | [80] |
| ON012380 |
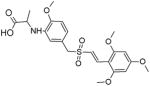
|
BCR-ABL | 10 | Substrate-competitive | [81] |
| LS104 |
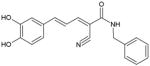
|
Jak2, FLT3 | 1500 (Jak2), 4000 (FLT3) | Substrate-competitive | [87,88] |
| ON044580 |
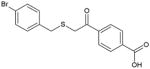
|
BCR-ABL, Jak2 | 3000 (BCR- ABL), 4000 (Jak2) | Substrate-competitive | [89] |
Similar to BCR-ABL, allosteric inhibitors are also being developed for other kinases including Akt and various MAP kinases such as BRAF and MEK [82, 83, 84, 85]. Another leading example of a drug that does not target the kinase domain is the rapamycin derivative, temsirolimus. It is an FDA approved anti-cancer drug that binds to the FKBP12/Rapamycin Binding (FRB) domain of the mTOR kinase and inhibits its function [86].
Thus, allosteric inhibitors have proven useful for improved selectivity and decreased toxicity resulting in improved clinical efficacy. These drugs also serve as good alternatives to ATP-competitive inhibitors that can sometimes lead to drug resistance. Therefore, lessons learnt from these examples may guide the development of Jak2 allosteric inhibitors which can act as suitable alternatives to the existing class of ATP mimetic inhibitors.
Allosteric Inhibitors for Jak2
While there are a number of ATP-competitive inhibitors for Jak2, to date, there are only two known non-ATP competitive inhibitors for this target.
LS104
LS104 is a hydroxystyryl-acrylonitrile compound, which is an analog of a non-specific Jak2 inhibitor, AG490. It has increased affinity and specificity for Jak2 when compared to AG490. LS104 has an elongated linker between the nitrile and 3,4-dihydroxyphenyl groups. It inhibits the growth of leukemic cell lines and primary cultures obtained from leukemia patients [87] (Table 3). It has been shown to inhibit BCR-ABL, Jak2, and FLT3, but does not have any effect on Src family kinases [88]. LS104 inhibits the Jak2 kinase activity, signaling distal to Jak2, and selectively induces apoptosis in Jak2-V617F transformed cells, in vitro. The combination of LS104 with ATP-competitive inhibitors induced apoptosis in Jak2-V617F transformed cells, synergistically. It prevents cytokine-independent colony growth of bone marrow cells derived from MPN patients, ex vivo. LS104 is predicted to be a substrate competitive inhibitor. However, the crystal structure of LS104 with Jak2 is not available to confirm its binding mode. Additionally, its clinical efficacy is yet to be determined.
ON044580
ON044580 is an α-benzoyl styryl benzyl sulfide, which is shown to inhibit the growth of leukemic cell lines and Jak2-V617F transformed cells [89]. Similar to ON012380, ON044580 was also designed to have a structure that is unrelated to ATP or any other purine or pyrimidine nucleosides such that it would not be ATP-competitive and still possess kinase inhibitory activity. ON044580 inhibits both Jak2 and BCR-ABL in a non-ATP competitive manner. It is predicted to be substrate-competitive inhibitor. Both BCR-ABL and Jak2 share a common substrate, STAT5, which in turn supports the substrate inhibitor hypothesis. ON044580 also inhibits the imatinib-resistant BCR-ABL-T315I mutant with higher sensitivity. Moreover, it was observed that ON044580 leads to the degradation of BCR-ABL, Jak2 and STAT5 possibly by destabilizing their multi-protein complex [90]. Interestingly, the Jak2 pseudokinase domain was found to be required for complete kinase inhibition by ON044580. This further suggests that ON044580 could be binding to Jak2 in its inactive conformation and that the pseudokinase domain is required to maintain the inactive form. No crystal structure information is available regarding the interaction of ON044580 with Jak2 or BCR-ABL so its exact binding mode is not known. This drug also effectively inhibits the growth and induces apoptosis in ex vivo cultures of bone marrow cells from patients in different stages of leukemia and monosomy 7 myelodysplastic syndrome [89]. Jak2 signaling is constitutively active in both these conditions and therefore, it is proposed that ON044580 could be used to treat both MDS and MPN.
Collectively, LS104 and ON044580 are two known allosteric inhibitors for Jak2, which effectively down regulate Jak2-dependent, transformed cell growth. Further studies are required to determine their binding interactions with Jak2, selectivity for Jak2 mutant over wild type, relative toxicity, and drug resistance. This information will be useful to perform a comparative analysis with their ATP-competitive counter parts and identify the benefits of allosteric inhibitor development for Jak2.
Possible targets on the Jak2 surface for Allosteric Inhibition
Jak2 is a multi-domain tyrosine kinase, which is made up of 1132 amino acids, weighs approximately 130 kDa and occupies a volume of about 158,000 cubic angstroms. Regulation of Jak2 activity can be achieved using different levels of coordination among its domains. This aspect will be useful in identifying allosteric pockets that could be targeted for Jak2 inhibition. During the development of ATP-competitive Jak2 inhibitors for instance, the target pocket is usually defined as that surrounding the ATP-binding pocket. However, in order to develop allosteric inhibitors for Jak2, it would be necessary to identify druggable pockets on the Jak2 surface that are remote to the active site. Exploring the vast surface area of Jak2 to identify such pockets may seem like a daunting task. However, this task is feasible when the recent advancements in molecular docking are combined with available Jak2 structure-function relationship data. The contribution of structural biology to the development of inhibitors, specifically for kinases, is notable and has been discussed in detail elsewhere [86]. Based on the available structural information, possible areas for allosteric targeting in Jak2 are discussed below.
Jak2 kinase domain
The ATP-binding site in the kinase domain is currently being targeted for ATP-competitive inhibitors that bind to the active conformation of the kinase. However, targeting the pockets in the inactive kinase conformation may provide more Jak2 selectivity. In the inactive conformation, when the catalytic loop is in the DFG-out conformation, it blocks ATP binding and this in turn may result in distinct pockets adjacent to the ATP binding site (Figure 3). Specifically, the type II inhibitor pocket formed between the C-helix and the activation segment could be a possible target. A classic example for this is Gleevec, which preferentially binds to and stabilizes the inactive conformation of BCR-ABL [91]. The crystal structure of the inactive form of Jak2 is not available and this information may be useful in identifying such pockets. Also, the Jak2 kinase domain has a unique insertion loop, which is not conserved in other kinases. This feature should be considered during the design of Jak2 allosteric inhibitors. Additionally, considering the case of LS104, targeting the substrate-binding site of Jak2 may also serve as an effective strategy for inhibition. There is also precedent for a peptide inhibitor, Tkip, which inhibits Jak2 by binding to the activation loop in its autophosphorylated state similar to that of SOCS binding [92]. Thus, targeting regions apart from the ATP binding site may also result in effective Jak2 inhibition through an allosteric mechanism.
FIGURE 3. Possible Targets for Jak2 Allosteric Inhibition.

Surface representation of a full length Jak2 homology model prepared using VMD 1.8.6. The kinase domain is shown in orange, pseudokinase domain in cyan, SH2-JH2 linker in yellow, SH2-like domain in red and the FERM domain in green. Other linker regions between the domains are shown in white.
Autoinhibitory Interface between the Kinase and Pseudokinase Domains
Among the non-receptor tyrosine kinases, Jak2 has a unique architecture of having a pseudokinase domain located adjacent to the kinase domain. The JH2 domain is classified to be a pseudokinase due to the lack of tyrosine kinase activity and the absence of a conserved HRD motif, which is replaced by HGN [93, 94]. Saharinen and Silvennoinen first reported the role of the JH2 domain being an autoinhibitory domain over the JH1 kinase domain [95, 96]. They also showed that residues 758-807 in the JH2 domain are important for the autoinhibition, while residues 619-757 enhance this inhibition. Therefore, Jak2 activation in response to ligand binding involves the relief of the kinase domain from the autoinhibition of JH2, possibly via the phosphorylation of its activation loop. The molecular mechanism for the constitutive activation of Jak2-V617F involves the disruption of inhibitory interactions at this interface [34, 35, 38]. The autoinhibitory regulation of Jak2 makes it an attractive drug target since it involves conformational changes as a part of its functional cycle [97]. Therapeutically successful inhibitors that stabilize the auto-inhibited fold of proteins have been identified for other targets including BCR-ABL (imatinib) and N-WASP (wiskostatin). Therefore, targeting Jak2 at its JH1-JH2 interface may stabilize the auto-inhibited form of the protein.
