

Treatment with everolimus in patients with extrapancreatic neuroendocrine tumors appears to be a promising strategy that is safe and well-tolerated. The use of this emerging opportunity needs to be validated with clinical trials specifically designed on this topic.
Keywords: Neuroendocrine tumors, Everolimus, Extrapancreatic neuroendocrine tumors, Efficacy, Safety
Abstract
Background.
Everolimus, an oral mTOR (mammalian target of rapamycin) inhibitor, is currently approved for the treatment of progressive pancreatic neuroendocrine tumors (NETs). Although promising, only scattered data, often from nondedicated studies, are available for extrapancreatic NETs.
Patients and Methods.
A systematic review of the published data was performed concerning the use of everolimus in extrapancreatic NET, with the aim of summarizing the current knowledge on its efficacy and tolerability. Moreover, the usefulness of everolimus was evaluated according to the different sites of the primary.
Results.
The present study included 22 different publications, including 874 patients and 456 extrapancreatic NETs treated with everolimus. Nine different primary sites of extrapancreatic NETs were found. The median progression-free survival ranged from 12.0 to 29.9 months. The median time to progression was not reached in a phase II prospective study, and the interval to progression ranged from 12 to 36 months in 5 clinical cases. Objective responses were observed in 7 prospective studies, 2 retrospective studies, and 2 case reports. Stabilization of the disease was obtained in a high rate of patients, ranging from 67.4% to 100%. The toxicity of everolimus in extrapancreatic NETs is consistent with the known safety profile of the drug. Most adverse events were either grade 1 or 2 and easy manageable with a dose reduction or temporary interruption and only rarely requiring discontinuation.
Conclusion.
Treatment with everolimus in patients with extrapancreatic NETs appears to be a promising strategy that is safe and well tolerated. The use of this emerging opportunity needs to be validated with clinical trials specifically designed on this topic.
Implications for Practice:
The present study reviewed all the available published data concerning the use of everolimus in 456 extrapancreatic neuroendocrine tumors (NETs) and summarized the current knowledge on the efficacy and safety of this drug, not yet approved except for pancreatic NETs. The progression-free survival rates and some objective responses seem promising and support the extension of the use of this drug. The site-by-site analysis seems to suggest that some subtypes of NETs, such as colorectal, could be more sensitive to everolimus than other primary NETs. No severe adverse events were usually reported and discontinuation was rarely required; thus, everolimus should be considered a valid therapeutic option for extrapancreatic NETs.
Introduction
In the latest years, a deep interest has arisen regarding neuroendocrine tumors (NETs). Their incidence has increased fivefold in the past 40 years [1, 2]. These tumors can develop from various primary sites, such as the small intestine, stomach, colon, pancreas, lung, thymus, and other organs. Almost one half of patients will have metastatic disease at diagnosis, and the 5-year survival rate has been estimated to be approximately 35% in metastatic patients [3]. In patients with advanced NETs, the survival rate has been better for those with well-differentiated tumors (low or intermediate grade) than for those with poorly differentiated disease and for those with locoregional disease than distant disease [1].
The role of medical therapy for well-differentiated NETs has been questioned. As chemotherapy is scarcely effective and biotherapy only affects progression-free survival (PFS) but not the objective response [4, 5], researchers have investigated the role of intracellular signaling pathways commonly deregulated in NETs, such as activation of the autocrine mammalian target of rapamycin (mTOR) signaling. mTOR is a serine/threonine protein kinase involved in the transduction of PI3K/AKT-dependent growth factor signaling and eventually regulates cell metabolism, proliferation, apoptosis, and angiogenesis [6–10]. Therefore, it can be used to advantage as target therapy. It comprises two major complexes: mTORC1 and mTORC2. mTORC1 mainly controls the cell energy balance, and its activation is inhibited by rapamycin. mTORC2 mainly modulates cell motility by influencing the actin cytoskeleton, and it is not sensitive to rapamycin [11, 12]. Rapamycin and its analogs, such as everolimus, bind the intracellular protein FKBP12, thus inactivating mTORC1 and its related downstream signaling [8]. Until now, everolimus (RAD 001), an oral mTOR inhibitor, has been used in therapy for pancreatic NETs [13, 14]. Everolimus is administered as monotherapy but a well-established molecular rationale exists for its use combined with octreotide LAR in the management of NETs.
To date, everolimus has been approved by the U.S. Food and Drug Administration (FDA) only for the treatment of progressive NETs of pancreatic origin (pNET) in patients with unresectable, locally advanced, or metastatic disease. Several phase II and III studies have recently documented the efficacy and safety of everolimus for the treatment of advanced NETs arising in sites other than the pancreas [15–18]. However, treatment with everolimus in these types of NET continues to be debated, and its use has not yet been approved. A recent retrospective Italian multicenter study collected the data from 85 patients with pNETs and 84 with extra-pNET from a compassionate use program [19]. Similar PFS and overall survival (OS) were reported in pNETs and extra-pNETs. In December 2015, the results from the expected phase III clinical trial on gastrointestinal and lung NETs, RADIANT-4 (RAD 001 for Advanced Neuroendocrine Tumors), were published [20]. Everolimus was significantly effective in nonfunctioning extrapancreatic NETs compared with placebo. Because of the relevance of this finding for the objectives of the present study, the results of the RADIANT-4 study will be considered in the discussion section [20].
In the present study, we reviewed all the available data concerning the use of everolimus in NETs other than pancreatic, with the aim of summarizing the current knowledge on the efficacy and tolerability of this mTOR-inhibiting drug.
Patients and Methods
We searched Medline (PubMed database) for reports published in English on the use of everolimus for the treatment of NETs other than pNETs. The terms considered in the search strategy were “everolimus” or “RAD001” and “neuroendocrine tumor” or “neuroendocrine carcinoma” or “carcinoid” or “medullary thyroid carcinoma” or “pheochromocytoma” or “paraganglioma.” Studies were selected if they included sufficient data to permit the evaluation of the everolimus treatment outcomes and/or safety. Different study types were considered, including prospective and retrospective studies and case reports. Studies focusing on combination therapy of everolimus plus somatostatin analogs or other tyrosine kinase (TK) inhibitors were considered, and those focusing on combinations with chemotherapy were excluded. No language restrictions were used. Data were also extrapolated from studies not exclusively focusing on the topic of the present report. Those exclusively focusing on pNETs were excluded. Reviews, editorials, and letters to the editor were also excluded. The search was last updated in May 2015.
