

Abstract
Vascular remodeling is a complex process critical to development of the mature vascular system. Astrocytes are known to be indispensable for initial formation of the retinal vasculature; our studies with the Nuc1 rat provide novel evidence that these cells are also essential in the retinal vascular remodeling process. Nuc1 is a spontaneous mutation in the Sprague–Dawley rat originally characterized by nuclear cataracts in the heterozygote and microphthalmia in the homozygote. We report here that the Nuc1 allele results from mutation of the βA3/A1-crystallin gene, which in the neural retina is expressed only in astrocytes. We demonstrate striking structural abnormalities in Nuc1 astrocytes with profound effects on the organization of intermediate filaments. While vessels form in the Nuc1 retina, the subsequent remodeling process required to provide a mature vascular network is deficient. Our data implicate βA3/A1-crystallin as an important regulatory factor mediating vascular patterning and remodeling in the retina.
Keywords: βA3/A1-crystallin, Astrocytes, Retinal vasculature, Remodeling, Intermediate filaments, Rat spontaneous mutation
Introduction
We previously described a naturally occurring mutation (Nuc1) in the Sprague–Dawley rat with a novel and unusual eye phenotype (Sinha et al., 2005; Hose et al., 2005; Zhang et al., 2005; Gehlbach et al., 2006). Nuc1 is inherited as a single Mendelian locus with viable homozygotes and an intermediate phenotype in heterozygotes. We now report that the mutation causing Nuc1 is a 27 base pair insertion in exon 6 of the βA3/A1-crystallin gene on rat chromosome 10.
Crystallins are highly abundant structural proteins of the lens of the eye that contribute to transparency and refractive power. Three major families of crystallins, α, β, and γ are expressed in all vertebrate lenses. While the β- and γ-crystallins were earlier considered to be unrelated, it is now known that they actually are evolutionarily and structurally related members of a single superfamily, the β,γ-crystallin superfamily. Once believed to be lens-specific, many, if not all, crystallins are present in other tissues and it is now believed that crystallins evolved for specialized function in the lens from pre-existing proteins with other functions. The evolutionary origins and non-lens functions of α–crystallin (a small heat shock protein) and of many “taxon-specific” crystallins (various enzymes) are known (Piatigorsky, 2007); however, such information is largely lacking for β,γ-crystallins. They are evolutionarily related to certain microorganism stress proteins and to vertebrate proteins associated with processes of cellular differentiation and morphological change (Wistow, 1995). Various β,γ-crystallins are present in low abundance in tissues other than the lens, most prominently in the retina (Andley, 2007) where their expression is reported to be upregulated following various injuries or stresses (Sakaguchi et al., 2003; Vazquez-Chona et al., 2004; Crabb et al., 2002). βB2-crystallin has recently been reported to be secreted by the retina in culture where it promotes the regrowth of retinal ganglion cell axons (Liedtke et al., 2007). βB2-crystallin is also expressed in sperm and its mutation in the Philly mouse is reported to cause reduced fertility (DuPrey et al., 2007). While these studies and others point to important “non-crystallin” functions for β-crystallins, the underlying mechanisms remain unknown.
The βA3/A1-crystallin gene is one of 6 β-crystallin genes present in many vertebrates. It is unique among them in generating two polypeptides (βA3 and βA1) by utilizing two separate start codons. Both polypeptides are affected by the Nuc1 mutation. Mutations in βA3/A1-crystallin are well known to cause cataract in both humans and mice (Kannabiran et al., 1998; Bateman et al., 2000; Qi et al., 2004; Ferrini et al., 2004; Reddy et al., 2004; Graw et al., 1999). In Nuc1 homozygotes, however, in addition to cataracts, severe retinal and other ocular abnormalities are apparent (Sinha et al., 2005; Hose et al., 2005; Zhang et al., 2005; Gehlbach et al., 2006). These rats manifest persistence of the fetal intraocular vessels even after development of the retinal vessels. Our recent studies indicate that the mutation also affects both the initial patterning of retinal vessels during development, as well as the subsequent remodeling which produces the mature vascular architecture.
In this report, we show that in the neural retina, the βA3/A1 polypeptides are present only in astrocytes and that mutation of these proteins disrupts the normal structure and function of astrocytes, leading to abnormalities in the development and maturation of the retinal vasculature. The retinal changes that we report here appear to be recessive (in contrast to the Nuc1 cataracts which are semi-dominant) and we believe that this phenotype reflects the loss of the “original” or “non-crystallin” function of βA3/A1-crystallin. Therefore, the Nuc1 mutation represents a valuable tool to further our understanding of blood vessel development and maturation in the developing eye and the regulatory role of astrocytes in the process. Moreover, the possibility that βA3/A1-crystallin may have a role in vascular remodeling is important, because such remodeling is fundamental to normal ocular development and to the pathogenesis of numerous diseases.
Results and discussion
To localize the Nuc1 mutation, a genome-wide linkage analysis screen using 207 markers was undertaken on backcross progeny from Lewis/Sprague–Dawley hybrids. With an initial group of 20 animals (10 wild-type and 10 Nuc1 heterozygotes), the gene was localized to rat chromosome 10p22-26. Genotyping of 80 additional rats narrowed the interval to a 5.2 Mb region between markers D10Rat195 and D10Rat29. This interval contained 49 genes identified through the reference sequence annotation database for the rat genome assembly, which we augmented by alignment with the mouse and human genomes. The βA3/A1-crystallin gene was a clear candidate for Nuc1 (see below), and sequencing of the Nuc1 allele revealed a 27 base pair insertion in exon 6 (Fig. 1).
Fig. 1.
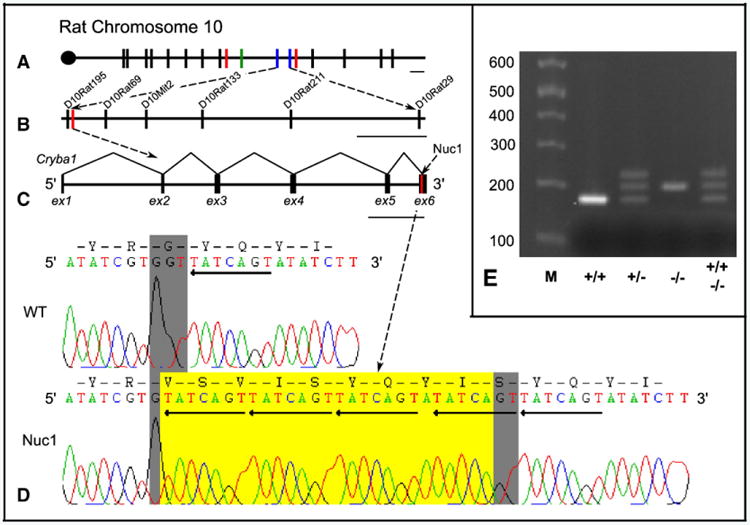
The positional cloning of the βA3/A1-crystallin gene to rat chromosome 10. Markers used are designated by bisecting lines and are drawn to scale by physical distance. Panel A shows the initial Nuc1 interval bounded by red lines (D10Rat125 and D10Rat98), D10Rat32 (green line) segregated perfectly with the disease and gave a LOD score of 6.02. The final Nuc1 interval is bounded by blue lines (D10Rat195 and D10Rat29) and this region is magnified in panel B. Scale bar=5 Mb. Panel B shows magnification of final linkage interval and markers, in which position of βA3/A1 gene (Cryba1) is marked with a red line. Scale bar=1 Mb. Panel C shows the genomic structure of Cryba1 with the site of the Nuc1 mutation highlighted in red. Scale bar=1 kb. Panel D displays the sequence at the site of mutation in +/+ and Nuc1/Nuc1 animals. Chromatograms display the sense strand of genomic amplimers. Grey highlight indicates the highly conserved glycine codon in normal Cryba1. Sequence inserted by the Nuc1 mutation is highlighted in yellow and the TGACTAT repeats are marked by reverse arrows. Agarose gel analysis of PCR products from the region of Cryba1 including the 27 base insertion is shown in panel E. Expected bands are observed for wild-type and homozygote. The 3rd (heavier) band seen in the heterozygote is the result of heteroduplex formation since it is also present in the wild-type plus homozygote heteroduplex control sample.