SH2-linker - JH2 interface
Function of the SH2-like domain located adjacent to the pseudokinase domain has not been well characterized. However, the linker connecting the SH2 domain to the pseudokinase domain has been implicated in Jak2 activation (Figure 3). Specifically, Zhao et al. reported that upon ligand binding, the SH2-pseudokinase linker flexes the hinge between the JH1 and JH2, thus leading to Jak2 activation [98]. Accordingly, several Jak2 exon 12 mutations that lead to constitutive Jak2 activation occur in the SH2-JH2 linker region. It is possible that these mutations are stabilizing the linker region in the ‘ON’ mode that is normally associated with Jak2 activation. However, this needs to be demonstrated experimentally. It is interesting to note that in another tyrosine kinase, ZAP-70, the linker between the SH2 and kinase domains auto-inhibits kinase activity by reducing the flexibility of the hinge region [99]. Targeting the pockets that might control conformational changes induced by the SH2-JH2 linker will be useful in regulating Jak2 kinase activity. Further, targeting different allosteric sites based on the location of the mutations may also provide mutant specific inhibition, as in the case of exon 14 vs. exon 12.
FERM domain
The Jak2 FERM domain has been known to be important for Jak-receptor association. Zhou et al. has shown that the Jak3 FERM domain physically interacts with the kinase domain and regulates both Jak3 receptor association and kinase activity [100]. Interestingly, in the case of Jak2, it was found that the FERM domain negatively regulates Jak2 wild type, but positively regulates Jak2-V617F kinase activity [32]. Further, phosphorylation of tyrosines in the Jak2 FERM domain has been reported to have both positive and negative regulatory effects on Jak2 kinase activity. Mutation of tyrosines 114, 119 and 317 in the FERM domain or deletion of the FERM domain itself has been shown to increase the kinase activity of Jak2-WT [36, 101, 102]. Recently, a potential activation mutation in the FERM domain, R340Q, was identified in Jak2-V617F negative MPN patients [103]. There is precedent for negative regulation through FERM domain in Focal Adhesion Kinase (FAK). The FAK kinase domain is auto-inhibited by the FERM domain via sterical blocking of the catalytic cleft, which is very similar to the predicted interaction between the JH1and JH2 domains in the JAKs [104].
Overall, Jak2 FERM domain seems to be regulating both Jak2 receptor association and kinase activity. Thus, targeting pockets in the FERM domain may help modulate Jak2 kinase activity. In this case, available information on the actual mode of FERM domain interactions with the receptor should be considered while assessing the pockets for targeting.
Identification of Druggable Allosteric Pockets using Virtual Screening
While the above-mentioned surfaces can be used as targets, identifying a druggable pocket within those surfaces that can bind a high affinity compound involves a different approach. Identification of lead compounds for targets using structure based virtual screening has become popular in the past decade and it has proved very useful in drug discovery. Its high throughput nature allows for the screening of a large number of compounds in a fast and inexpensive manner, which is not feasible experimentally. Virtual screening involves the molecular docking of various inhibitors to a defined pocket on the target and ranking the ligands based on parameters such as binding energy, geometrical complementarity, and bonding interactions. The scoring function used to rank the compounds varies based on the docking program used, such as a force field-based function in DOCK, an empirical function in Glide and a shape based function in LigandFit [105]. Ranking of compounds using current docking programs is very crude and therefore a large number of compounds have to be tested biologically using high throughput screening methods in order to identify a possible lead compound. This limitation arises due to unreliable scoring methods, which sometimes fail to account for the conformational flexibility of the protein and hence do not reflect the nature of ligand-protein interactions accurately.
Due to a high degree of conservation among kinase structures, even in the absence of crystal structures for certain kinases, homology models are being used for virtual screening of inhibitors. For example, before the crystal structure of the Jak2 kinase domain was solved in 2006 [106], this method was employed for the development of ATP-competitive Jak2 inhibitors, In addition, given the vast amount of Jak2 surface available for targeting, the homology model can also be adopted for Jak2 allosteric inhibitor design. Identification of allosteric binding sites can be tricky and use of molecular dynamics may be required. Since allosteric sites are linked to remote functional sites indirectly, they can be identified using cooperative coupling methods. This may require a combination of approaches that are based on both sequence, as in the Statistical Coupling Analysis (SCA) and protein structure, as in the COREX algorithm [107]. This virtual screening process can in turn be guided by the available biochemical data on Jak2 structure-function relationships. Further, Haan et al., has suggested a chemical genetics based approach to achieve more specificity while targeting Jak kinases [108].
Collectively, using allosteric inhibitor design as an alternative approach to ATP-competitive inhibitors for Jak2 may result in more effective inhibitors that are specific to mutant Jak2 and also prevent drug resistant mutations. Such allosteric inhibitor design can be guided by the existing information on Jak2 structure-function, though a crystal structure of full length Jak2 will certainly prove more useful.
Conclusion
The convergence of the identification of Jak2-V617F in MPNs and the recent developments in kinase targeting has catalyzed efforts on targeting Jak2 for the treatment of MPN. Many ATP-competitive Jak2 inhibitors that exhibited good pre-clinical results are now being tested in clinical trials. The most important ones will be those that can reduce the mutant allelic burden in the bone marrow. While these inhibitors may come to fruition, an alternative approach is to target Jak2 via allosteric design. Though the active conformation of protein kinases is highly conserved, ATP-competitive inhibitors can still be specific. The extended region of the drug that interacts outside the active site may provide specificity and significant effort has been expended to achieve such specificity using structure-activity relationship (SAR) studies of lead compounds. Still, non-specific, off-target effects have been unavoidable in several cases. While such non-specificity has proved toxic during some treatments, it has also been deemed favorable in the combinatorial targeting of multiple kinases involved in tumorigenesis [109]. For Jak2 targeting, non-specific effects with other Jak kinases or even on wild type Jak2 should be avoided due to its critical role in hematopoiesis. Therefore, in cases like Jak2 where selectivity is required, alternative approaches such as allosteric inhibitor design may be useful. Since allosteric inhibitor design utilizes the principles of autoinhibition adopted by nature and applies it to synthetic inhibitors, its efficiency could be greater. Using a combination of the available structural information and improved molecular docking methodologies, druggable allosteric pockets on Jak2 surface and high affinity drugs for the target pockets can be identified. We have made an attempt to point out the possible surfaces for Jak2 allosteric inhibition in this review. However, a systematic dissection of the Jak2 surface using computational approaches will be required to identify druggable allosteric sites. Identification of the Jak2 allosteric sites would require the use of cooperative coupling methods and analysis of the geometrical and physiochemical properties of multiple pockets on the Jak2 surface in various conformations as identified using molecular dynamics simulations. Further, the predicted allosteric sites can be cross-referenced to available biological data from the Jak2 structure-function studies for validation. It is our hope that future research in the area of Jak2 drug development would focus on identifying and targeting the allosteric sites on Jak2 surface, in order to improve the specificity during inhibition. While no judgments can be made on the current ATP-competitive inhibitors for Jak2, allosteric inhibitor design may simply provide an alternative approach that could lead to selective and more effective inhibitors for MPN treatment. Moreover, a combination of allosteric and ATP-competitive inhibitors could prove more effective than the individual inhibitors alone.
Acknowledgments
We would like to thank Dr. Kroemer for the PDB co-ordinates of the full length Jak2 homology model. This research was supported by NIH R01-HL67277 and AHA #0855361E.