Results
Of the 341 reports found in the initial search, 43 were judged to be potentially eligible by their title and abstract and the full text was reassessed. Overall, 22 studies fulfilled all the inclusion criteria (Table 1). These publications covered 2008–2015 and included different study designs. In particular, 2 were phase I studies [21, 22], 4 were phase II [14, 15, 23, 24], 1 was phase III, 2 were subanalyses of that phase III study [16–18], 1 was a prospective cohort study [25], 2 were retrospective studies [19, 26], and 10 were case reports [27–36] (Table 1). Altogether, the selected studies included 874 patients, and 456 of whom had NETs (non-pNETs) evaluable for treatment with everolimus. The efficacy and safety outcomes were obtained for the different types of NETs, including pNETs in six of the prospective phase I–III trials and 2 retrospective studies [14, 15, 18, 19, 21–23, 26], a population of NET and non-NET thyroid neoplasms in one prospective study [24]. The two other prospective studies, which were subanalyses of the phase III study, focused on a specific population of patients with colon/rectum and lung NETs, respectively [16, 17]. One of the three retrospective studies, including both extra-pNETs and pNETs, reported the PFS for extra-pNETs and pNETs separately, and the tumor response rates and safety outcomes were reported for the whole population [19].
Table 1.
Summary of studies evaluating NET patients (extra-pNET) treated with everolimus

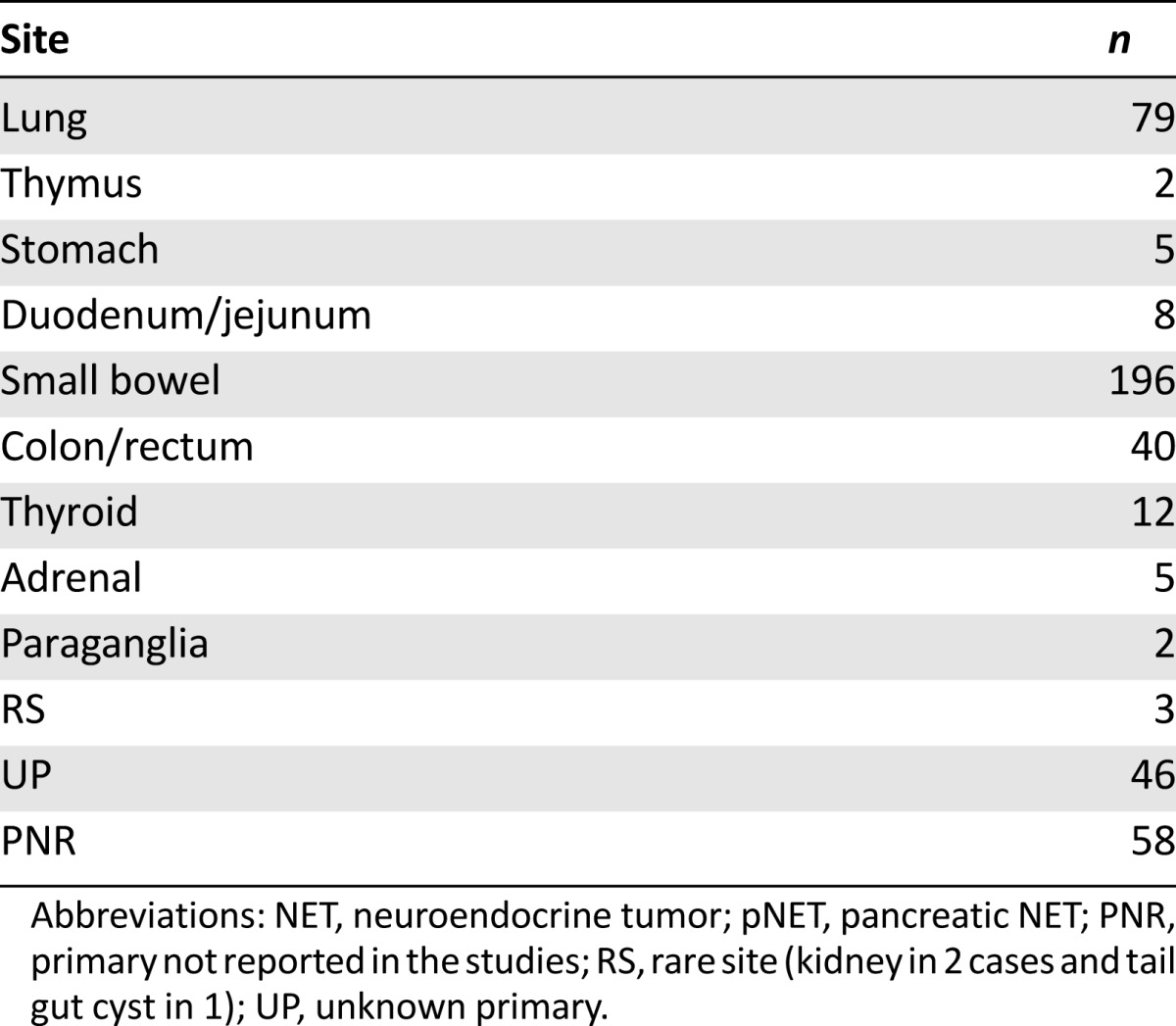
Nine different primary sites of extra-pNET were found. The most frequent were the small bowel, lung, and colon/rectum. An unknown primary was reported in 38 cases, and the primary was known but not reported in 58 cases (Table 2). Excluding thyroid, adrenal, and paraganglia NETs, the degree of neuroendocrine differentiation was reported in 330 of 437 cases. The NETs were well-, moderately, and poorly differentiated in 269, 60, and 1 case, respectively. The Ki-67 level was not reported in most cases.
Table 2.
Number of NET patients treated with everolimus according to site (excluding pNET) from selected reports
Previous treatment variably included surgery, chemotherapy, somatostatin analogs, interferon, target therapies, peptide receptor radionuclide therapy (PRRT), locoregional treatment for hepatic metastases, and radiotherapy (supplemental online Table 1). Chemotherapy included capecitabine/oxaliplatin, 5-fluorouracil/streptozotocin, 5-fluorouracil/dacarbazine/epirubicin, cisplatin/etoposide, cisplatin/irinotecan, Adriamycin, paclitaxel, gemcitabine, and vincristine/Adriamycin/cyclophosphamide. Somatostatin analogs included octreotide and lanreotide. Tyrosine kinase inhibitors included everolimus, temsirolimus, pazopanib, vandetanib, sunitinib, sorafenib, and bevacizumab. Locoregional treatment for hepatic metastases included embolization, chemoembolization, radioembolization, radiofrequency, and cryotherapy. Peptide radioreceptor therapy included 90Y- and 167Lu-radiolabeled therapy in alternating cycles.
Treatment
Everolimus 10 mg/day orally was the starting therapy in 18 of the 22 studies, and everolimus 10 mg/day and everolimus 5 mg/day were both reported as starting therapy in 4 other studies. These two different everolimus schedules were compared between different groups in one phase II randomized trial. Everolimus was administered as monotherapy in 6 studies, in association with octreotide LAR 30 mg every 28 days i.m. in 11, in association with octreotide LAR 30 mg every 28 days i.m. or lanreotide autogel 120 mg every 28 days s.c. in 2, with pasireotide 40–80 mg every 28 days i.m. in 1, with pasireotide 40–80 mg every 28 days i.m. or everolimus 10 mg/day orally in monotherapy in 1, and with sorafenib 400–600 mg/day orally in 1. The median treatment duration ranged from 4 to 17 months among the prospective and retrospective series. The longest treatment duration was 46 months.