βA3/A1-crystallin belongs to the β,γ-crystallin superfamily of proteins, members of which are abundantly expressed in the ocular lens of all vertebrates. β,γ-crystallins are also expressed at low levels in other tissues, where their functions are unknown. The 27 base pair insertion is composed of near-perfect tandem repeats of a 7 base pair sequence, TGACTAT. This in-frame insertion, results in the loss of a universally conserved glycine residue in exon 6 and its replacement with 10 new amino acids. There is one known mouse mutation in βA3/A1-crystallin, a point mutation in exon 6, which causes replacement of a tryptophan residue by arginine. This mutation results in a semi-dominant nuclear cataract phenotype (Graw et al., 1999). There are five reports of human mutations in βA3/A1-crystallin, all with dominant or semi-dominant cataract phenotypes (Kannabiran et al., 1998; Bateman et al., 2000; Qi et al., 2004; Ferrini et al., 2004; Reddy et al., 2004). Two of the mutations are at the five prime (donor) splice site of intron 3. The other three cause in-frame deletion of the highly conserved glycine 91 (exon 4). No retinal abnormalities have been reported for any of these human mutations; however, the patients studied were heterozygotes, whereas retinal abnormalities are apparent only in the homozygotes of Nuc1 rats. Interestingly, in a family with dominant cataracts associated with a βB2-crystallin mutation (Litt et al., 1997), there was a single individual who was homozygous for the mutation, and this patient lost all visual perception by adolescence due to microphthalmia and severe retinal abnormalities (I.H. Maumenee, personal communication).
Our previous study demonstrated expression of βA3/A1-crystallin mRNA in the retina (Zhang et al., 2005). To determine its cellular localization within the retina, laser-capture microdissection was employed. As measured by quantitative RT-PCR analysis of various dissected retinal tissue layers, βA3/A1 mRNA was detected only in the innermost retina, in the retinal ganglion cell/nerve fiber layer (Fig. 2, top panel). Immunohistochemistry confirmed that βA3/A1-crystallin protein is present only in this region and that it co-localizes strongly with glial fibrillary acidic protein (GFAP), a marker for astrocytes (Fig. 2, middle panel). Moreover, in cultured cerebral astrocytes from both wild-type and Nuc1 homozygous rats, βA3/A1-crystallin appears to be present in the cell nucleus as well as in the cytoplasm (Fig. 2, bottom panel). The astrocytes from Nuc1 homozygous rats are morphologically abnormal, with striking differences in the organization and expression of intermediate filament (IF) arrays when compared to normal astrocytes (Fig. 2, bottom panel).
Fig. 2.
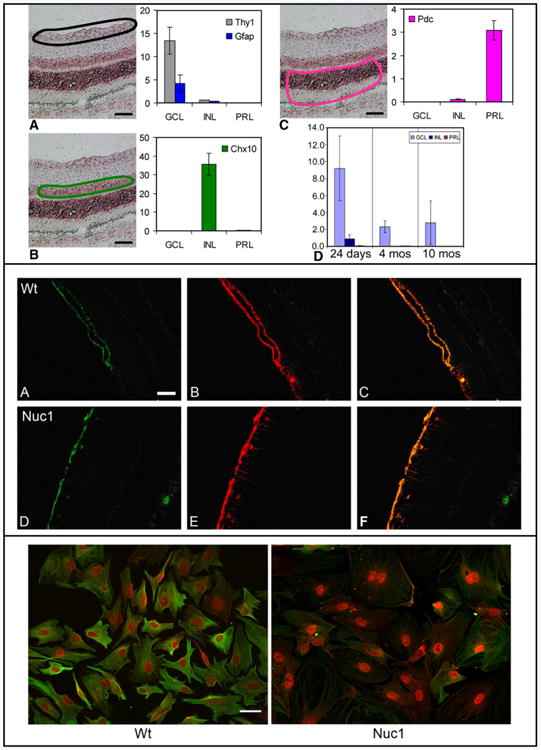
Localization of βA3/A1-crystallin in the retina. Top panel: Laser-capture microdissection studies. Panels A–C respectively show the location of samples taken from the ganglion cell layer (GCL), outlined in black, the inner nuclear layer (INL) outlined in green, and the photoreceptor layer (PRL) outlined in pink. Scale bar=50 μm. For each region quantitative RT-PCR demonstrates the purity of the dissected tissue by measuring specific markers for each cell type. In panel D, quantitative RT-PCR shows the relative expression of βA3/A1-crystallin in the 3 retinal regions of wild-type Sprague–Dawley rat at 24 days, 4 months, and 10 months of age. Data shown as mean±SD. Middle panel: Confocal microscopy showing GFAP positive staining at the internal limiting membrane of 4 week old wild-type (A) and Nuc1 (D) retinas. Panels B and E show βA3/A1-crystallin staining on the same section, while panels C and F are merged images showing co-localization of GFAP and βA3/A1-crystallin in the wild-type and Nuc1 retinas, respectively. The fluorescence is enhanced because of the reduced amount of GFAP present in the mutant cells. Scale bar=50 μm. Bottom panel: Wild-type and Nuc1 astrocytes cultured from brain of neonatal rats. Note βA3/A1-crystallin (red) is expressed strongly in the cell nuclei of both normal and mutant astrocytes but also in the cytoplasm. The astrocytes from Nuc1 homozygous rats show striking changes in cell morphology; they are larger in size with disrupted processes and reduced expression of GFAP (green). Scale bar=40 μm.
Astrocytes are one of the two types of macroglial cells found in mammalian retinas. Unlike Muller cells, which span the entire thickness of the retina and are present in all mammals, astrocytes are mainly confined to the retina's inner surface (Ling et al., 1989; Watanabe and Raff, 1988) and are closely associated with retinal blood vessels. Avascular retinas contain no astrocytes (Stone and Dreher, 1987; Schnitzer, 1988). Retinas that are diffusely vascularized contain diffusely distributed astrocytes (Stone and Dreher, 1987; Schnitzer, 1988), and those that are vascularized in a restricted region contain astrocytes only in that region (Stone and Dreher, 1987; Schnitzer, 1988). In fetal humans, astrocyte differentiation occurs in association with fetal retinal vasculature (Chan-Ling et al., 2004). Astrocytes originate outside of the retina, arising from the neuroepithelial cells that form the optic stalk, the primordium of the optic nerve (Small et al., 1987). They migrate from the optic nerve head into the inner retina. Astrocytes first appear in the developing rat optic nerve at E (embryonic day) 16 and increase in number until 6 weeks after birth (Miller et al., 1985). They form a corona of processes around the optic nerve head at E18, cover approximately 35% of the retina at birth, and reach the periphery of the retina by P (postnatal day) 8 (Ling et al., 1989).