Abbreviations
- Jak
Janus Kinase
- JH
Janus Kinase Homology
- STAT
Signal Transducers and Activators of Transcription
- FERM
4.1 protein/ezrin/radixin/moesin
- SH2
Src Homology 2
- MPN
Myeloproliferative Neoplasms
- PV
Polycythemia vera
- ET
Essential thrombocythemia
- PMF
Primary Myelofibrosis
- SCID
Severe Combined Immunodeficiency
- HPV
Human Papilloma Virus
- HCMV
Human cytomegalovirus
- ALL
Acute Lymphoid Leukemia
- DS
Down Syndrome
- CML
Chronic Myelogenous Leukemia
- SOCS
Suppressor of Cytokine Signaling
- cHL
classical Hodgkin Lymphoma
- BCR
B-cell Receptor
Footnotes
Conflict of Interest: None.
References
- 1.Rodig SJ, Meraz MA, White JM, Lampe PA, Riley JK, Arthur CD, King KL, Sheehan KC, Yin L, Pennica D, Johnson EM, Schreiber RD. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell. 1998;93(3):373–83. doi: 10.1016/s0092-8674(00)81166-6. [DOI] [PubMed] [Google Scholar]
- 2.Parganas E, Wang D, Stravopodis D, Topham DJ, Marine JC, Teglund S, Vanin EF, Bodner S, Colamonici OR, van Deursen JM, Grosveld G, Ihle JN. Jak2 is essential for signaling through a variety of cytokine receptors. Cell. 1998;93(3):385–95. doi: 10.1016/s0092-8674(00)81167-8. [DOI] [PubMed] [Google Scholar]
- 3.Neubauer H, Cumano A, Muller M, Wu H, Huffstadt U, Pfeffer K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell. 1998;93(3):397–409. doi: 10.1016/s0092-8674(00)81168-x. [DOI] [PubMed] [Google Scholar]
- 4.Nosaka T, van Deursen JM, Tripp RA, Thierfelder WE, Witthuhn BA, McMickle AP, Doherty PC, Grosveld GC, Ihle JN. Defective lymphoid development in mice lacking Jak3. Science. 1995;270(5237):800–2. doi: 10.1126/science.270.5237.800. [DOI] [PubMed] [Google Scholar]
- 5.Park SY, Saijo K, Takahashi T, Osawa M, Arase H, Hirayama N, Miyake K, Nakauchi H, Shirasawa T, Saito T. Developmental defects of lymphoid cells in Jak3 kinase-deficient mice. Immunity. 1995;3(6):771–82. doi: 10.1016/1074-7613(95)90066-7. [DOI] [PubMed] [Google Scholar]
- 6.Thomis DC, Gurniak CB, Tivol E, Sharpe AH, Berg LJ. Defects in B lymphocyte maturation and T lymphocyte activation in mice lacking Jak3. Science. 1995;270(5237):794–7. doi: 10.1126/science.270.5237.794. [DOI] [PubMed] [Google Scholar]
- 7.Russell SM, Tayebi N, Nakajima H, Riedy MC, Roberts JL, Aman MJ, Migone TS, Noguchi M, Markert ML, Buckley RH, O’Shea JJ, Leonard WJ. Mutation of Jak3 in a patient with SCID: essential role of Jak3 in lymphoid development. Science. 1995;270(5237):797–800. doi: 10.1126/science.270.5237.797. [DOI] [PubMed] [Google Scholar]
- 8.Macchi P, Villa A, Giliani S, Sacco MG, Frattini A, Porta F, Ugazio AG, Johnston JA, Candotti F, O’Shea JJ. Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID) Nature. 1995;377(6544):65–8. doi: 10.1038/377065a0. [DOI] [PubMed] [Google Scholar]
- 9.Karaghiosoff M, Neubauer H, Lassnig C, Kovarik P, Schindler H, Pircher H, McCoy B, Bogdan C, Decker T, Brem G, Pfeffer K, Müller M. Partial impairment of cytokine responses in Tyk2-deficient mice. Immunity. 2000;13(4):549–60. doi: 10.1016/s1074-7613(00)00054-6. [DOI] [PubMed] [Google Scholar]
- 10.Shimoda K, Kato K, Aoki K, Matsuda T, Miyamoto A, Shibamori M, Yamashita M, Numata A, Takase K, Kobayashi S, Shibata S, Asano Y, Gondo H, Sekiguchi K, Nakayama K, Nakayama T, Okamura T, Okamura S, Niho Y. Tyk2 plays a restricted role in IFN alpha signaling, although it is required for IL-12-mediated T cell function. Immunity. 2000;13(4):561–71. doi: 10.1016/s1074-7613(00)00055-8. [DOI] [PubMed] [Google Scholar]
- 11.Duhé RJ, Wang LH, Farrar WL. Negative regulation of Janus kinases. Cell Biochem Biophys. 2001;34(1):17–59. doi: 10.1385/CBB:34:1:17. [DOI] [PubMed] [Google Scholar]
- 12.Bogoyevitch MA, Fairlie DP. A new paradigm for protein kinase inhibition: blocking phosphorylation without directly targeting ATP binding. Drug Discov Today. 2007;12(15–16):622–33. doi: 10.1016/j.drudis.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 13.Girault JA, Labesse G, Mornon JP, Callebaut I. Janus kinases and focal adhesion kinases play in the 4.1 band: a superfamily of band 4.1 domains important for cell structure and signal transduction. Mol Med. 1998;4(12):751–69. [PMC free article] [PubMed] [Google Scholar]
- 14.Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264(5164):1415–21. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 15.Feng J, Witthuhn BA, Matsuda T, Kohlhuber F, Kerr IM, Ihle JN. Activation of Jak2 catalytic activity requires phosphorylation of Y1007 in the kinase activation loop. Mol Cell Biol. 1997;17(5):2497–501. doi: 10.1128/mcb.17.5.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saharinen P, Vihinen M, Silvennoinen O. Autoinhibition of Jak2 tyrosine kinase is dependent on specific regions in its pseudokinase domain. Mol Biol Cell. 2003;14(4):1448–59. doi: 10.1091/mbc.E02-06-0342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gery S, Cao Q, Gueller S, Xing H, Tefferi A, Koeffler HP. Lnk inhibits myeloproliferative disorder-associated JAK2 mutant, JAK2V617F. J Leukoc Biol. 2009;85(6):957–65. doi: 10.1189/jlb.0908575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thompson BR, Mazurkiewicz-Muñoz AM, Suttles J, Carter-Su C, Bernlohr DA. Interaction of adipocyte fatty acid-binding protein (AFABP) and JAK2: AFABP/aP2 as a regulator of JAK2 signaling. J Biol Chem. 2009;284(20):13473–80. doi: 10.1074/jbc.M900075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levine RL, Pardanani A, Tefferi A, Gilliland DG. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer. 2007;7(9):673–83. doi: 10.1038/nrc2210. [DOI] [PubMed] [Google Scholar]
- 20.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, Adelsperger J, Koo S, Lee JC, Gabriel S, Mercher T, D’Andrea A, Frohling S, Dohner K, Marynen P, Vandenberghe P, Mesa RA, Tefferi A, Griffin JD, Eck MJ, Sellers WR, Meyerson M, Golub TR, Lee SJ, Gilliland DG. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–97. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 21.James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, Garcon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N, Vainchenker W. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–8. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 22.Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–90. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 23.Zhao R, Xing S, Li Z, Fu X, Li Q, Krantz SB, Zhao ZJ. Identification of an acquired JAK2 mutation in polycythemia vera. J Biol Chem. 2005;280(24):22788–92. doi: 10.1074/jbc.C500138200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N, Scott MA, Erber WN, Green AR. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–61. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 25.Kralovics R, Teo SS, Buser AS, Brutsche M, Tiedt R, Tichelli A, Passamonti F, Pietra D, Cazzola M, Skoda RC. Altered gene expression in myeloproliferative disorders correlates with activation of signaling by the V617F mutation of Jak2. Blood. 2005;106(10):3374–6. doi: 10.1182/blood-2005-05-1889. [DOI] [PubMed] [Google Scholar]