Outcomes
Tumor progression before everolimus was documented in all prospective and retrospective studies but two and in all case reports but one, which reported clinical progression (Table 3).
Table 3.
Progression-free survival, time to progression, and overall survival in NET patients treated with everolimus

To evaluate everolimus efficacy, PFS data were available from 11 studies, in 8 of them as the median PFS [14, 16–19, 23, 24, 26] and in 3 as the 6- or 12-month PFS [21, 22, 25]. The median time to progression (TTP) was available from one prospective study [25], and TTP was available from four case reports [27, 28, 30, 31]. Progression had not occurred in three case reports during the observation time with everolimus, and data on progression were unavailable from three case reports. The median OS was available from five studies [14–16, 18, 24]. In six studies (five prospective and one retrospective), data on pNETs were also included [15, 18, 21–23, 26].
The median PFS ranged from 12 months in the real-world retrospective study of 84 patients with digestive, lung, or unknown primary NETs [19] to 29.9 months in the subgroup analysis of 19 patients with colorectal NETs from the RADIANT-2 study [16]. However, in the phase II study by Oh et al. [23], when the 7 patients with pheochromocytoma or paraganglioma were considered separately from those with other NETs, the median PFS was 3.8 months. When evaluated in different series of digestive, lung, and unknown primary NETs, the 6-month PFS was 79% and 76% in two phase I studies [21, 22], and the 12-month PFS was 65% in a phase I study [22] and 64% in a prospective cohort study [25]. The median TTP was not reached in a phase II prospective study of 50 patients with different types of NETs that also included 14 pNETs [15]. The TTP ranged from 12 to 36 months in five clinical cases experiencing tumor progression [27, 28, 30, 31]. Where available, the median OS was not reached in four studies (three phase II and one phase III study [14, 15, 18, 24]) and was 32 months in the real-world retrospective study [19].
Objective responses were observed in seven prospective phase I–III studies, two retrospective studies, and two case reports. A complete response was reported in one nonfunctioning metastatic ileal NET treated with everolimus plus octreotide in a first-line phase II study [15] and in another NET, the primary site of which was unspecified, in a retrospective study [19]. Forty-three patients had a partial response (PR). The primary site of the tumor was reported in nine cases: the ileum in three, the stomach, duodenum, and lung in one each, and an unknown primary in three. The objective response rates ranged from 2.5% in a phase III study [18] to 18% in the first-line phase II study [15]. Stable disease (SD) was observed in 480 patients, ranging from 67.4% in the real-world retrospective study [19] to 100% in the prospective cohort study [25], both of which included digestive, thoracic, and unknown primary NETs. Progressive disease (PD) was observed in 68 patients, ranging from 3.3% in the phase II study by Yao et al. [14] in digestive, thoracic, and unknown primary NETs to 28.6% in a series of patients with pheochromocytoma or paraganglioma [23]. These findings for all 22 selected studies are listed in Table 4. A similar percentage of PR, SD, and PD was observed when we evaluated only the 15 of 22 studies that reported results on extra-pNETs.
Table 4.
Tumor response according to RECIST in NET patients treated with everolimus

Safety
Data on the safety and toxicity of everolimus were reported in 19 of the 22 reports; in 9 studies, data on pNETs were also included (Table 5). Drug-related adverse events were mainly classified in accordance with the National Cancer Institute’s Common Terminology Criteria for Adverse Events, version 3.0, although in some studies, the classification system was not specified. The toxicity of everolimus in non-pNETs is consistent with the known safety profile of the drug, as proved by the relatively low rate of drug discontinuation due to adverse events (19% in the largest series including pNETs) [18]. Dose reduction or temporary interruption of drug administration were reported in 15 of 22 studies and drug discontinuation in 14, although it was difficult to infer whether these could be specifically attributed to everolimus, especially in those studies allowing drug combinations, in which the toxicity could have resulted from cumulative effects (Table 5). Moreover, in phase I studies [21, 22], the spectrum and intensity of drug-related adverse events can be widely different according to the diverse dose administered. The highest rate of dose reduction, together with temporary interruption (140 patients, 56%), was reported in the RADIANT-2 study [18]; however, that study referred to any cause and not only to drug toxicity. In some studies, dose reduction, temporary interruption, and/or complete discontinuation of everolimus were allowed; however, the number or percentage of patients involved was not specified [14, 16, 17, 23, 24]. No directly treatment-related deaths were reported, but two cases of lethal pneumonitis after everolimus therapy were described [19]. The commonly reported adverse events are pooled into two groups: mild to moderate (grade 1–2; Table 6) and severe (grade 3–4; Table 7), although in some studies, data on the adverse events were reported as a whole [16–19, 23, 24, 26], with a separate analysis only for grade 3–4. Most adverse events were either grade 1 or 2 and easy manageable with a dose reduction or temporary interruption and only rarely required discontinuation. One case of severe hepatic steatosis was the only unexpected adverse event [35]. The most common grade 1–2 nonhematological drug-related adverse events were mucositis and stomatitis, reported in 11 studies with high rates (>40% of patients in 9 studies). Other common and frequent adverse events were rash, hyperglycemia, diarrhea, and pulmonary events, including pneumonitis, interstitial lung disease, lung infiltrations, and pulmonary fibrosis. The grade 1–2 hematological adverse events were thrombocytopenia and anemia, reported in 12 and 11 studies, respectively, both with high rates. Grade 3–4 mucositis and stomatitis were reported in 11 studies, although with lower rates. Grade 3–4 diarrhea was reported in 10 studies and thrombocytopenia in 9.
Table 5.
Safety assessment, treatment variations due to adverse events, and treatment-related deaths in NET patients treated with everolimus

Table 6.
Everolimus adverse events grade 1–2

Table 7.
Everolimus adverse events grade 3–4

The only study evaluating a correlation between toxicity and tumor subtype found no statistically significant difference among patients with grade 3–4 toxicities or the most common overall toxicities [22]. However, the safety profile of everolimus might be affected by previous therapies, because a 12-fold increased risk of severe toxicity has been reported in patients who had already received PRRT and chemotherapy [19]. In contrast, everolimus has been advocated as a safe option after tumor progression following 177Lu-octreotate radionuclide therapy [26].
In summary, everolimus appears to be quite a safe therapeutic option, with a good and manageable safety profile, even in NETs other than pancreatic.
Lung NETs
Lung NETs represent approximately 25% of all lung cancers, with an annual incidence of 1.35 patients per 100,000 in the U.S. [2, 37]. Lung NETs were classified by Travis et al. [38] into four types: two with highly aggressive biological behavior (i.e., small-cell neuroendocrine carcinomas, which represent slightly 20% of all lung cancers, and large-cell neuroendocrine carcinomas, the rarest subtype) and two with low- to intermediate-grade malignancy (typical and atypical carcinoids, representing 5% of all lung cancers). The therapeutic options are limited for lung NETs with unresectable or metastatic disease, because no standard treatment exists [37, 39].