In Nuc1 homozygotes, the astrocytes begin to exhibit an abnormal pattern and organization during postnatal development of the retina. At 6 days after birth (P6), obvious abnormalities can be seen in the Nuc1 astrocytes. Confocal microscopy of retinal flatmounts double-labeled with GFAP and Isolectin B4, which labels blood vessels in the rat (Ashwell et al., 1989), clearly showed changes in process length in Nuc1 astrocytes. The compact stellate astrocyte structure normally seen in the wild-type was missing in the Nuc1 retina. In the normal P6 retina, astrocytes have made a highly structured, honeycomb-like template on which blood vessels will begin to develop (Fig. 3A). In the Nuc1 retina, this template is less dense and irregular in pattern (Fig. 3B). At 10 weeks (Fig. 3C), when the astrocytes in the wild-type have covered the retina and the superficial vascular plexus has formed, an abnormal network of astrocytes is evident in Nuc1 mutants (Fig. 3D). The normal stellate appearance of astrocytes with delicate processes (Fig. 3C) was not observed in the Nuc1 retina. Instead, the astrocytes had short, thick processes, and their number was reduced (Fig. 3D).
Fig. 3.
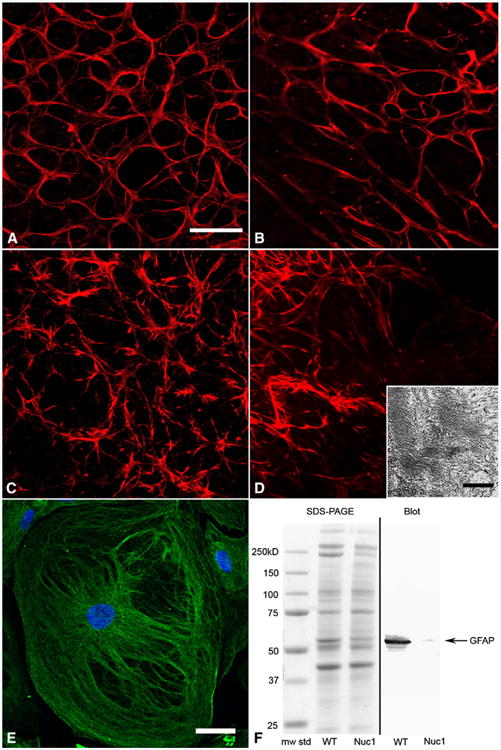
βA3/A1-crystallin in wild-type and Nuc1 astrocytes. Confocal microscopy of retinal flat mounts from wild-type 6 day (A), Nuc1 6 day (B), wild-type 10 week (C), and Nuc1 10 week (D) rats. Astrocytes are labeled with GFAP (red). Nuc1 astrocytes at P6 lack the compact stellate structure and the honeycomb-like template present in the wild-type retina. By 10 weeks the network of astrocytes in the Nuc1 retina has become more abnormal with short, thick processes. Scale bar=20 μm. Inset in panel D: transmission electron microscopy of an astrocyte from a fixed Nuc1 retina at 3 months of age showing disorganized arrays of intermediate filaments (scale bar=250 nm). Panel E shows a cultured brain astrocyte from a neonatal Nuc1 rat pup. Cells are very large with disorganized intermediate filaments labeled with GFAP (green). The fluorescence is enhanced because of the reduced amount of GFAP present in the mutant cells. Nuclei are stained with DAPI (blue), scale bar=20 μm. Panel F shows an analysis of protein extracts from astrocyte cell cultures. Left side: Coomassie blue-stained SDS-PAGE with molecular-weight markers, extract from wild-type astrocytes and extract from Nuc1 astrocytes, respectively. Right side: Western blotting with anti-GFAP (DAKO). A marked decrease in immunoreactivity is evident in the Nuc1 extract consistent with the immunohistochemistry data shown in Fig. 2 (bottom panel).
Thus, while astrocytes in Nuc1 homozygous rats do migrate from the optic nerve head into the retina, their number and morphology are clearly affected by the mutation of βA3/A1-crystallin. In addition, transmission electron microscopy (TEM) of retinal astrocytes from wild-type and Nuc1 homozygous rats shows that in the mutants the intermediate filament (IF) arrays have abnormal size and organization (Fig. 3D, inset). The cultured astrocytes from the Nuc1 homozygous cerebral cortex also show striking changes in organization and ability to assemble an IF network (Fig. 3E) similar to IFs in the retinal astrocytes as indicated by TEM.
Intermediate filaments are thought to be instrumental in the maintenance of mechanical integrity of cells and tissues; however, their specific function in astrocytes remains obscure. Astrocytes produce three differentially regulated IF proteins, GFAP, vimentin, and nestin (Chu et al., 2001). Vimentin and nestin are characteristic of immature astrocytes. Vimentin and GFAP are expressed in mature astrocytes. Nestin cannot form IFs on its own, but vimentin may form IFs with either nestin or GFAP as obligatory partners. GFAP is the only IF protein that may form filaments on its own (Eliasson et al., 1999). The lack of expression of either GFAP or vimentin inhibits the mean cell speed of astrocytes, whereas the persistence in direction remains unchanged (Lepekhin et al., 2001). Removal of both of these proteins, leading to a complete absence of IFs in astrocytes, decreases the mean cell speed further, without any effect on persistence time (Lepekhin et al., 2001). These studies indicate that IFs are an integral part of the cell motility machinery in astrocytes.
Interestingly, the level of GFAP protein in Nuc1 astrocytes is greatly reduced (Figs. 3F and 2, bottom panel), showing that the mutation has a profound effect on IFs. In contrast, vimentin protein expression remains unchanged (data not shown). This is interesting because we previously showed that filensin, a lens-specific IF protein, is markedly downregulated in the Nuc1 lens (Sinha et al., 2005). Moreover, as in astrocytes, lens fiber cells do not orient properly and are also structurally abnormal (Sinha et al., 2005).
Moreover, confocal microscopy shows that aberrant astrocyte development in the Nuc1 retina is associated with abnormal retinal vascular patterning, perhaps due to weakened astrocyte–blood vessel interactions. During normal development of the superficial vascular plexus, retinal vessels are intimately associated with astrocytes (Dorrell and Friedlander, 2006) (Fig. 4A). The capillaries follow the same honeycomb pattern seen in the astrocyte template (Fig. 3A). In the Nuc1 homozygous retina at P6, the capillary plexus is less dense, and the honeycomb pattern is stretched and irregular (Fig. 4B). The vascular remodeling that normally produces the capillary free zone (De Schaepdrijver et al., 1995; Ashton, 1970) and removes excess endothelial cells does not occur in the Nuc1 retinas (Fig. 4B, inset). By 10 weeks of age, the vasculature of a normal retina has been remodeled, and the thin processes of the stellate astrocytes enwrap the blood vessels (Fig. 4C). In Nuc1 10 week retinas, however, the number of astrocytes and their processes are greatly reduced. Excess endothelial cells remain in the Nuc1 retina, suggesting a deficiency in the apoptotic process normally associated with vascular remodeling.