- 26.Puigdecanet E, Espinet B, Lozano JJ, Sumoy L, Bellosillo B, Arenillas L, Alvarez-Larrán A, Solé F, Serrano S, Besses C, Florensa L. Gene expression profiling distinguishes JAK2V617F-negative from JAK2V617F-positive patients in essential thrombocythemia. Leukemia. 2008;22(7):1368–76. doi: 10.1038/leu.2008.112. [DOI] [PubMed] [Google Scholar]
- 27.Levine RL. Mechanisms of mutations in myeloproliferative neoplasms. Best Pract Res Clin Haematol. 2009;22(4):489–94. doi: 10.1016/j.beha.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 28.Xing S, Wanting TH, Zhao W, Ma J, Wang S, Xu X, Li Q, Fu X, Xu M, Zhao ZJ. Transgenic expression of JAK2V617F causes myeloproliferative disorders in mice. Blood. 2008;111(10):5109–17. doi: 10.1182/blood-2007-05-091579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olcaydu D, Harutyunyan A, Jäger R, Berg T, Gisslinger B, Pabinger I, Gisslinger H, Kralovics R. A common JAK2 haplotype confers susceptibility to myeloproliferative neoplasms. Nat Genet. 2009;41(4):450–4. doi: 10.1038/ng.341. [DOI] [PubMed] [Google Scholar]
- 30.Jones AV, Chase A, Silver RT, Oscier D, Zoi K, Wang YL, Cario H, Pahl HL, Collins A, Reiter A, Grand F, Cross NC. JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nat Genet. 2009;41(4):446–9. doi: 10.1038/ng.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kilpivaara O, Mukherjee S, Schram AM, Wadleigh M, Mullally A, Ebert BL, Bass A, Marubayashi S, Heguy A, Garcia-Manero G, Kantarjian H, Offit K, Stone RM, Gilliland DG, Klein RJ, Levine RL. A germline JAK2 SNP is associated with predisposition to the development of JAK2(V617F)-positive myeloproliferative neoplasms. Nat Genet. 2009;41(4):455–9. doi: 10.1038/ng.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao L, Ma Y, Seemann J, Huang LJ. A regulating role of the JAK2 FERM domain in hyperactivation of JAK2(V617F) Biochem J. 2010;426(1):91–8. doi: 10.1042/BJ20090615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Erdmann D, Allard B, Bohn J, De Pover A, Floersheimer A, Fontana P, Gerspacher M, Hau JC, Hofmann F, Radimerski T, Wille R, Zimmermann C, Chene P. Kinetic study of human full-length wild-type JAK2 and V617F mutant proteins. Open Enzym Inhib J. 2008;1:80–84. [Google Scholar]
- 34.Gnanasambandan K, Magis A, Sayeski PP. The constitutive activation of Jak2-V617F is mediated by a π stacking mechanism involving phenylalanines 595 and 617. Biochemistry. 2010;49(46):9972–84. doi: 10.1021/bi1014858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dusa A, Mouton C, Pecquet C, Herman M, Constantinescu SN. JAK2 V617F constitutive activation requires JH2 residue F595: a pseudokinase domain target for specific inhibitors. PLoS One. 2010;5(6):e11157. doi: 10.1371/journal.pone.0011157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wernig G, Gonneville JR, Crowley BJ, Rodrigues MS, Reddy MM, Hudon HE, Walz C, Reiter A, Podar K, Royer Y, Constantinescu SN, Tomasson MH, Griffin JD, Gilliland DG, Sattler M. The Jak2V617F oncogene associated with myeloproliferative diseases requires a functional FERM domain for transformation and for expression of the Myc and Pim proto-oncogenes. Blood. 2008;111(7):3751–9. doi: 10.1182/blood-2007-07-102186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gorantla SP, Dechow TN, Grundler R, Illert AL, Zum Büschenfelde CM, Kremer M, Peschel C, Duyster J. Oncogenic JAK2V617F requires an intact SH2-like domain for constitutive activation and induction of a myeloproliferative disease in mice. Blood. 2010;116(22):4600–11. doi: 10.1182/blood-2009-07-236133. [DOI] [PubMed] [Google Scholar]
- 38.Lee TS, Ma W, Zhang X, Giles F, Kantarjian H, Albitar M. Mechanisms of constitutive activation of Janus kinase 2-V617F revealed at the atomic level through molecular dynamics simulations. Cancer. 2009;115(8):1692–700. doi: 10.1002/cncr.24183. [DOI] [PubMed] [Google Scholar]
- 39.Lee TS, Ma W, Zhang X, Kantarjian H, Albitar M. Structural effects of clinically observed mutations in JAK2 exons 13-15: comparison with V617F and exon 12 mutations. BMC Struct Biol. 2009;9:58. doi: 10.1186/1472-6807-9-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lindauer K, Loerting T, Liedl KR, Kroemer RT. Prediction of the structure of human Janus kinase 2 (JAK2) comprising the two carboxy-terminal domains reveals a mechanism for autoregulation. Protein Eng. 2001;14(1):27–37. doi: 10.1093/protein/14.1.27. [DOI] [PubMed] [Google Scholar]
- 41.Giordanetto F, Kroemer RT. Prediction of the structure of human Janus kinase 2 (JAK2) comprising JAK homology domains 1 through 7. Protein Eng. 2002;15(9):727–37. doi: 10.1093/protein/15.9.727. [DOI] [PubMed] [Google Scholar]
- 42.Fourouclas N, Li J, Gilby DC, Campbell PJ, Beer PA, Boyd EM, Goodeve AC, Bareford D, Harrison CN, Reilly JT, Green AR, Bench AJ. Methylation of the suppressor of cytokine signaling 3 gene (SOCS3) in myeloproliferative disorders. Haematologica. 2008;93(11):1635–44. doi: 10.3324/haematol.13043. [DOI] [PubMed] [Google Scholar]
- 43.Hookham MB, Elliott J, Suessmuth Y, Staerk J, Ward AC, Vainchenker W, Percy MJ, McMullin MF, Constantinescu SN, Johnston JA. The myeloproliferative disorder-associated JAK2 V617F mutant escapes negative regulation by suppressor of cytokine signaling 3. Blood. 2007;109(11):4924–9. doi: 10.1182/blood-2006-08-039735. [DOI] [PubMed] [Google Scholar]
- 44.Wang Y, Fiskus W, Chong DG, Buckley KM, Natarajan K, Rao R, Joshi A, Balusu R, Koul S, Chen J, Savoie A, Ustun C, Jillella AP, Atadja P, Levine RL, Bhalla KN. Cotreatment with panobinostat and JAK2 inhibitor TG101209 attenuates JAK2V617F levels and signaling and exerts synergistic cytotoxic effects against human myeloproliferative neoplastic cells. Blood. 2009;114(24):5024–33. doi: 10.1182/blood-2009-05-222133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lacout C, Pisani DF, Tulliez M, Gachelin FM, Vainchenker W, Villeval JL. JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood. 2006;108(5):1652–60. doi: 10.1182/blood-2006-02-002030. [DOI] [PubMed] [Google Scholar]