Lung NETs have demonstrated overactivation of the mTOR signaling pathway in vitro [40]. In preclinical data obtained from experiments in cell lines derived from human lung NETs, everolimus demonstrated antiproliferative effects, with mTOR expression significantly higher in “responsive” human bronchial carcinoma cultures than in nonresponsive tissues [41]. Moreover, low- to intermediate-grade lung NETs show preferential sensitivity to everolimus compared with high-grade lung NETs [41]. The efficacy and safety of everolimus plus octreotide LAR in lung NET has been evaluated by Fazio et al. [17] as exploratory subanalysis of the RADIANT-2 study. In that cohort of 44 patients with low- to intermediate-grade advanced lung NETs (10.3% of the overall RADIANT-2 population study), the analysis demonstrated that the median PFS was increased by 8 months, from 5.6 months in patients receiving placebo plus octreotide LAR to 13.6 months in patients receiving everolimus plus octreotide LAR, corresponding to a 28.8% reduction in the risk of progression. Although this difference was not statistically significant (p = .228), this clinically meaningful improvement in PFS represented a benefit similar to that seen in the overall RADIANT-2 population (a PFS increase of 5.1 months), despite the generally poorer prognosis of those with lung NETs compared with other types of NETs [18]. Moreover, tumor shrinkage was observed in a higher proportion of patients treated with everolimus plus octreotide LAR (66.7%) than in those treated with placebo plus octreotide LAR (27.3%). The ITMO (Italian Trials in Medical Oncology) study suggested a possible role of everolimus plus octreotide as first-line treatment in patients with gastroenteropancreatic (GEP) and lung NET [15]. The overall response rate was 18%, and 74% achieved stable disease. The study conclusion was that the everolimus-octreotide LAR combination was active and well tolerated in these previously untreated patients with advanced NETs.
Gastric NETs
Although gastric NETs (g-NETs) represent approximately 20% of all digestive NETs, they are often underrepresented in clinical trials, and data have mainly been obtained from case reports. The optimal therapeutic management of g-NETs depends on several factors. In the case of advanced disease, several medical treatments, including cytotoxic chemotherapy, have been proposed; however, the rate of objective response has not been satisfying.
In preclinical data obtained from experiments in cell lines derived from human lung NETs, everolimus demonstrated antiproliferative effects, with mTOR expression significantly higher in “responsive” human bronchial carcinoma cultures than in nonresponsive tissues. Moreover, low- to intermediate-grade lung NETs show preferential sensitivity to everolimus compared with high-grade lung NETs.
Few data are available regarding everolimus for g-NETs. Bariani et al. [27] reported the case of a 64-year-old male patient with metastatic type 3 g-NET. The patient experienced a clinical benefit from everolimus administration and after 6 months of treatment, the liver metastases had decreased by 17%.
Ileal NETs
Almost one third (30.8%) of all GEP-NETs arise from the small intestine [42]. Terminal ileum NETs are frequently diagnosed at an advanced stage, with liver and/or regional lymph node metastases in up to 70% of patients [43]. Also, approximately 20% of total ileal NETs are associated with carcinoid syndrome [44].
Surgery represents the first therapeutic approach in most cases [45, 46]. However, in advanced unresectable disease, medical treatment is required [45]. Somatostatin analogs induce partial stabilization of the disease and control of secretory symptoms in patients with carcinoid syndrome [47]. Owing to the low rate of proliferation of ileal NETs, chemotherapy will not be able to induce tumor shrinkage in most cases. Together with the frequent development of potentially severe adverse events, chemotherapy is generally recommended for patients with a poor prognosis and not responding to less aggressive therapies [14, 48–50]. In this scenario, therapy with everolimus might represent a good compromise between effectiveness and tolerability. Capdevila et al. [29] reported the case of a 50-year-old woman with carcinoid syndrome from a well-differentiated neuroendocrine carcinoma of the ileum with diffuse liver metastases. After treatment with everolimus plus octreotide, her symptoms improved, and a 50% decrease in liver metastases was obtained. This schedule has also been tested in a phase II study by Bajetta et al., demonstrating a high objective response plus stable disease rate when everolimus and octreotide were used in a first-line setting [15]. In particular, the only complete response reported in their study was in a patient with a nonfunctioning metastatic ileal NET. In a phase I study, Chan et al. [22] reported the use of everolimus combined with pasireotide (SOM230) in 21 patients with advanced NETs, 66.7% of which originated from the small bowel. Most treated patients (95.2%) had a partial response or stable disease using the RECIST, and after 12 months, 65% were progression free. The investigators reported no differences in efficacy according to tumor type.
In order to investigate the possible synergistic effect of the inhibition of both mTOR and vascular endothelial growth factor pathways in patients with advanced NETs, Chan et al. [21] evaluated the efficacy of everolimus combined with sorafenib in 21 patients, 52.4% of which originated from the small bowel. They observed a 62% objective response rate, mainly corresponding to stable disease. In the RADIANT-2 trial, 52.2% of the overall 429 enrolled patients had a small bowel primary NET. In that subgroup of patients, the combined treatment with everolimus plus octreotide, compared with placebo plus octreotide, led to a 23% reduction in the estimated risk of progression and an additional prolongation of PFS by 4.6 months. Despite these clinically meaningful findings, no statistically significant difference was found between everolimus and placebo [18].
Colorectal NETs
Among all GEP-NETs, colorectal NETs are the most frequent in terms of prevalence [51]. Moreover, epidemiological data have revealed an increase in their incidence. Patients with colorectal NETs have a poor prognosis, as many of them already have advanced disease at diagnosis. The median survival for those with metastatic colorectal NETs has been 5 months compared with 24 and 56 months for those with NETs of the pancreas and small bowel, respectively [2]. To date, chemotherapy and biotherapy have not significantly improved the prognosis of these neoplasms, owing to the low efficacy and often relevant toxicity; thus, effective therapeutic options are lacking.
Everolimus has been evaluated for NETs arising from the colon and rectum in some large, multicenter, single or double arm, clinical trials that included NETs of different origin, well or moderately differentiated, functioning and nonfunctioning, with local, locally advanced, or metastatic disease [14, 16, 18, 23]. In the RADIANT-2 study, 6.5% of the 429 enrolled patients had a primary NET of the colon. In that subgroup of patients, the combined treatment with everolimus and octreotide, compared with placebo plus octreotide, led to a 61% reduction in the estimated risk of progression and an additional prolongation of PFS of 16.9 months. Despite these encouraging findings, the difference between the everolimus and placebo groups was not statistically significant, mainly because of the small number of patient with colon NETs. Castellano et al. [16] performed an exploratory post hoc subgroup analysis of the RADIANT-2 study by including patients with both colonic and rectal NETs as the primary site (39 of 429; 9.1%). They observed that patients with colorectal NETs treated with everolimus plus octreotide LAR had significantly longer median PFS compared with those who had received placebo plus octreotide LAR (29.9 vs. 6.6 months, respectively).