Fig. 4.
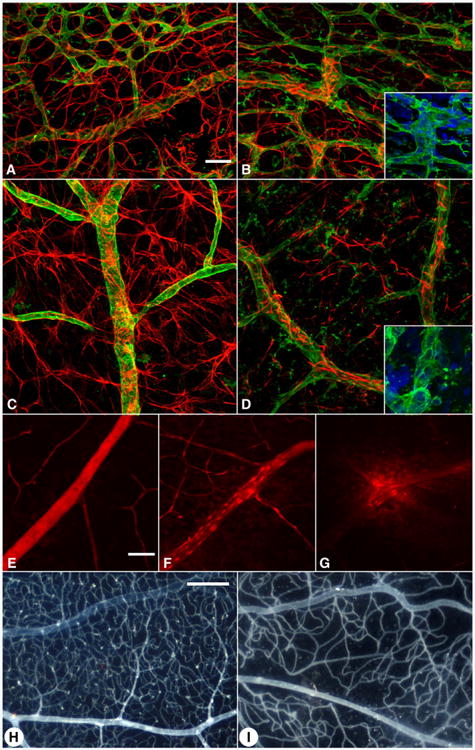
Retinal blood vessel abnormalities in Nuc1 homozygous rats. GFAP (red) and GSA lectin blood vessel staining (green) in wild-type (A, C) and Nuc1 retinas (B, D) at postnatal day 6 (A, B) and 10 weeks of age (C, D). In the wild-type (A), there is a dense capillary plexus (top) and a capillary-free zone around arteries. The capillary-free zones are missing in Nuc1 (B) and the capillary plexus pattern is abnormal. By 10 weeks, the wild-type retina is remodeled and the astrocyte processes are extensive and enwrapping the blood vessels (C). In Nuc1, the number of astrocytes and their processes are greatly reduced (D) suggesting fewer interactions between astrocytes and endothelial cells. In the insets of panels B and D, it is apparent that the Nuc1 blood vessels have too many endothelial cells suggesting that remodeling during maturation of the retinal vasculature has not occurred. Evans blue injections at 4 weeks of age demonstrate that the wild-type retinal vasculature does not leak (E) but the Nuc1 retinal blood vessels do leak (F and G at higher magnification). Using the ADPase flat-embedding technique, the normal vascular pattern is apparent in the 10 month old wild-type (H) while the Nuc1 vascular pattern is sparse, appears to be lacking a deep capillary plexus, and has an abnormal pattern (I). Scale bars=20 μm (A–G) and 0.25 mm (H–I).
The intimate relationship between blood vessels and astrocytes is critical for maintaining a tight blood-retinal barrier (Fig. 4E) (Jiang et al., 1995). When this tight relationship is disrupted as in Nuc1, vascular permeability in the retina increases, as exhibited by Evan's blue leakage in Nuc1 rat retinas (Figs. 4F–G). By 10 months of age, the number of retinal vessels in Nuc1 rats is greatly reduced compared to wild-type, and the secondary, or deep, capillary net is sparse (Figs. 4H–I).
Thus, our studies provide evidence that βA3/A1-crystallin affects both the initial patterning of retinal vessels during development, as well as the subsequent remodeling which produces the mature vascular architecture. The mutant retina appears to be fully vascularized by 3 weeks of age; however, gross abnormalities in the patterning of the vessels, blood flow, and increased vessel leakage (Figs. 4F–G) are found at that time.
In the Nuc1 mutant, genetic impairment of function in the lens is semi-dominant, while impairment of the glial function of βA3/A1-crystallin appears fully recessive. In the heterozygote, dense nuclear cataracts are present at birth, presumably because 50% abnormal protein is able to cause protein aggregation which causes a cataract of the lens. In contrast, heterozygotes do not have obvious retinal abnormalities; these become apparent only in the homozygotes. This suggests that these effects result from total loss of the original or “non-crystallin” function of βA3/A1-crystallin in the astrocyte. Our previous report demonstrating abnormalities in the maturation and orientation of differentiating lens fibers in Nuc1 homozygotes, but not heterozygotes, suggests that this “non-crystallin” regulatory function of βA3/A1-crystallin is also necessary in the lens. However, the nature of this biochemical function is totally unknown.
While βA3/A1-crystallin is entirely cytoplasmic in lens fiber cells, the protein is largely nuclear in astrocytes. This raises the possibility that the glial function may involve regulation of mRNA expression and that cytoskeletal effects in Nuc1 could result from altered gene expression. Alternatively, βA3/A1-crystallin protein might also affect intermediate filaments through direct interaction, a function which could be lost in the mutant protein. Molecular modeling of the mutant βA3/A1-crystallin protein suggests that the insertion creates a loop at the C-terminus that protrudes out of the normal structure (Figs. 5A–D). Application of molecular dynamics to the model of the mutant C-terminal domain (Fig. 5D) in water predicts a stable and compact structure with the addition of 7 hydrophobic residues increasing the hydrophobic potential at the surface of the mutant C-terminal domain (Fig. 5B). This loop would be expected to sterically affect protein-protein interactions at this site. Consistent with this hypothesis, gel chromatography studies show aggregation of the mutant βA3/A1-crystallin in the lens (Fig. 5E). Specifically, βA3/A1-crystallin from Nuc1 homozygote lenses was confined to the void volume peak and was not seen in the β-crystallin peaks, as in wild-type lens extracts (Fig. 5E, inset).
Fig. 5.