- 46.Li J, Spensberger D, Ahn JS, Anand S, Beer PA, Ghevaert C, Chen E, Forrai A, Scott LM, Ferreira R, Campbell PJ, Watson SP, Liu P, Erber WN, Huntly BJ, Ottersbach K, Green AR. JAK2 V617F impairs hematopoietic stem cell function in a conditional knock-in mouse model of JAK2 V617F-positive essential thrombocythemia. Blood. 2010;116(9):1528–38. doi: 10.1182/blood-2009-12-259747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, Futreal PA, Erber WN, McMullin MF, Harrison CN, Warren AJ, Gilliland DG, Lodish HF, Green AR. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356(5):459–68. doi: 10.1056/NEJMoa065202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Olcaydu D, Skoda RC, Looser R, Li S, Cazzola M, Pietra D, Passamonti F, Lippert E, Carillo S, Girodon F, Vannucchi A, Reading NS, Prchal JT, Ay C, Pabinger I, Gisslinger H, Kralovics R. The ‘GGCC’ haplotype of JAK2 confers susceptibility to JAK2 exon 12 mutation-positive polycythemia vera. Leukemia. 2009;23(10):1924–6. doi: 10.1038/leu.2009.110. [DOI] [PubMed] [Google Scholar]
- 49.Pietra D, Li S, Brisci A, Passamonti F, Rumi E, Theocharides A, Ferrari M, Gisslinger H, Kralovics R, Cremonesi L, Skoda R, Cazzola M. Somatic mutations of JAK2 exon 12 in patients with JAK2 (V617F)-negative myeloproliferative disorders. Blood. 2008;111(3):1686–9. doi: 10.1182/blood-2007-07-101576. [DOI] [PubMed] [Google Scholar]
- 50.Zou H, Yan D, Mohi G. Differential biological activity of disease-associated JAK2 mutants. FEBS Lett. 2011;585(7):1007–13. doi: 10.1016/j.febslet.2011.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ma W, Kantarjian H, Zhang X, Yeh CH, Zhang ZJ, Verstovsek S, Albitar M. Mutation profile of JAK2 transcripts in patients with chronic myeloproliferative neoplasias. J Mol Diagn. 2009;11(1):49–53. doi: 10.2353/jmoldx.2009.080114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lacronique V, Boureux A, Valle VD, Poirel H, Quang CT, Mauchauffé M, Berthou C, Lessard M, Berger R, Ghysdael J, Bernard OA. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science. 1997;278(5341):1309–12. doi: 10.1126/science.278.5341.1309. [DOI] [PubMed] [Google Scholar]
- 53.Peeters P, Raynaud SD, Cools J, Wlodarska I, Grosgeorge J, Philip P, Monpoux F, Van Rompaey L, Baens M, Van den Berghe H, Marynen P. Fusion of TEL, the ETS-variant gene 6 (ETV6): to the receptor-associated kinase JAK2 as a result of t(9;12) in a lymphoid and t(9;15;12) in a myeloid leukemia. Blood. 1997;90(7):2535–40. [PubMed] [Google Scholar]
- 54.Griesinger F, Hennig H, Hillmer F, Podleschny M, Steffens R, Pies A, Wörmann B, Haase D, Bohlander SK. A BCR-JAK2 fusion gene as the result of a t(9;22)(p24;q11.2) translocation in a patient with a clinically typical chronic myeloid leukemia. Genes Chromosome Canc. 2005;44(3):329–33. doi: 10.1002/gcc.20235. [DOI] [PubMed] [Google Scholar]
- 55.Cirmena G, Aliano S, Fugazza G, Bruzzone R, Garuti A, Bocciardi R, Bacigalupo A, Ravazzolo R, Ballestrero A, Sessarego M. A BCR-JAK2 fusion gene as the result of a t(9;22)(p24;q11) in a patient with acute myeloid leukemia. Cancer Genet Cytogenet. 2008;183(2):105–8. doi: 10.1016/j.cancergencyto.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 56.Reiter A, Walz C, Watmore A, Schoch C, Blau I, Schlegelberger B, Berger U, Telford N, Aruliah S, Yin JA, Vanstraelen D, Barker HF, Taylor PC, O’Driscoll A, Benedetti F, Rudolph C, Kolb HJ, Hochhaus A, Hehlmann R, Chase A, Cross NC. The t(8;9)(p22;p24) is a recurrent abnormality in chronic and acute leukemia that fuses PCM1 to JAK2. Cancer Res. 2005;65(7):2662–7. doi: 10.1158/0008-5472.CAN-04-4263. [DOI] [PubMed] [Google Scholar]
- 57.Murati A, Gelsi-Boyer V, Adélaïde J, Perot C, Talmant P, Giraudier S, Lodé L, Letessier A, Delaval B, Brunel V, Imbert M, Garand R, Xerri L, Birnbaum D, Mozziconacci MJ, Chaffanet M. PCM1-JAK2 fusion in myeloproliferative disorders and acute erythroid leukemia with t(8;9) translocation. Leukemia. 2005;19(9):1692–6. doi: 10.1038/sj.leu.2403879. [DOI] [PubMed] [Google Scholar]
- 58.Bousquet M, Quelen C, De Mas V, Duchayne E, Roquefeuil B, Delsol G, Laurent G, Dastugue N, Brousset P. The t(8;9)(p22;p24) translocation in atypical chronic myeloid leukaemia yields a new PCM1-JAK2 fusion gene. Oncogene. 2005;24(48):7248–52. doi: 10.1038/sj.onc.1208850. [DOI] [PubMed] [Google Scholar]
- 59.Adélaïde J, Pérot C, Gelsi-Boyer V, Pautas C, Murati A, Copie-Bergman C, Imbert M, Chaffanet M, Birnbaum D, Mozziconacci MJ. A t(8;9) translocation with PCM1-JAK2 fusion in a patient with T-cell lymphoma. Leukemia. 2006;20(3):536–7. doi: 10.1038/sj.leu.2404104. [DOI] [PubMed] [Google Scholar]
- 60.Nebral K, Denk D, Attarbaschi A, König M, Mann G, Haas OA, Strehl S. Incidence and diversity of PAX5 fusion genes in childhood acute lymphoblastic leukemia. Leukemia. 2009;23(1):134–43. doi: 10.1038/leu.2008.306. [DOI] [PubMed] [Google Scholar]
- 61.Coyaud E, Struski S, Prade N, Familiades J, Eichner R, Quelen C, Bousquet M, Mugneret F, Talmant P, Pages MP, Lefebvre C, Penther D, Lippert E, Nadal N, Taviaux S, Poppe B, Luquet I, Baranger L, Eclache V, Radford I, Barin C, Mozziconacci MJ, Lafage-Pochitaloff M, Antoine-Poirel H, Charrin C, Perot C, Terre C, Brousset P, Dastugue N, Broccardo C. Wide diversity of PAX5 alterations in B-ALL: a Groupe Francophone de Cytogenetique Hematologique study. Blood. 2010;115(15):3089–97. doi: 10.1182/blood-2009-07-234229. [DOI] [PubMed] [Google Scholar]
- 62.Dawson MA, Bannister AJ, Göttgens B, Foster SD, Bartke T, Green AR, Kouzarides T. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature. 2009;461(7265):819–22. doi: 10.1038/nature08448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bousquet M, Broccardo C, Quelen C, Meggetto F, Kuhlein E, Delsol G, Dastugue N, Brousset P. A novel PAX5-ELN fusion protein identified in B-cell acute lymphoblastic leukemia acts as a dominant negative on wild-type PAX5. Blood. 2007;109(8):3417–23. doi: 10.1182/blood-2006-05-025221. [DOI] [PubMed] [Google Scholar]
- 64.Poitras JL, Dal Cin P, Aster JC, Deangelo DJ, Morton CC. Novel SSBP2-JAK2 fusion gene resulting from a t(5;9)(q14.1;p24.1) in pre-B acute lymphocytic leukemia. Genes Chromosome Canc. 2008;47(10):884–9. doi: 10.1002/gcc.20585. [DOI] [PubMed] [Google Scholar]
- 65.Van Roosbroeck K, Cox L, Tousseyn T, Lahortiga I, Gielen O, Cauwelier B, De Paepe P, Verhoef G, Marynen P, Vandenberghe P, De Wolf-Peeters C, Cools J, Wlodarska I. JAK2 rearrangements, including the novel SEC31A-JAK2 fusion, are recurrent in classical Hodgkin lymphoma. Blood. 2011;117(15):4056–64. doi: 10.1182/blood-2010-06-291310. [DOI] [PubMed] [Google Scholar]