Medullary Thyroid Cancer
Medullary thyroid cancer (MTC) accounts for approximately 4% of all thyroid cancers. Although the 10-year survival rate for MTC patients is 80%, it decreases to 31% when distant metastases are present. The first therapeutic option is thyroidectomy plus lymphadenectomy. However, a high rate of tumors relapse after surgery. In patients with progressive metastatic disease, the only effective therapeutic options are the TK inhibitors vandetanib and cabozantinib; chemotherapy is ineffective. If these compounds are effective as antitumor agents, they could be responsible for common grade 3–4 side effects, which implies their use only in the case of RECIST progression [52, 53]. In 2012, Faggiano et al. demonstrated in an in vivo and in vitro study that everolimus is active in MTC [31]. In particular, everolimus, combined with octreotide, might be effective as antitumor therapy in patients with progressive metastatic MTC. Lim et al. [24], in a multicenter phase II trial, investigated the efficacy of everolimus in 40 patients with locally advanced or metastatic thyroid cancer (including 9 patients with MTC). The overall objective response rate (complete response plus partial response) was not satisfying (5%); however, the primary endpoint of the study (i.e., the disease control rate; defined by the investigators as the partial response plus stable disease of ≥12 weeks) was achieved in all 9 MTC patients.
Pheochromocytoma and Paraganglioma
Pheochromocytomas and paragangliomas are rare NETs with an estimated annual incidence in the U.S. of 500–1,600 cases annually [54]. Surgery has been the primary treatment, although systemic treatment has been reported in cases of malignant unresectable disease or in the presence of distant metastases. The most used chemotherapy regimen is a combination of cyclophosphamide, vincristine, and dacarbazine, which provides a partial response in only 30% of patients [55].
Oh et al. [23] evaluated the efficacy of treatment with everolimus (median duration of 3.8 months) in 5 patients with pheochromocytoma and 2 with paraganglioma. Five of these patients (71.4%) achieved stable disease and two developed progression; the median PFS was 3.8 months.
Discussion
Although perceived as rare, the incidence and prevalence of NETs have been increasing. Also, NETs are heterogeneous in terms of biology, natural history, and therapeutic options [1, 2]. In particular, pNETs can be distinguished from those arising elsewhere, and medical therapy has been dichotomized between these two groups [56, 57]. The knowledge of molecular pathways involved in human carcinogenesis has allowed in the past decade the development of new therapeutic agents that specifically act against the deregulated molecular signal implicated in cancer progression, thus representing an interesting and promising option for cancer therapy [58]. Such is the case with the mTOR pathway, which is hyperactivated in several cancers, including NETs [31, 59–61]. Many studies have evaluated whether NET patients might benefit from targeted agents. At present, everolimus is the only mTOR inhibitor drug approved by both the FDA and the European Medicines Agency for the treatment of advanced grade 1–2 pNETs. Significant differences exist in the molecular genetics between pancreatic and extrapancreatic NETs. In the present study, we discuss the current data concerning the use of everolimus for the treatment of extrapancreatic NETs.
We identified 22 clinical studies investigating the use of everolimus in 456 extrapancreatic NETs that originated from nine different primary sites, including typical NETs from the gastrointestinal and thoracic tract and neuroectodermal NETs from the thyroid, adrenal glands, and paraganglia. Less common sites and tumors with an unknown primary were also included. Although the efficacy of everolimus has been documented regardless of the primary site [19, 22], not all studies allowed the extrapolation of the data regarding the outcome of extrapancreatic NET patients treated with everolimus. However, in 20 of 22 studies, the disease was progressing before everolimus treatment (Table 4), and the best tumor response obtained after everolimus administration was to stabilize the disease progression in a high percentage of patients, ranging from 67.4% to 100% (Table 5). Although some studies also included patients with pNETs, our results were still confirmed when we analyzed only those studies with data available on patients with extrapancreatic NETs [14, 22, 24, 25]. Moreover, the efficacy of everolimus in extrapancreatic NETs has been tested in placebo-controlled phase III trials. In two exploratory subanalyses of the RADIANT-2 study [16, 17], the median PFS for patients with lung and colorectal NETs was 13.6 and 29.9 months compared with 16.4 months in the RADIANT-2 study and 11.0 months in pNETs evaluated in the RADIANT-3 study [14, 18]. In December 2015, the results of the expected phase III trial RADIANT-4 focusing on nonfunctioning extrapancreatic NETs were published [20]. That study reinforced previous data regarding the effectiveness of everolimus in extrapancreatic grade 1–2 NETs. In particular, NETs of the lung, ileum, rectum, and unknown primary were well represented in the study population, and other digestive NETs were outnumbered. The median PFS in the RADIANT-4 study was 11.0 months for patients treated with everolimus and 3.9 for the placebo group. Safety was similar to that in previous studies. No difference in median PFS was observed after stratification for primary tumor site or for tumor grade.
In 20 of 22 studies, the disease was progressing before everolimus treatment, and the best tumor response obtained after everolimus administration was to stabilize the disease progression in a high percentage of patients, ranging from 67.4% to 100%.
Despite the promising results obtained through therapy with everolimus, in the real-world setting, the occurrence of resistance to mTOR inhibitor drugs is not rare. Compensatory feedback loops and cross-talk between the PI3K/AKT/mTOR cascade and other pathways have been identified [62, 63]. For instance, mTOR inhibition induces upstream activation of AKT signaling [64], thus potentially leading to resistance to rapamycin and its analogs. Resistance to mTOR inhibition could also result from upstream activation of PI3K/AKT signaling by a mutated and constitutively activated RAS/MAPK pathway. Chiu et al. reported restored sensitivity to everolimus by combined therapy with erlotinib (an inhibitor of the epithelial growth factor receptor) in a genetically engineered mouse model of pNET [65]. Resistance to everolimus was reported by Di Nicolantonio et al. in metastatic cancer patients with KRAS-mutated tumors [66]. Moreover, in human cancer cells with both PIK3CA and KRAS mutations, sensitivity to everolimus was restored after genetically deleting the KRAS mutations.
Another relevant issue concerns the use of everolimus in the treatment of poorly differentiated grade 3 neuroendocrine carcinomas (NECs). Evidence is lacking regarding whether everolimus might be advisable for the treatment of high-grade NECs, because most patients enrolled in the cited studies had a low- to intermediate-grade NET (grade 1 or 2). Bollard et al. [67] observed strong expression of the two major effectors (phospho-p70S6K and phospho-4E-BP1) of mTOR in six human tissue samples of NECs. Moreover, they investigated the effect of everolimus in a xenograft model of two neuroendocrine cell lines (STC-1 and GluTag) in nude mice. The tumors derived from these cell lines mimicked NEC in vivo. After treating the xenografted mice with everolimus, a significant reduction in the tumor volume was obtained [67]. Therefore, several clinical trials have been designed to investigate the use of everolimus in poorly differentiated NECs as first-line therapy combined with temozolomide (ClinicalTrials.gov identifier NCT02248012) or with paclitaxel plus carboplatin (ClinicalTrials.gov identifier NCT01317615) and as second-line treatment after failure of first-line platinum-based chemotherapy (ClinicalTrials.gov identifier NCT02113800). An intriguing hypothesis is that everolimus could be of great benefit in well-differentiated moderately proliferating grade 3 disease, a recently identified subcategory of grade 3 tumors that seems to have longer survival and lower chemosensitivity than poorly differentiated, highly proliferating grade 3 tumors [68]. Further studies are required to test this potential indication for everolimus.