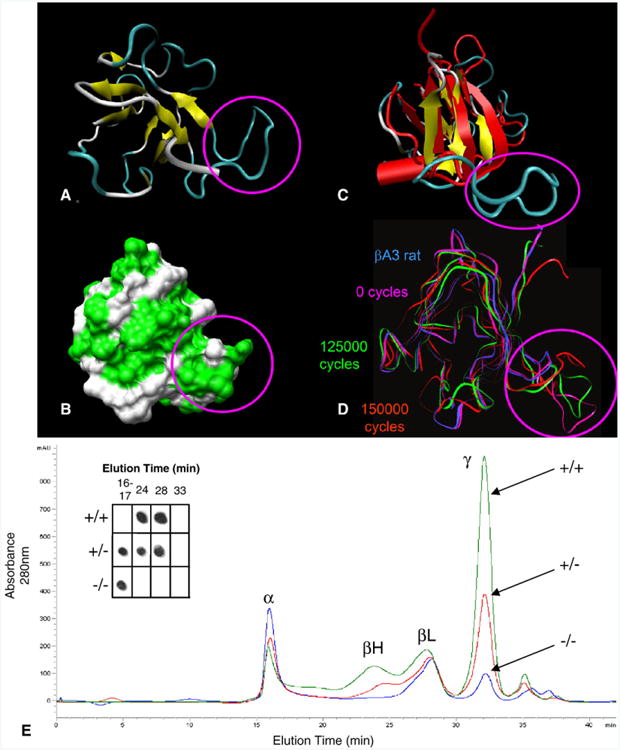
Molecular modeling and gel-chromatography patterns of normal and mutant βA3/A1-crystallin protein. Top panel: The 3D ribbon structure (A) and the surface area (B) of the βA3/A1-crystallin mutant are shown with the location of the insertion highlighted by the magenta circle. In panel B, hydrophobic residues at the protein surface are labeled with bright green. In panel C, the wild-type (red) and mutant (yellow and blue) βA3/A1-crystallin C-terminals are superimposed clearly displaying the extended loop created by the insertion (circled in magenta). Arrows represent β-sheets and the red cylinder represents an α-helix. Molecular dynamics simulations performed on the C-terminal domain of the βA3/A1-crystallin mutant are shown in panel D. Ribbon trajectories represent the C-terminal domain of the wild-type βA3/A1-crystallin (blue) and the mutant βA3/A1-crystallin at 0 (magenta), 125,000 (green), and 150,000 (red) 1-fs cycles. Bottom panel: The effects of the βA3/A1-crystallin mutation on the composition of soluble crystallins in the lens. The elution patterns show trends in the distribution of soluble proteins (+/+ = green, +/− =red, −/− =blue). The pattern from normal lens has four major peaks: a void volume peak which contains α-crystallin plus any heavy molecular weight protein aggregates, two peaks of β-crystallins of different size which have been described in many species (βH and βL), and γ-crystallin which is the dominant peak in the rat lens. In the cataract lenses, the relative proportion of the void fraction increases from about 10% in the wild-type lens to 18% in the heterozygote lens and to 32% in the homozygote. γ-crystallin, which constitutes 47% of the soluble protein in the wild-type lens, is reduced to 37% in the heterozygote and to 16% in the homozygote. As a percentage of the soluble protein, β-crystallin is quite constant in all phenotypes; however, there is a dramatic shift to the lower molecular weight species (dimers) in the heterozygote and especially the homozygous lens. The inset shows a dot blot in which an antibody specific for βA3/A1-crystallin (reactive with both normal and mutant forms) is used to localize that polypeptide in the chromatographically separated peaks. In the wild-type lens, the protein is present only in the β-crystallin peaks as expected (fractions 24 and 28). In the heterozygote lens, reactivity is present in those fractions, but also in the void peak. In the homozygote, reactivity is essentially limited to the void peak. This indicates that the mutant protein aggregates and does not participate in the formation of stable oligomers as does normal βA3/A1-crystallin.
Interestingly, our yeast two-hybrid studies identified vimentin as one of the proteins with the greatest affinity for β,γ-crystallins (data not shown). It is possible that GFAP filament organization in Nuc1 astrocytes is affected due to disruption in the association between vimentin and βA3/A1-crystallin, since it has been shown that the GFAP network is disrupted in vimentin knockout mice (Galou et al., 1996). Our microarray studies show that GFAP mRNA actually increases (unpublished observation) in Nuc1 homozygotes. This suggests that the low level of GFAP protein in Nuc1 could be due to decreased translation or increased protein degradation.
Effects of the mutation on IFs may provide insight into how Nuc1 disrupts the normal cellular structure and function of retinal astrocytes. IFs are now known to be critical elements in a variety of regulatory processes by serving as scaffolds to sequester or organize molecules of signaling pathways (Pallari and Eriksson, 2006). One major regulatory function in which IFs have been implicated is the control of cell death via apoptosis. We have suggested previously that impaired apoptosis may be the key factor in the Nuc1 phenotype. We based this on several observations. First, in Nuc1 homozygotes the mutation selectively arrests the loss of nuclei, but not other organelles, from lens fiber cells (Sinha et al., 2005). During normal differentiation, lens fiber cells lose their nuclei and organelles, a process essential to development of lens transparency. We believe that the remodeling of lens fibers during the “apoptotic-like” lens denucleation process is affected in Nuc1 (Sinha et al., 2005). Second, the mutation results in decreased apoptosis in the developing retina (Sinha et al., 2005). Remodeling is critical to retinal development and maturation, by generating the different cell types in proper ratios and positions, which then migrate to correct layers before finally forming synaptic connections. Third, we have recently shown persistence of the fetal vasculature in this model (Zhang et al., 2005). Regression of the fetal vasculature is an essential element of eye development, apoptosis is vital to this process (Lobov et al., 2005).
Our data suggest, for the first time, that βA3/A1-crystallin is essential for the normal morphology of retinal astrocytes and has a critical role in mediating vascular remodeling in the developing eye of the rat. In addition, the involvement of βA3/A1-crystallin in normal astrocyte function and subsequent retinal vascular development suggests a previously unknown non-lens function for β/γ crystallins. Experiments under way in our laboratory may help us to better understand the non-lens function by using the Cre-loxP system to delete the βA3/A1-crystallin gene selectively from retinal astrocytes early in development.
Experimental methods
Animals
The colony of Sprague–Dawley rats used in this study originated from Taconic Farms (Hamilton, NY) and has been maintained by the investigators at the National Eye Institute (Bethesda, MD) and more recently at Spring Valley Laboratories (Woodbine, MD). Lewis rats were obtained directly from Harlan (Indianapolis, IN). All procedures were approved by the Animal Care and Use Committees of the National Eye Institute and Johns Hopkins University and were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (National Academy Press). Invasive procedures were not used in this study.
Genotyping
The Sprague–Dawley (SD) and Lewis (Lew) rat strains are known to differ extensively across the genome at many polymorphic loci and so Lew/SD hybrids were created by crossing Nuc1/Nuc1 SD rats with +/+ Lew rats. The offspring from these matings were +/Nuc1 and designated the F1 generation. These heterozygotes were backcrossed with +/+ Lew rats to produce the F2 generation consisting of a 1:1 mix of +/+ and +/Nuc1 offspring and therefore Lew/Lew and Lew/SD at the disease locus, respectively. Ten +/+ and another 10 +/Nuc1 offspring were randomly chosen from the F2 generation as the initial population for genome scanning. Additional F2 rats were used for subsequent linkage analysis.
To perform the genotyping, DNA was extracted from tissues (liver and tail) obtained from euthanized rats using the Qiagen DNA Extraction Kit. A genome-wide scan, excluding chromosomes X and Y, was performed to detect genetic linkage. PCR amplification was performed using the fluorescently labeled Rat Map Pairs, Genome-Wide screening set (Invitrogen Corporation, USA). Amplicon size was then determined using the Applied Biosystems 3100 and GeneMapper software according to the protocols provided by the manufacturer (Applied Biosystems, Foster City, CA). A total of 207 markers were genotyped, of which 140 were informative in the cross. Statistical analysis was carried out using R/qtl, an add-on package to the general statistical software R. With the assumption of complete penetrance for the Nuc1 mutation, single-marker analysis was carried out by simply counting recombinants. The recombination fraction between a marker and the Nuc1 locus was estimated by the fraction of recombinants observed among the 20 chosen F2 generation rats. A LOD score was calculated by the following formula, where n is the total number of backcross rats, and x is the number of observed recombinants.