- 66.Najfeld V, Cozza A, Berkofsy-Fessler W, Prchal J, Scalise A. Numerical gain and structural rearrangements of JAK2, identified by FISH, characterize both JAK2617V>F-positive and -negative patients with Ph-negative MPD, myelodysplasia, and B-lymphoid neoplasms. Exp Hematol. 2007;35(11):1668–76. doi: 10.1016/j.exphem.2007.08.025. [DOI] [PubMed] [Google Scholar]
- 67.Mark HF, Sotomayor EA, Nelson M, Chaves F, Sanger WG, Kaleem Z, Caughron SK. Chronic idiopathic myelofibrosis (CIMF) resulting from a unique 3;9 translocation disrupting the janus kinase 2 (JAK2) gene. Exp Mol Pathol. 2006;81(3):217–23. doi: 10.1016/j.yexmp.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 68.Tirado CA, Chen W, Huang LJ, Laborde C, Hiemenz MC, Valdez F, Ho K, Winick N, Lou Z, Koduru P. Novel JAK2 rearrangement resulting from a t(9;22)(p24;q11.2) in B-acute lymphoblastic leukemia. Leuk Res. 2010;34(12):1674–6. doi: 10.1016/j.leukres.2010.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Patnaik MM, Knudson RA, Gangat N, Hanson CA, Pardanani A, Tefferi A, Ketterling RP. Chromosome 9p24 abnormalities: prevalence, description of novel JAK2 translocations, JAK2V617F mutation analysis and clinicopathologic correlates. Eur J Haematol. 2010;84(6):518–24. doi: 10.1111/j.1600-0609.2010.01428.x. [DOI] [PubMed] [Google Scholar]
- 70.Smith CA, Fan G. The saga of JAK2 mutations and translocations in hematologic disorders: pathogenesis, diagnostic and therapeutic prospects, and revised World Health Organization diagnostic criteria for myeloproliferative neoplasms. Hum Pathol. 2008;39(6):795–810. doi: 10.1016/j.humpath.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 71.Mercher T, Wernig G, Moore SA, Levine RL, Gu TL, Fröhling S, Cullen D, Polakiewicz RD, Bernard OA, Boggon TJ, Lee BH, Gilliland DG. JAK2T875N is a novel activating mutation that results in myeloproliferative disease with features of megakaryoblastic leukemia in a murine bone marrow transplantation model. Blood. 2006;108(8):2770–9. doi: 10.1182/blood-2006-04-014712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wadleigh M, Tefferi A. Preclinical and clinical activity of ATP mimetic JAK2 inhibitors. Clin Adv Hematol Oncol. 2010;8(8):557–63. [PubMed] [Google Scholar]
- 73.Majumer A, Sayeski PP. Tyrosine-protein kinase Jak2 Inhibitors for the treatment of myeloproliferative disorders. Drugs Fut. 2010;35(8):651–60. [Google Scholar]
- 74.Taylor SS, Knighton DR, Zheng J, Ten Eyck LF, Sowadski JM. Structural framework for the protein kinase family. Annu Rev Cell Biol. 1992;8:429–62. doi: 10.1146/annurev.cb.08.110192.002241. [DOI] [PubMed] [Google Scholar]
- 75.Hornakova T, Springuel L, Devreux J, Dusa A, Constantinescu S, Knoops L, Renauld JC. Oncogenic JAK1 and JAK2-activating mutations resistant to ATP-competitive inhibitors. Haematologica. 2011 doi: 10.3324/haematol.2010.036350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9(1):28–39. doi: 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.An X, Tiwari AK, Sun Y, Ding PR, Ashby CR, Chen ZS. BCR-ABL tyrosine kinase inhibitors in the treatment of Philadelphia chromosome positive chronic myeloid leukemia: a review. Leuk Res. 2010;34(10):1255–68. doi: 10.1016/j.leukres.2010.04.016. [DOI] [PubMed] [Google Scholar]
- 78.Deng X, Okram B, Ding Q, Zhang J, Choi Y, Adrian FJ, Wojciechowski A, Zhang G, Che J, Bursulaya B, Cowan-Jacob SW, Rummel G, Sim T, Gray NS. Expanding the diversity of allosteric bcr-abl inhibitors. J Med Chem. 2010;53(19):6934–46. doi: 10.1021/jm100555f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang J, Adrián FJ, Jahnke W, Cowan-Jacob SW, Li AG, Iacob RE, Sim T, Powers J, Dierks C, Sun F, Guo GR, Ding Q, Okram B, Choi Y, Wojciechowski A, Deng X, Liu G, Fendrich G, Strauss A, Vajpai N, Grzesiek S, Tuntland T, Liu Y, Bursulaya B, Azam M, Manley PW, Engen JR, Daley GQ, Warmuth M, Gray NS. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature. 2010;463(7280):501–6. doi: 10.1038/nature08675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chan WW, Wise SC, Kaufman MD, Ahn YM, Ensinger CL, Haack T, Hood MM, Jones J, Lord JW, Lu WP, Miller D, Patt WC, Smith BD, Petillo PA, Rutkoski TJ, Telikepalli H, Vogeti L, Yao T, Chun L, Clark R, Evangelista P, Gavrilescu LC, Lazarides K, Zaleskas VM, Stewart LJ, Van Etten RA, Flynn DL. Conformational Control Inhibition of the BCR-ABL1 Tyrosine Kinase, Including the Gatekeeper T315I Mutant, by the Switch-Control Inhibitor DCC-2036. Cancer Cell. 2011;19(4):556–68. doi: 10.1016/j.ccr.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gumireddy K, Baker SJ, Cosenza SC, John P, Kang AD, Robell KA, Reddy MV, Reddy EP. A non-ATP-competitive inhibitor of BCR-ABL overrides imatinib resistance. Proc Natl Acad Sci U S A. 2005;102(6):1992–7. doi: 10.1073/pnas.0408283102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Barnett SF, Defeo-Jones D, Fu S, Hancock PJ, Haskell KM, Jones RE, Kahana JA, Kral AM, Leander K, Lee LL, Malinowski J, McAvoy EM, Nahas DD, Robinson RG, Huber HE. Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific Akt inhibitors. Biochem J. 2005;385(Pt 2):399–408. doi: 10.1042/BJ20041140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lindsley CW, Barnett SF, Layton ME, Bilodeau MT. The PI3K/Akt pathway: recent progress in the development of ATP-competitive and allosteric Akt kinase inhibitors. Curr Cancer Drug Targets. 2008;8(1):7–18. doi: 10.2174/156800908783497096. [DOI] [PubMed] [Google Scholar]
- 84.Murphy EA, Shields DJ, Stoletov K, Dneprovskaia E, McElroy M, Greenberg JI, Lindquist J, Acevedo LM, Anand S, Majeti BK, Tsigelny I, Saldanha A, Walsh B, Hoffman RM, Bouvet M, Klemke RL, Vogt PK, Arnold L, Wrasidlo W, Cheresh DA. Disruption of angiogenesis and tumor growth with an orally active drug that stabilizes the inactive state of PDGFRbeta/B-RAF. Proc Natl Acad Sci U S A. 2010;107(9):4299–304. doi: 10.1073/pnas.0909299107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee CS, Duesbery NS. Highly selective MEK inhibitors. Curr Enz Inhib. 2010;6:146–157. [Google Scholar]
- 86.Johnson LN. Protein kinase inhibitors: contributions from structure to clinical compounds. Q Rev Biophys. 2009;42(1):1–40. doi: 10.1017/S0033583508004745. [DOI] [PubMed] [Google Scholar]
- 87.Lipka DB, Hoffmann LS, Heidel F, Markova B, Blum MC, Breitenbuecher F, Kasper S, Kindler T, Levine RL, Huber C, Fischer T. LS104, a non-ATP-competitive small-molecule inhibitor of JAK2, is potently inducing apoptosis in JAK2V617F-positive cells. Mol Cancer Ther. 2008;7(5):1176–84. doi: 10.1158/1535-7163.MCT-07-2215. [DOI] [PubMed] [Google Scholar]