At present, to better validate the role of everolimus monotherapy or combined therapy for patients with extra-pNETs, some clinical trials are recruiting participants or have completed recruitment, and the results are awaited. These studies are investigating the safety and efficacy of everolimus alone for advanced NETs as first-line treatment (ClinicalTrials.gov identifier NCT01648465) or in pretreated patients with progressive disease (ClinicalTrials.gov identifier NCT01524783). Other ongoing trials are investigating the efficacy of combined therapies, including everolimus plus pasireotide, in patients with advanced NETs of the lung and thymus (ClinicalTrials.gov identifier NCT01563354) and everolimus plus erlotinib in patients with grade 1 or 2 NETs.
Conclusion
We examined the evolving therapeutic landscape and discussed the available evidence suggesting everolimus as a valid therapeutic option for NETs other than pancreatic. Moreover, some subtypes of NETs, such as colorectal, could be more sensitive to everolimus than the pancreatic type. Finally, a new emerging and attractive opportunity requiring validation is the use of everolimus for poorly differentiated NETs. Everolimus is well tolerated, with most side effects mild or moderate in severity. However, careful clinical and biochemical monitoring is needed for the early diagnosis of hematological and metabolic abnormalities or pneumonitis.
See http://www.TheOncologist.com for supplemental material available online.
This article is available for continuing medical education credit at CME.TheOncologist.com.
Supplementary Material
Author Contributions
Conception/design: Antongiulio Faggiano, Pasqualino Malandrino, Roberta Modica, Annamaria Colao
Provision of study material or patients: Antongiulio Faggiano, Pasqualino Malandrino, Roberta Modica, Daniela Agrimi, Maurizio Aversano, Vincenzo Bassi, Ernesto A. Giordano, Valentina Guarnotta, Francesco A. Logoluso, Erika Messina, Vincenzo Nicastro, Vincenzo Nuzzo, Marcello Sciaraffia, Annamaria Colao
Collection and/or assembly of data: Antongiulio Faggiano, Pasqualino Malandrino, Roberta Modica, Daniela Agrimi, Maurizio Aversano, Vincenzo Bassi, Ernesto A. Giordano, Valentina Guarnotta, Francesco A. Logoluso, Erika Messina, Vincenzo Nicastro, Vincenzo Nuzzo, Marcello Sciaraffia, Annamaria Colao
Data analysis and interpretation: Antongiulio Faggiano, Pasqualino Malandrino, Roberta Modica, Annamaria Colao
Manuscript writing: Antongiulio Faggiano, Pasqualino Malandrino, Roberta Modica, Annamaria Colao
Final approval of manuscript: Antongiulio Faggiano, Pasqualino Malandrino, Roberta Modica, Annamaria Colao
Disclosures
The authors indicated no financial relationships.
References
- 1.Fraenkel M, Kim M, Faggiano A, et al. Incidence of gastroenteropancreatic neuroendocrine tumours: A systematic review of the literature. Endocr Relat Cancer. 2014;21:R153–R163. doi: 10.1530/ERC-13-0125. [DOI] [PubMed] [Google Scholar]
- 2.Yao JC, Hassan M, Phan A, et al. One hundred years after “carcinoid”: Epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26:3063–3072. doi: 10.1200/JCO.2007.15.4377. [DOI] [PubMed] [Google Scholar]
- 3.Faggiano A, Ferolla P, Grimaldi F, et al. Natural history of gastro-entero-pancreatic and thoracic neuroendocrine tumors. Data from a large prospective and retrospective Italian epidemiological study: The NET management study. J Endocrinol Invest. 2012;35:817–823. doi: 10.3275/8102. [DOI] [PubMed] [Google Scholar]
- 4.Rinke A, Müller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: A report from the PROMID Study Group. J Clin Oncol. 2009;27:4656–4663. doi: 10.1200/JCO.2009.22.8510. [DOI] [PubMed] [Google Scholar]
- 5.Saltz L, Trochanowski B, Buckley M, et al. Octreotide as an antineoplastic agent in the treatment of functional and nonfunctional neuroendocrine tumors. Cancer. 1993;72:244–248. doi: 10.1002/1097-0142(19930701)72:1<244::aid-cncr2820720143>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 6.Bjornsti MA, Houghton PJ. The TOR pathway: A target for cancer therapy. Nat Rev Cancer. 2004;4:335–348. doi: 10.1038/nrc1362. [DOI] [PubMed] [Google Scholar]
- 7.Faivre S, Kroemer G, Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov. 2006;5:671–688. doi: 10.1038/nrd2062. [DOI] [PubMed] [Google Scholar]
- 8.Meric-Bernstam F, Gonzalez-Angulo AM. Targeting the mTOR signaling network for cancer therapy. J Clin Oncol. 2009;27:2278–2287. doi: 10.1200/JCO.2008.20.0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Reilly T, McSheehy PM. Biomarker development for the clinical activity of the mTOR inhibitor everolimus (rad001): Processes, limitations, and further proposals. Transl Oncol. 2010;3:65–79. doi: 10.1593/tlo.09277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manfredi GI, Dicitore A, Gaudenzi G, et al. PI3K/Akt/mTOR signaling in medullary thyroid cancer: A promising molecular target for cancer therapy. Endocrine. 2015;48:363–370. doi: 10.1007/s12020-014-0380-1. [DOI] [PubMed] [Google Scholar]
- 11.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 12.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 13.Yao JC, Lombard-Bohas C, Baudin E, et al. Daily oral everolimus activity in patients with metastatic pancreatic neuroendocrine tumors after failure of cytotoxic chemotherapy: A phase II trial. J Clin Oncol. 2010;28:69–76. doi: 10.1200/JCO.2009.24.2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yao JC, Phan AT, Chang DZ, et al. Efficacy of RAD001 (everolimus) and octreotide LAR in advanced low- to intermediate-grade neuroendocrine tumors: Results of a phase II study. J Clin Oncol. 2008;26:4311–4318. doi: 10.1200/JCO.2008.16.7858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bajetta E, Catena L, Fazio N, et al. Everolimus in combination with octreotide long-acting repeatable in a first-line setting for patients with neuroendocrine tumors: An ITMO group study. Cancer. 2014;120:2457–2463. doi: 10.1002/cncr.28726. [DOI] [PubMed] [Google Scholar]