Gene candidate selection
Gene candidate selection was performed after strong evidence for linkage to marker D10Rat32 (LOD score = 6.02) was found and additional genotyping revealed a 5.2 Mb interval between markers D10Rat195 and D10Rat29. This interval contained 49 genes identified through the reference sequence annotation database for the rat genome assembly, which we augmented by alignment with the mouse and human genomes. One of the genes was the βA3/A1-crystallin gene, which has been shown to cause congenital cataracts when mutated (Kannabiran et al., 1998; Bateman et al., 2000; Qi et al., 2004; Ferrini et al., 2004; Reddy et al., 2004; Graw et al., 1999). This gene was sequenced and the mutation was identified as a 27 base insertion in the 6th exon.
Laser capture microdissection
Enucleated eyes were cryoprotected with an increasing concentration gradient of sucrose at 4 °C. Eyes were embedded in Tissue-Tek O.C.T compound (Sakura Finetek, Inc., Torrance, CA) with snap-freezing on dry ice then stored at −80 °C. Sections (7 μm) were cut with a Microm cryostat and placed on PEN-membrane LCM slides (Leica Microsystems, Bannockburn, IL). Sections were dehydrated and stained with Mayer's hematoxylin solution (Sigma-Aldrich, St. Louis, MO) for visualization of cell nuclei. Cells from three layers of the retina (ganglion cell layer: GCL, inner nuclear layer: INL, photoreceptor layer: PRL) were cut by a Leica LMD6000 instrument. Samples were collected in 0.5 ml tube caps in 45 μl RLT lysis buffer (Qiagen, Valencia, CA) then stored at −80 °C until processed.
RNA isolation
Total RNA was purified with the RNeasy Micro kit (Qiagen) according to the manufacturer's protocol.
cDNA synthesis
The purified total RNA was reverse transcribed in a 20 μl reaction containing 100 ng random hexamer (Invitrogen), 0.4 μl 25 mM dNTP Mix, 4 μl 5× First Strand Buffer (Invitrogen), 2 μl 0.1 M DTT, 1 μl RNaseOut ribonuclease inhibitor (40 U/μl) (Invitrogen), and 1 μl SuperScripIII (200 U/μl) (Invitrogen). The reaction mix was incubated at 50 °C for 1 h then inactivated at 70 °C for 15 min. The reaction mix was diluted 10-fold and used as template in Q-RTPCR reactions.
Q-RTPCR
A Bio-Rad IQ5 Multicolor Real-Time PCR Detection System was used for analysis. Reactions (20 μl) contained 6 pmol of each primer, 10 μl of 2× iQ Sybr Green Supermix (Bio-Rad) and 5 μl cDNA template. Cycling conditions were: 95 °C for 10 s, 59 °C for 30 s, 72 °C for 30 s. Detection was carried out at 72 °C after the extension step. The integrity of PCR products was verified with melting curve analysis. Relative expression was calculated using the expression of HPRT as a reference gene (Pfaffl, 2001).
Immunohistochemistry
Immunofluorescence was performed on frozen sections and astrocytes cultured from brain tissue. The sections or cultured cells were incubated with phosphate-buffered saline, containing 5% normal donkey or goat serum, for 30 min prior to being incubated overnight with primary antibodies at 4 °C, washed in PBS, incubated for 1 h at room temperature with secondary antibodies, and washed again with PBS. Sections and cultured cells were mounted using DAKO Paramount (DAKO Corporation, Carpinteria, CA). The primary monoclonal mouse antibodies used were GFAP (1:200; Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Primary polyclonal antibodies used were GFAP (1:400; DAKO Corporation, Carpinteria, CA), βA3/A1-crystallin (1:300; from Dr. J. Fielding Hejtmancik of the National Eye Institute). Secondary antibodies used for sections and cultured cells were Donkey Anti-Mouse IgG conjugated with Cy-3 (1:400; Jackson Immunoresearch, West Grove, PA) for GFAP and Donkey Anti-Rabbit IgG conjugated with Cy-3 (1:400; Jackson Immunoresearch, West Grove, PA) for GFAP and βA3/A1-crystallin. The slides were then examined on a Zeiss Axioskop II. Confocal microscopy was done on a Zeiss LSM510.
Astrocyte culture from brain tissue
Two day old rats were anesthetized and euthanized in an ether chamber. The brain was removed using sterile techniques, transferred to a 60×15 mm tissue culture dish, and gently washed twice with Hank's buffer. The cerebellum and the meninges were removed and discarded. The cortical lobes were placed into a 15 ml conical tube. The cortical tissue was dissociated and digested into cell suspension by using mechanical digestion. Cells were counted and plated in 75 cm2 tissue culture flasks (approximately 15×106 cells in 15 ml of DMEM/F12 culturing media, containing 10% fetal bovine serum and 1% Antibiotic-Antimycotic solution). Flasks were incubated at 37 °C in a 5% CO2 incubator for 48–72 h. The medium was changed every 48–72 h. The cultures were maintained for approximately 7–9 days. The flasks were then wrapped with parafilm and placed on a shaker platform where they were shaken for 6 h at 280 rpm to separate the oligodendrocytes from the astrocytes. The medium was replaced with 15 ml of fresh new medium and put back into the incubator. After approximately 12 h, the shaking process was repeated. The detached cells were discarded. The astrocyte cultures are normally 90% pure as determined by GFAP staining.
Retinal flat mounts
Eyes to be used for flat mounts were fixed for 1 h in 4% paraformaldehyde, transferred to PBS, and stored at 4 °C until needed. To isolate the retina, the anterior segment was removed and the retina teased away from the sclera using a fine camels hair brush. The whole retina was then washed/blocked in ICC buffer (PBS, pH 7.3 plus 0.5% BSA, 0.2% Tween 20, and 0.05% sodium azide) plus 5% normal goat serum for 24 h at 4 °C on a rotating platform. The blocking solution was removed and the samples were incubated overnight in primary antibody diluted appropriately in ICC buffer plus 2% blocking serum at 4 °C on the rotating platform. Samples were then washed 3× for 15 min each and once for 1 h in ICC buffer at 4 °C. The retinas were then incubated for 4 h in secondary antibody at 4 °C. Typically the following mixture prepared in ICC buffer was used: Alexa 555 labeled goat anti-rabbit IgG (1:300), Alexa 488 labeled isolectin B4 (1:100), and DAPI (1:1000) (all from Molecular Probes, Eugene, OR). Samples were subsequently washed as for the primary antibody and mounted with the ganglion cell layer up on superfrost slides using Gel/Mount (Biomeda Corp, Foster City, CA). Relaxing cuts were used to make the retinas lay flat on the slide. Cover slips were applied and the edges sealed with clear nail polish. Samples were analyzed by confocal microscopy.
Electron microscopy
For transmission electron microscopy, eyes were fixed in 2.5% glutaraldehyde, 4% sucrose, 2 mM calcium chloride in 50 mM cacodylate buffer, pH 7.2 for 24 h, then transferred to 10% buffered formalin. Ultrathin sections were collected on 300 mesh grids and stained with uranyl acetate and lead citrate. Images were captured with a JEM-100 CX electron microscope (JEOL USA, Inc., Peabody, MA).