- 88.Kasper S, Breitenbuecher F, Hoehn Y, Heidel F, Lipka DB, Markova B, Huber C, Kindler T, Fischer T. The kinase inhibitor LS104 induces apoptosis, enhances cytotoxic effects of chemotherapeutic drugs and is targeting the receptor tyrosine kinase FLT3 in acute myeloid leukemia. Leuk Res. 2008;32(11):1698–708. doi: 10.1016/j.leukres.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 89.Jatiani SS, Cosenza SC, Reddy MV, Ha JH, Baker SJ, Samanta AK, Olnes MJ, Pfannes L, Sloand EM, Arlinghaus RB, Reddy EP. A Non-ATP-Competitive Dual Inhibitor of JAK2 and BCR-ABL Kinases: Elucidation of a Novel Therapeutic Spectrum Based on Substrate Competitive Inhibition. Genes Cancer. 2010;1(4):331–345. doi: 10.1177/1947601910371337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Samanta AK, Chakraborty SN, Wang Y, Schlette E, Reddy EP, Arlinghaus RB. Destabilization of Bcr-Abl/Jak2 Network by a Jak2/Abl Kinase Inhibitor ON044580 Overcomes Drug Resistance in Blast Crisis Chronic Myelogenous Leukemia (CML) Genes Cancer. 2010;1(4):346–359. doi: 10.1177/1947601910372232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289(5486):1938–42. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 92.Flowers LO, Johnson HM, Mujtaba MG, Ellis MR, Haider SM, Subramaniam PS. Characterization of a peptide inhibitor of Janus kinase 2 that mimics suppressor of cytokine signaling 1 function. J Immunol. 2004;172(12):7510–8. doi: 10.4049/jimmunol.172.12.7510. [DOI] [PubMed] [Google Scholar]
- 93.Wilks AF, Harpur AG, Kurban RR, Ralph SJ, Zürcher G, Ziemiecki A. Two novel protein-tyrosine kinases, each with a second phosphotransferase-related catalytic domain, define a new class of protein kinase. Mol Cell Biol. 1991;11(4):2057–65. doi: 10.1128/mcb.11.4.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Frank SJ, Yi W, Zhao Y, Goldsmith JF, Gilliland G, Jiang J, Sakai I, Kraft AS. Regions of the JAK2 tyrosine kinase required for coupling to the growth hormone receptor. J Biol Chem. 1995;270(24):14776–85. doi: 10.1074/jbc.270.24.14776. [DOI] [PubMed] [Google Scholar]
- 95.Saharinen P, Takaluoma K, Silvennoinen O. Regulation of the Jak2 tyrosine kinase by its pseudokinase domain. Mol Cell Biol. 2000;20(10):3387–95. doi: 10.1128/mcb.20.10.3387-3395.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Saharinen P, Silvennoinen O. The pseudokinase domain is required for suppression of basal activity of Jak2 and Jak3 tyrosine kinases and for cytokine-inducible activation of signal transduction. J Biol Chem. 2002;277(49):47954–63. doi: 10.1074/jbc.M205156200. [DOI] [PubMed] [Google Scholar]
- 97.Peterson JR, Golemis EA. Autoinhibited proteins as promising drug targets. J Cell Biochem. 2004;93(1):68–73. doi: 10.1002/jcb.20184. [DOI] [PubMed] [Google Scholar]
- 98.Zhao L, Dong H, Zhang CC, Kinch L, Osawa M, Iacovino M, Grishin NV, Kyba M, Huang LJ. A JAK2 interdomain linker relays Epo receptor engagement signals to kinase activation. J Biol Chem. 2009;284(39):26988–98. doi: 10.1074/jbc.M109.011387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Deindl S, Kadlecek TA, Brdicka T, Cao X, Weiss A, Kuriyan J. Structural basis for the inhibition of tyrosine kinase activity of ZAP-70. Cell. 2007;129(4):735–46. doi: 10.1016/j.cell.2007.03.039. [DOI] [PubMed] [Google Scholar]
- 100.Zhou YJ, Chen M, Cusack NA, Kimmel LH, Magnuson KS, Boyd JG, Lin W, Roberts JL, Lengi A, Buckley RH, Geahlen RL, Candotti F, Gadina M, Changelian PS, O’Shea JJ. Unexpected effects of FERM domain mutations on catalytic activity of Jak3: structural implication for Janus kinases. Mol Cell. 2001;8(5):959–69. doi: 10.1016/s1097-2765(01)00398-7. [DOI] [PubMed] [Google Scholar]
- 101.Funakoshi-Tago M, Pelletier S, Matsuda T, Parganas E, Ihle JN. Receptor specific downregulation of cytokine signaling by autophosphorylation in the FERM domain of Jak2. EMBO J. 2006;25(20):4763–72. doi: 10.1038/sj.emboj.7601365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Robertson SA, Koleva RI, Argetsinger LS, Carter-Su C, Marto JA, Feener EP, Myers MG. Regulation of Jak2 function by phosphorylation of Tyr317 and Tyr637 during cytokine signaling. Mol Cell Biol. 2009;29(12):3367–78. doi: 10.1128/MCB.00278-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Aranaz P, Ormazábal C, Hurtado C, Erquiaga I, Calasanz MJ, García-Delgado M, Novo FJ, Vizmanos JL. A new potential oncogenic mutation in the FERM domain of JAK2 in BCR/ABL1-negative and V617F-negative chronic myeloproliferative neoplasms revealed by a comprehensive screening of 17 tyrosine kinase coding genes. Cancer Genet Cytogenet. 2010;199(1):1–8. doi: 10.1016/j.cancergencyto.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 104.Lietha D, Cai X, Ceccarelli DF, Li Y, Schaller MD, Eck MJ. Structural basis for the autoinhibition of focal adhesion kinase. Cell. 2007;129(6):1177–87. doi: 10.1016/j.cell.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Muegge I, Enyedy IJ. Virtual screening for kinase targets. Curr Med Chem. 2004;11(6):693–707. doi: 10.2174/0929867043455684. [DOI] [PubMed] [Google Scholar]
- 106.Lucet IS, Fantino E, Styles M, Bamert R, Patel O, Broughton SE, Walter M, Burns CJ, Treutlein H, Wilks AF, Rossjohn J. The structural basis of Janus kinase 2 inhibition by a potent and specific pan-Janus kinase inhibitor. Blood. 2006;107(1):176–83. doi: 10.1182/blood-2005-06-2413. [DOI] [PubMed] [Google Scholar]
- 107.Henrich S, Salo-Ahen OM, Huang B, Rippmann FF, Cruciani G, Wade RC. Computational approaches to identifying and characterizing protein binding sites for ligand design. J Mol Recognit. 2010;23(2):209–19. doi: 10.1002/jmr.984. [DOI] [PubMed] [Google Scholar]
- 108.Haan C, Behrmann I, Haan S. Perspectives for the use of structural information and chemical genetics to develop inhibitors of Janus kinases. J Cell Mol Med. 2010;14(3):504–27. doi: 10.1111/j.1582-4934.2010.01018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ghoreschi K, Laurence A, O’Shea JJ. Selectivity and therapeutic inhibition of kinases: to be or not to be? Nat Immunol. 2009;10(4):356–60. doi: 10.1038/ni.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mulligan C, Geertsma ER, Severi E, Kelly DJ, Poolman B, Thomas GH. The substrate-binding protein imposes directionality on an electrochemical sodium gradient-driven TRAP transporter. Proc Natl Acad Sci U S A. 2009;106(6):1778–83. doi: 10.1073/pnas.0809979106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bercovich D, Ganmore I, Scott LM, Wainreb G, Birger Y, Elimelech A, Shochat C, Cazzaniga G, Biondi A, Basso G, Cario G, Schrappe M, Stanulla M, Strehl S, Haas OA, Mann G, Binder V, Borkhardt A, Kempski H, Trka J, Bielorei B, Avigad S, Stark B, Smith O, Dastugue N, Bourquin JP, Tal NB, Green AR, Izraeli S. Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down’s syndrome. Lancet. 2008;372(9648):1484–92. doi: 10.1016/S0140-6736(08)61341-0. [DOI] [PubMed] [Google Scholar]