- 16.Castellano D, Bajetta E, Panneerselvam A, et al. Everolimus plus octreotide long-acting repeatable in patients with colorectal neuroendocrine tumors: A subgroup analysis of the phase III RADIANT-2 study. The Oncologist. 2013;18:46–53. doi: 10.1634/theoncologist.2012-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fazio N, Granberg D, Grossman A, et al. Everolimus plus octreotide long-acting repeatable in patients with advanced lung neuroendocrine tumors: Analysis of the phase 3, randomized, placebo-controlled RADIANT-2 study. Chest. 2013;143:955–962. doi: 10.1378/chest.12-1108. [DOI] [PubMed] [Google Scholar]
- 18.Pavel ME, Hainsworth JD, Baudin E, et al. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): A randomised, placebo-controlled, phase 3 study. Lancet. 2011;378:2005–2012. doi: 10.1016/S0140-6736(11)61742-X. [DOI] [PubMed] [Google Scholar]
- 19.Panzuto F, Rinzivillo M, Fazio N, et al. Real-world study of everolimus in advanced progressive neuroendocrine tumors. The Oncologist. 2014;19:966–974. doi: 10.1634/theoncologist.2014-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yao JC, Fazio N, Singh S, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): A randomised, placebo-controlled, phase 3 study. Lancet. 2016;387:968–977. doi: 10.1016/S0140-6736(15)00817-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chan JA, Mayer RJ, Jackson N, et al. Phase I study of sorafenib in combination with everolimus (RAD001) in patients with advanced neuroendocrine tumors. Cancer Chemother Pharmacol. 2013;71:1241–1246. doi: 10.1007/s00280-013-2118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan JA, Ryan DP, Zhu AX, et al. Phase I study of pasireotide (SOM 230) and everolimus (RAD001) in advanced neuroendocrine tumors. Endocr Relat Cancer. 2012;19:615–623. doi: 10.1530/ERC-11-0382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oh DY, Kim TW, Park YS, et al. Phase 2 study of everolimus monotherapy in patients with nonfunctioning neuroendocrine tumors or pheochromocytomas/paragangliomas. Cancer. 2012;118:6162–6170. doi: 10.1002/cncr.27675. [DOI] [PubMed] [Google Scholar]
- 24.Lim SM, Chang H, Yoon MJ, et al. A multicenter, phase II trial of everolimus in locally advanced or metastatic thyroid cancer of all histologic subtypes. Ann Oncol. 2013;24:3089–3094. doi: 10.1093/annonc/mdt379. [DOI] [PubMed] [Google Scholar]
- 25.van Asselt SJ, Oosting SF, Brouwers AH, et al. Everolimus reduces 89zr-bevacizumab tumor uptake in patients with neuroendocrine tumors. J Nucl Med. 2014;55:1087–1092. doi: 10.2967/jnumed.113.129056. [DOI] [PubMed] [Google Scholar]
- 26.Kamp K, Gumz B, Feelders RA, et al. Safety and efficacy of everolimus in gastrointestinal and pancreatic neuroendocrine tumors after (177)Lu-octreotate. Endocr Relat Cancer. 2013;20:825–831. doi: 10.1530/ERC-13-0254. [DOI] [PubMed] [Google Scholar]
- 27.Bariani GM, Carvalheira JB, Riechelmann RP. Antitumor effect of everolimus in a patient with type 3 gastric neuroendocrine tumor. Onkologie. 2013;36:502–504. doi: 10.1159/000354637. [DOI] [PubMed] [Google Scholar]
- 28.Beck T, Mantooth R. Long-term management of a patient with well-differentiated pulmonary neuroendocrine carcinoma: A case report. Case Rep Oncol. 2013;6:209–215. doi: 10.1159/000350745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Capdevila J, Diez Miranda I, Obiols G, et al. Control of carcinoid syndrome with everolimus. Ann Oncol. 2011;22:237–239. doi: 10.1093/annonc/mdq670. [DOI] [PubMed] [Google Scholar]
- 30.Druce M, Chung TT, Grozinsky-Glasberg S, et al. Preliminary report of the use of everolimus in a patient with progressive medullary thyroid carcinoma. Clin Endocrinol (Oxf) 2012;77:154–155. doi: 10.1111/j.1365-2265.2011.04296.x. [DOI] [PubMed] [Google Scholar]
- 31.Faggiano A, Ramundo V, Dicitore A, et al. Everolimus is an active agent in medullary thyroid cancer: A clinical and in vitro study. J Cell Mol Med. 2012;16:1563–1572. doi: 10.1111/j.1582-4934.2011.01438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mitsuyama T, Kubota M, Nakamura Y, et al. Neuroendocrine tumor arising from tailgut cyst with spinal cord tethering: Case report and literature review. Spine J. 2015;15:e1–e8. doi: 10.1016/j.spinee.2014.09.027. [DOI] [PubMed] [Google Scholar]
- 33.Parikh D, Shinder R. Primary renal carcinoid metastatic to the orbit. Ophthal Plast Reconstr Surg. 2015;31:e37–e38. doi: 10.1097/IOP.0000000000000061. [DOI] [PubMed] [Google Scholar]
- 34.Pusceddu S, Milione M, Procopio G. Compassionate use of everolimus in a patient with a neuroendocrine tumor: A case report and discussion of the literature. Oncol Res. 2011;19:403–406. doi: 10.3727/096504011x13123323849799. [DOI] [PubMed] [Google Scholar]
- 35.Schieren G, Bölke E, Scherer A, et al. Severe everolimus-induced steatohepatis: A case report. Eur J Med Res. 2013;18:22. doi: 10.1186/2047-783X-18-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sibertin-Blanc C, Norguet E, Duluc M, et al. Severe hypersensitivity pneumonitis associated with everolimus therapy for neuroendocrine tumour: A case report. BMC Res Notes. 2013;6:471. doi: 10.1186/1756-0500-6-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rekhtman N. Neuroendocrine tumors of the lung: An update. Arch Pathol Lab Med. 2010;134:1628–1638. doi: 10.5858/2009-0583-RAR.1. [DOI] [PubMed] [Google Scholar]
- 38.Travis WD, Linnoila RI, Tsokos MG, et al. Neuroendocrine tumors of the lung with proposed criteria for large-cell neuroendocrine carcinoma: An ultrastructural, immunohistochemical, and flow cytometric study of 35 cases. Am J Surg Pathol. 1991;15:529–553. doi: 10.1097/00000478-199106000-00003. [DOI] [PubMed] [Google Scholar]
- 39.Phan AT, Oberg K, Choi J, et al. NANETS consensus guideline for the diagnosis and management of neuroendocrine tumors: Well-differentiated neuroendocrine tumors of the thorax (includes lung and thymus) Pancreas. 2010;39:784–798. doi: 10.1097/MPA.0b013e3181ec1380. [DOI] [PubMed] [Google Scholar]
- 40.Righi L, Volante M, Rapa I, et al. Mammalian target of rapamycin signaling activation patterns in neuroendocrine tumors of the lung. Endocr Relat Cancer. 2010;17:977–987. doi: 10.1677/ERC-10-0157. [DOI] [PubMed] [Google Scholar]
- 41.Zatelli MC, Minoia M, Martini C, et al. Everolimus as a new potential antiproliferative agent in aggressive human bronchial carcinoids. Endocr Relat Cancer. 2010;17:719–729. doi: 10.1677/ERC-10-0097. [DOI] [PubMed] [Google Scholar]