Protein analysis
Lenses were dissected intact from 3 week old wild-type and Nuc1 heterozygous rats immediately following euthanasia. Since the lens ruptures before birth in the Nuc1 homozygote, it was not possible to obtain intact lenses. We were able to get the most complete sampling of lens material by freezing the freshly enucleated eyes, bisecting them with a scalpel, and then removing the white lens material while still frozen. The lenses (or lens material) were homogenized in 50 mM Tris, pH 7.4 containing 0.1 M KCl, 1 mM EDTA, 10 mM 2-mercaptoethanol, and 0.02% sodium azide at 40 mg tissue weight per ml. After centrifugation to remove insoluble material, the supernatants were filtered (0.02 μm) and fractionated using a Superose 12 gel exclusion column (GE Healthcare, Uppsala, Sweden) on an Agilent Model 1100 HPLC system. Fractions were collected and the composition of protein peaks assessed by SDS-PAGE on 4–12% NuPage Bis–Tris minigels (Invitrogen, Carlsbad, CA). For western blotting, proteins were transferred directly from gels to nitrocellulose using the XCell II Blot Module (Invitrogen). Blots were blocked in milk diluent (Kirkegard and Perry, Gaithersburg, MD), reacted with primary antibody overnight at 4 °C, washed 3× in TBS, reacted with secondary antibody (HRP labeled goat anti-rabbit IgG, Kirkegard and Perry) for 1 h at 37 °C, washed as above, and developed with 4-CN substrate (Kirkegard and Perry). For dot blotting, samples (1 μl) were spotted onto nitrocellulose and allowed to dry. The nitrocellulose was then processed as outlined above for the western blots. In some instances blots of either type were visualized using the Western Lightning Chemiluminescence Reagent (Perkin Elmer, Boston, MA). Primary antibodies used were directed against βA3/A1-crystallin (gift of Dr. J.F. Hejtmancik), anti-GFAP (DAKO Corp., Carpinteria, CA), and antivimentin (Santa Cruz Biotechnology, Santa Cruz, CA).
Evans blue dye injection
Rats were anesthetized by intraperitoneal injection using 100/20 mg/kg body weight ketamine/xylazine. Evans blue dye (Sigma, St. Louis, MO) was injected through the femoral vein at a dosage of 45 mg/kg with a 30-gauge needle. Immediately after Evans blue infusion, the rat turned visibly blue, confirming the uptake and distribution of the dye. Five minutes after the injection of Evans blue, the animal was sacrificed with an overdose of anesthesia and eyeballs were enucleated. Retinas were flat mounted and visualized for vascular leakage by confocal microscopy (Zeiss LSM510).
Adenosine diphosphatase (ADPase) enzyme histochemistry for assessment of retinal vasculature
To evaluate retinal blood vessel morphology, the dissected retinas were fixed in 2% wt/vol paraformaldehyde in phosphate-buffered saline (PBS), pH 7.4, at 4 °C overnight and processed for magnesium-activated ADPase staining as described by Lutty and McLeod (1992). ADPase-stained retinas were temporarily flatmounted on microscope slides in phosphate-buffered saline with a coverslip and photographed. In all retinas, the ADPase reaction product was confined almost exclusively to the vasculature, with arteries having more reaction product than veins or capillaries.
Molecular modeling
The model of the rat βA3-crystallin (βA3) was built by homology modeling using crystal coordinates of the bovine βB2-crystallin (Brookhaven protein database (PDB) file: 1blb.pdb) as the structural template. The primary protein sequences of βA3 and 1blb were aligned and incorporated in the program Look, version 3.5.2, for the 3-dimensional structure prediction. Finally, the dimeric βA3 was built using the automatic segment matching method in the program Look followed by 500 cycles of energy minimization. The same program was used to generate the conformation of the insertion in the βA3-Nuc1 mutant protein and to refine it by self-consistent ensemble optimization which applies the statistical mechanical mean-force approximation iteratively to achieve the global energy minimum structure. The geometry of predicted structures was tested using the program Procheck.
The structure of the rat βA3 dimer was minimized in the presence of 1164 water molecules by 100 cycles of steepest descent followed by the 500 cycles of conjugate gradient. The structure of the C-terminal domain of the βA3-Nuc1 mutant was refined in a similar way. The stability of the βA3-Nuc1 C-terminal domain in water was examined by the method of molecular dynamics (MD) using the impact dynamics module incorporated in the program package Maestro v40217 (Schrodinger LLC). The molecular volume and the surface changes were analyzed using the Crysol program, version 1.01.
Acknowledgments
The assistance of Dr. Bhaja K Padhi (Health Canada, Tunney's Pasture, Ottawa) for genetic consultation is gratefully acknowledged. We thank Drs. Eric Wawrousek (National Eye Institute, Bethesda, Maryland), Nader Sheibani (University of Wisconsin-Madison, Wisconsin), and James Handa (The Johns Hopkins University School of Medicine, Baltimore, Maryland) for critically reading the manuscript. The help of Dr. Xiaodong Jiao with genotyping and Ms. Mary Alice Crawford and Dr. Chi-Chao Chan with electron microscopy is gratefully acknowledged. This work was supported in part by Research to Prevent Blindness (an unrestricted grant to Wilmer Eye Institute) and grants from the National Institutes of Health (NIH) EY009769 (to DZ), EY009357 (to GAL), GM074244 (to KWB), P01MH070306, and P01MH70056 (to AN) and by the Intramural Research Program of the National Eye Institute, NIH (to JFH, RF, and SZ).