- 112.Kearney L, Gonzalez De Castro D, Yeung J, Procter J, Horsley SW, Eguchi-Ishimae M, Bateman CM, Anderson K, Chaplin T, Young BD, Harrison CJ, Kempski H, So CW, Ford AM, Greaves M. Specific JAK2 mutation (JAK2R683) and multiple gene deletions in Down syndrome acute lymphoblastic leukemia. Blood. 2009;113(3):646–8. doi: 10.1182/blood-2008-08-170928. [DOI] [PubMed] [Google Scholar]
- 113.Gaikwad A, Rye CL, Devidas M, Heerema NA, Carroll AJ, Izraeli S, Plon SE, Basso G, Pession A, Rabin KR. Prevalence and clinical correlates of JAK2 mutations in Down syndrome acute lymphoblastic leukaemia. Br J Haematol. 2009;144(6):930–2. doi: 10.1111/j.1365-2141.2008.07552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Malinge S, Ben-Abdelali R, Settegrana C, Radford-Weiss I, Debre M, Beldjord K, Macintyre EA, Villeval JL, Vainchenker W, Berger R, Bernard OA, Delabesse E, Penard-Lacronique V. Novel activating JAK2 mutation in a patient with Down syndrome and B-cell precursor acute lymphoblastic leukemia. Blood. 2007;109(5):2202–4. doi: 10.1182/blood-2006-09-045963. [DOI] [PubMed] [Google Scholar]
- 115.Schnittger S, Bacher U, Kern W, Schroder M, Haferlach T, Schoch C. Report on two novel nucleotide exchanges in the JAK2 pseudokinase domain: D620E and E627E. Leukemia. 2006;20(12):2195–7. doi: 10.1038/sj.leu.2404325. [DOI] [PubMed] [Google Scholar]
- 116.Lee JW, Kim YG, Soung YH, Han KJ, Kim SY, Rhim HS, Min WS, Nam SW, Park WS, Lee JY, Yoo NJ, Lee SH. The JAK2 V617F mutation in de novo acute myelogenous leukemias. Oncogene. 2006;25(9):1434–6. doi: 10.1038/sj.onc.1209163. [DOI] [PubMed] [Google Scholar]
- 117.Kratz CP, Boll S, Kontny U, Schrappe M, Niemeyer CM, Stanulla M. Mutational screen reveals a novel JAK2 mutation, L611S, in a child with acute lymphoblastic leukemia. Leukemia. 2006;20(2):381–3. doi: 10.1038/sj.leu.2404060. [DOI] [PubMed] [Google Scholar]
- 118.Tono C, Xu G, Toki T, Takahashi Y, Sasaki S, Terui K, Ito E. JAK2 Val617Phe activating tyrosine kinase mutation in juvenile myelomonocytic leukemia. Leukemia. 2005;19(10):1843–4. doi: 10.1038/sj.leu.2403903. [DOI] [PubMed] [Google Scholar]
- 119.Remacha AF, Nomdedeu JF, Puget G, Estivill C, Sarda MP, Canals C, Aventin A. Occurrence of the JAK2 V617F mutation in the WHO provisional entity: myelodysplastic/myeloproliferative disease, unclassifiable-refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Haematologica. 2006;91(5):719–20. [PubMed] [Google Scholar]
- 120.Szpurka H, Tiu R, Murugesan G, Aboudola S, Hsi ED, Theil KS, Sekeres MA, Maciejewski JP. Refractory anemia with ringed sideroblasts associated with marked thrombocytosis (RARS-T), another myeloproliferative condition characterized by JAK2 V617F mutation. Blood. 2006;108(7):2173–81. doi: 10.1182/blood-2006-02-005751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Renneville A, Quesnel B, Charpentier A, Terriou L, Crinquette A, Lai JL, Cossement C, Lionne-Huyghe P, Rose C, Bauters F, Preudhomme C. High occurrence of JAK2 V617 mutation in refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Leukemia. 2006;20(11):2067–70. doi: 10.1038/sj.leu.2404405. [DOI] [PubMed] [Google Scholar]
- 122.Pich A, Riera L, Sismondi F, Godio L, Davico Bonino L, Marmont F, Francia di Celle P. JAK2V617F activating mutation is associated with the myeloproliferative type of chronic myelomonocytic leukaemia. J Clin Pathol. 2009;62(9):798–801. doi: 10.1136/jcp.2009.065904. [DOI] [PubMed] [Google Scholar]
- 123.Grunebach F, Bross-Bach U, Kanz L, Brossart P. Detection of a new JAK2 D620E mutation in addition to V617F in a patient with polycythemia vera. Leukemia. 2006;20(12):2210–1. doi: 10.1038/sj.leu.2404419. [DOI] [PubMed] [Google Scholar]
- 124.Zhang SJ, Li JY, Li WD, Song JH, Xu W, Qiu HX. The investigation of JAK2 mutation in Chinese myeloproliferative diseases-identification of a novel C616Y point mutation in a PV patient. Intl J Lab Hematol. 2007;29(1):71–2. doi: 10.1111/j.1365-2257.2006.00864.x. [DOI] [PubMed] [Google Scholar]
- 125.Yoo JH, Park TS, Maeng HY, Sun YK, Kim YA, Kie JH, Cho EH, Song J, Lee KA, Suh B, Choi JR. JAK2 V617F/C618R mutation in a patient with polycythemia vera: a case study and review of the literature. Cancer Genet Cytogenet. 2009;189(1):43–7. doi: 10.1016/j.cancergencyto.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 126.Ma W, Kantarjian H, Zhang X, Wang X, Zhang Z, Yeh CH, O’Brien S, Giles F, Bruey JM, Albitar M. JAK2 exon 14 deletion in patients with chronic myeloproliferative neoplasms. PLoS One. 2010;5(8):e12165. doi: 10.1371/journal.pone.0012165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sayyah J, Magis A, Ostrov DA, Allan RW, Braylan RC, Sayeski PP. Z3, a novel Jak2 tyrosine kinase small-molecule inhibitor that suppresses Jak2-mediated pathologic cell growth. Mol Cancer Ther. 2008;7(8):2308–18. doi: 10.1158/1535-7163.MCT-08-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Passamonti F, Elena C, Schnittger S, Skoda RC, Green AR, Girodon F, Kiladjian JJ, McMullin MF, Ruggeri M, Besses C, Vannucchi AM, Lippert E, Gisslinger H, Rumi E, Lehmann T, Ortmann CA, Pietra D, Pascutto C, Haferlach T, Cazzola M. Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood. 2011;117(10):2813–6. doi: 10.1182/blood-2010-11-316810. [DOI] [PubMed] [Google Scholar]
- 129.Schnittger S, Bacher U, Haferlach C, Geer T, Muller P, Mittermuller J, Petrides P, Schlag R, Sandner R, Selbach J, Slawik HR, Tessen HW, Wehmeyer J, Kern W, Haferlach T. Detection of JAK2 exon 12 mutations in 15 patients with JAK2V617F negative polycythemia vera. Haematologica. 2009;94(3):414–8. doi: 10.3324/haematol.13223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Percy MJ, Scott LM, Erber WN, Harrison CN, Reilly JT, Jones FG, Green AR, McMullin MF. The frequency of JAK2 exon 12 mutations in idiopathic erythrocytosis patients with low serum erythropoietin levels. Haematologica. 2007;92(12):1607–14. doi: 10.3324/haematol.11643. [DOI] [PubMed] [Google Scholar]
- 131.Williams DM, Kim AH, Rogers O, Spivak JL, Moliterno AR. Phenotypic variations and new mutations in JAK2 V617F-negative polycythemia vera, erythrocytosis, and idiopathic myelofibrosis. Exp Hematol. 2007;35(11):1641–6. doi: 10.1016/j.exphem.2007.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Butcher CM, Hahn U, To LB, Gecz J, Wilkins EJ, Scott HS, Bardy PG, D’Andrea RJ. Two novel JAK2 exon 12 mutations in JAK2V617F-negative polycythaemia vera patients. Leukemia. 2008;22(4):870–3. doi: 10.1038/sj.leu.2404971. [DOI] [PubMed] [Google Scholar]