- 42.Frilling A, Akerström G, Falconi M, et al. Neuroendocrine tumor disease: An evolving landscape. Endocr Relat Cancer. 2012;19:R163–R185. doi: 10.1530/ERC-12-0024. [DOI] [PubMed] [Google Scholar]
- 43.Pasieka JL. Carcinoid tumors. Surg Clin North Am. 2009;89:1123–1137. doi: 10.1016/j.suc.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 44.Klöppel G, Perren A, Heitz PU. The gastroenteropancreatic neuroendocrine cell system and its tumors: The WHO classification. Ann N Y Acad Sci. 2004;1014:13–27. doi: 10.1196/annals.1294.002. [DOI] [PubMed] [Google Scholar]
- 45.Bendelow J, Apps E, Jones LE, et al. Carcinoid syndrome. Eur J Surg Oncol. 2008;34:289–296. doi: 10.1016/j.ejso.2007.07.202. [DOI] [PubMed] [Google Scholar]
- 46.Ramage JK, Davies AH, Ardill J, et al. Guidelines for the management of gastroenteropancreatic neuroendocrine (including carcinoid) tumours. Gut. 2005;54(suppl 4):iv1–iv16. doi: 10.1136/gut.2004.053314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arnold R, Rinke A, Klose KJ, et al. Octreotide versus octreotide plus interferon-alpha in endocrine gastroenteropancreatic tumors: A randomized trial. Clin Gastroenterol Hepatol. 2005;3:761–771. doi: 10.1016/s1542-3565(05)00481-7. [DOI] [PubMed] [Google Scholar]
- 48.Di Bartolomeo M, Bajetta E, Bochicchio AM, et al. A phase II trial of dacarbazine, fluorouracil and epirubicin in patients with neuroendocrine tumours. A study by the Italian trials in medical oncology (I.T.M.O.) group. Ann Oncol. 1995;6:77–79. doi: 10.1093/oxfordjournals.annonc.a059049. [DOI] [PubMed] [Google Scholar]
- 49.Kouvaraki MA, Ajani JA, Hoff P, et al. Fluorouracil, doxorubicin, and streptozotocin in the treatment of patients with locally advanced and metastatic pancreatic endocrine carcinomas. J Clin Oncol. 2004;22:4762–4771. doi: 10.1200/JCO.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 50.Moertel CG, Kvols LK, O’Connell MJ, et al. Treatment of neuroendocrine carcinomas with combined etoposide and cisplatin: Evidence of major therapeutic activity in the anaplastic variants of these neoplasms. Cancer. 1991;68:227–232. doi: 10.1002/1097-0142(19910715)68:2<227::aid-cncr2820680202>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 51.Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97:934–959. doi: 10.1002/cncr.11105. [DOI] [PubMed] [Google Scholar]
- 52.Marotta V, Franzese MD, Del Prete M, et al. Targeted therapy with kinase inhibitors in aggressive endocrine tumors. Expert Opin Pharmacother. 2013;14:1187–1203. doi: 10.1517/14656566.2013.796931. [DOI] [PubMed] [Google Scholar]
- 53.Marotta V, Sciammarella C, Vitale M, et al. The evolving field of kinase inhibitors in thyroid cancer. Crit Rev Oncol Hematol. 2015;93:60–73. doi: 10.1016/j.critrevonc.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 54.Chen H, Sippel RS, O’Dorisio MS, et al. The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: Pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas. 2010;39:775–783. doi: 10.1097/MPA.0b013e3181ebb4f0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Averbuch SD, Steakley CS, Young RC, et al. Malignant pheochromocytoma: Effective treatment with a combination of cyclophosphamide, vincristine, and dacarbazine. Ann Intern Med. 1988;109:267–273. doi: 10.7326/0003-4819-109-4-267. [DOI] [PubMed] [Google Scholar]
- 56.Halperin DM, Kulke MH, Yao JC. A tale of two tumors: Treating pancreatic and extrapancreatic neuroendocrine tumors. Annu Rev Med. 2015;66:1–16. doi: 10.1146/annurev-med-061813-012908. [DOI] [PubMed] [Google Scholar]
- 57.Reidy-Lagunes D, Thornton R. Pancreatic neuroendocrine and carcinoid tumors: What’s new, what’s old, and what’s different? Curr Oncol Rep. 2012;14:249–256. doi: 10.1007/s11912-012-0232-1. [DOI] [PubMed] [Google Scholar]
- 58.Oberg K, Casanovas O, Castano JP, et al. Molecular pathogenesis of neuroendocrine tumors: Implications for current and future therapeutic approaches. Clin Cancer Res. 2013;19:2842–2849. doi: 10.1158/1078-0432.CCR-12-3458. [DOI] [PubMed] [Google Scholar]
- 59.Cingarlini S, Bonomi M, Corbo V, et al. Profiling mTOR pathway in neuroendocrine tumors. Target Oncol. 2012;7:183–188. doi: 10.1007/s11523-012-0226-9. [DOI] [PubMed] [Google Scholar]
- 60.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zitzmann K, De Toni EN, Brand S, et al. The novel mTOR inhibitor RAD001 (everolimus) induces antiproliferative effects in human pancreatic neuroendocrine tumor cells. Neuroendocrinology. 2007;85:54–60. doi: 10.1159/000100057. [DOI] [PubMed] [Google Scholar]
- 62.Burris HA., III Overcoming acquired resistance to anticancer therapy: Focus on the PI3K/AKT/mTOR pathway. Cancer Chemother Pharmacol. 2013;71:829–842. doi: 10.1007/s00280-012-2043-3. [DOI] [PubMed] [Google Scholar]
- 63.Markman B, Dienstmann R, Tabernero J. Targeting the PI3K/Akt/mTOR pathway—Beyond rapalogs. Oncotarget. 2010;1:530–543. doi: 10.18632/oncotarget.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.O’Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chiu CW, Nozawa H, Hanahan D. Survival benefit with proapoptotic molecular and pathologic responses from dual targeting of mammalian target of rapamycin and epidermal growth factor receptor in a preclinical model of pancreatic neuroendocrine carcinogenesis. J Clin Oncol. 2010;28:4425–4433. doi: 10.1200/JCO.2010.28.0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Di Nicolantonio F, Arena S, Tabernero J, et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J Clin Invest. 2010;120:2858–2866. doi: 10.1172/JCI37539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bollard J, Couderc C, Blanc M, et al. Antitumor effect of everolimus in preclinical models of high-grade gastroenteropancreatic neuroendocrine carcinomas. Neuroendocrinology. 2013;97:331–340. doi: 10.1159/000347063. [DOI] [PubMed] [Google Scholar]
- 68.Vélayoudom-Céphise FL, Duvillard P, Foucan L, et al. Are G3 ENETS neuroendocrine neoplasms heterogeneous? Endocr Relat Cancer. 2013;20:649–657. doi: 10.1530/ERC-13-0027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.