References
- Andley UP. Crystallins in the eye: function and pathology. Prog Retin Eye Res. 2007;26:78–98. doi: 10.1016/j.preteyeres.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Ashton N. Retinal angiogenesis in the human embryo. Br Med Bull. 1970;26:103–106. doi: 10.1093/oxfordjournals.bmb.a070758. [DOI] [PubMed] [Google Scholar]
- Ashwell KW, Hollander H, Streit W, Stone J. The appearance and distribution of microglia in the developing retina of the rat. Vis Neurosci. 1989;2:437–448. doi: 10.1017/s0952523800012335. [DOI] [PubMed] [Google Scholar]
- Bateman JB, et al. A new betaA1-crystallin splice junction mutation in autosomal dominant cataract. Invest Ophthalmol Vis Sci. 2000;41:3278–3285. [PubMed] [Google Scholar]
- Chan-Ling T, et al. Astrocyte–endothelial cell relationships during human retinal vascular development. Invest Ophthalmol Vis Sci. 2004;45:2020–2032. doi: 10.1167/iovs.03-1169. [DOI] [PubMed] [Google Scholar]
- Chu Y, Hughes S, Chan-Ling T. Differentiation and migration of astrocyte precursor cells and astrocytes in human fetal retina: relevance to optic nerve coloboma. FASEB J. 2001;15:2013–2315. doi: 10.1096/fj.00-0868fje. [DOI] [PubMed] [Google Scholar]
- Crabb JW, et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. PNAS. 2002;99:14682–14687. doi: 10.1073/pnas.222551899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Schaepdrijver L, Simoens P, Lauwers H. Development of the retinal circulation in the pig. Anat Embryol (Berl) 1995;192:527–536. doi: 10.1007/BF00187183. [DOI] [PubMed] [Google Scholar]
- Dorrell MI, Friedlander M. Mechanisms of endothelial cell guidance and vascular patterning in the developing mouse retina. Prog Retin Eye Res. 2006;25:277–295. doi: 10.1016/j.preteyeres.2006.01.001. [DOI] [PubMed] [Google Scholar]
- DuPrey KM, et al. Subfertility in mice harboring a mutation in βB2-crystallin. Mol Vis. 2007;13:366–373. [PMC free article] [PubMed] [Google Scholar]
- Eliasson C, et al. Intermediate filament protein partnership in astrocytes. J Biol Chem. 1999;274:23996–24006. doi: 10.1074/jbc.274.34.23996. [DOI] [PubMed] [Google Scholar]
- Ferrini W, et al. CRYBA3/A1 gene mutation associated with suture-sparing autosomal dominant congenital nuclear cataract: a novel phenotype. Invest Ophthalmol Vis Sci. 2004;45:1436–1441. doi: 10.1167/iovs.03-0760. [DOI] [PubMed] [Google Scholar]
- Galou M, et al. Disrupted glial fibrillary acidic protein network in astrocytes from vimentin knockout mice. J Cell Biol. 1996;133:853–863. doi: 10.1083/jcb.133.4.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehlbach P, et al. Developmental abnormalities in the Nuc1 rat retina: a spontaneous mutation that affects neuronal and vascular remodeling and retinal function. Neuroscience. 2006;137:447–461. doi: 10.1016/j.neuroscience.2005.08.084. [DOI] [PubMed] [Google Scholar]
- Graw J, et al. Mutation in the betaA3/A1-crystallin encoding gene Cryba1 causes a dominant cataract in the mouse. Genomics. 1999;62:67–73. doi: 10.1006/geno.1999.5974. [DOI] [PubMed] [Google Scholar]
- Hose S, Zigler JS, Jr, Sinha D. A novel rat model to study the functions of macrophages during normal development and pathophysiology of the eye. Immunol Lett. 2005;96:299–302. doi: 10.1016/j.imlet.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Jiang B, Bezhadian MA, Caldwell RB. Astrocytes modulate retinal vasculogenesis: effects on endothelial cell differentiation. Glia. 1995;15:1–10. doi: 10.1002/glia.440150102. [DOI] [PubMed] [Google Scholar]
- Kannabiran C, et al. Autosomal dominant zonular cataract with sutural opacities is associated with a splice mutation in the betaA3/A1-crystallin gene. Mol Vis. 1998;4:21. [PubMed] [Google Scholar]
- Lepekhin EA, et al. Intermediate filaments regulate astrocyte motility. J Neurochem. 2001;79:617–625. doi: 10.1046/j.1471-4159.2001.00595.x. [DOI] [PubMed] [Google Scholar]
- Liedtke T, Schwamborn JC, Schroer U, Thanos S. Elongation of axons during regeneration involves retinal crystallin β b2 (crybb2) Mol Cell Proteomics. 2007;6:895–907. doi: 10.1074/mcp.M600245-MCP200. [DOI] [PubMed] [Google Scholar]
- Ling TL, Mitrofanis J, Stone J. Origin of retinal astrocytes in the rat: evidence of migration from the optic nerve. J Comp Neurol. 1989;286:345–352. doi: 10.1002/cne.902860305. [DOI] [PubMed] [Google Scholar]
- Litt M, et al. Autosomal dominant cerulean cataract is associated with a chain termination mutation in the human beta-crystallin gene CRYBB2. Hum Mol Genet. 1997;6:665–668. doi: 10.1093/hmg/6.5.665. [DOI] [PubMed] [Google Scholar]
- Lobov IB, et al. WNT7b mediates macrophage-induced programmed cell death in patterning of the vasculature. Nature. 2005;437:417–421. doi: 10.1038/nature03928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutty GA, McLeod DS. A new technique for visualization of the human retinal vasculature. Arch Ophthalmol. 1992;110:267–276. doi: 10.1001/archopht.1992.01080140123039. [DOI] [PubMed] [Google Scholar]
- Miller RH, David S, Patel R, Abney ER, Raff MC. A quantitative immunohistochemical study of macroglial cell development in the rat optic nerve: in vivo evidence for two distinct astrocyte lineages. Dev Biol. 1985;111:35–41. doi: 10.1016/0012-1606(85)90432-4. [DOI] [PubMed] [Google Scholar]
- Pallari HM, Eriksson JE. Intermediate filaments as signaling platforms. Sci STKE. 2006:pe53. doi: 10.1126/stke.3662006pe53. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatigorsky J. Gene Sharing and Evolution: The Diversity of Protein Function. Harvard University Press; Cambridge, MA: 2007. [Google Scholar]
- Qi Y, et al. A deletion mutation in the betaA1/A3 crystallin gene (CRYBA1/A3) is associated with autosomal dominant congenital nuclear cataract in a Chinese family. Hum Genet. 2004;114:192–197. doi: 10.1007/s00439-003-1049-7. [DOI] [PubMed] [Google Scholar]
- Reddy MA, et al. Characterization of the G91del CRYBA1/3-crystallin protein: a cause of human inherited cataract. Hum Mol Genet. 2004;13:945–953. doi: 10.1093/hmg/ddh110. [DOI] [PubMed] [Google Scholar]
- Sakaguchi H, et al. Intense light exposure changes the crystallin content in retina. Exp Eye Res. 2003;76:131–133. doi: 10.1016/s0014-4835(02)00249-x. [DOI] [PubMed] [Google Scholar]
- Schnitzer J. Astrocytes in the guinea pig, horse, and monkey retina: their occurrence coincides with the presence of blood vessels. Glia. 1988;1:74–89. doi: 10.1002/glia.440010109. [DOI] [PubMed] [Google Scholar]
- Sinha D, et al. A spontaneous mutation affects programmed cell death during development of the rat eye. Exp Eye Res. 2005;80:323–335. doi: 10.1016/j.exer.2004.09.014. [DOI] [PubMed] [Google Scholar]
- Small RK, Riddle P, Noble M. Evidence for migration of oligodendrocyte-type-2 astrocyte progenitor cells into the developing rat optic nerve. Nature. 1987;328:155–157. doi: 10.1038/328155a0. [DOI] [PubMed] [Google Scholar]
- Stone J, Dreher Z. Relationship between astrocytes, ganglion cells and vasculature of the retina. J Comp Neurol. 1987;255:35–49. doi: 10.1002/cne.902550104. [DOI] [PubMed] [Google Scholar]
- Vazquez-Chona F, Song BK, Geisert EE., Jr Temporal changes in gene expression after injury in the rat retina. Invest Ophthalmol Vis Sci. 2004;45:2737–2746. doi: 10.1167/iovs.03-1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Raff MC. Retinal astrocytes are immigrants from the optic nerve. Nature. 1988;332:834–837. doi: 10.1038/332834a0. [DOI] [PubMed] [Google Scholar]
- Wistow G. Molecular Biology and Evolution of Crystallins: Gene Recruitment and Multifunctional Proteins in the Eye Lens. Springer; New York: 1995. [Google Scholar]
- Zhang C, et al. A potential role for beta- and gamma-crystallins in the vascular remodeling of the eye. Dev Dyn. 2005;234:36–47. doi: 10.1002/dvdy.20494. [DOI] [PubMed] [Google Scholar]