

Abstract
Human APOBEC3B (A3B) is a member of the APOBEC3 (A3) family of cytidine deaminases, which function as DNA mutators and restrict viral pathogens and endogenous retrotransposons. Recently, A3B was identified as a major source of genetic heterogeneity in several human cancers. Here, we determined the solution NMR structure of the catalytically active C-terminal domain (CTD) of A3B and performed detailed analyses of its deaminase activity. The core of the structure comprises a central five-stranded β-sheet with six surrounding helices, common to all A3 proteins. The structural fold is most similar to that of A3A and A3G-CTD, with the most prominent difference found in loop 1. The catalytic activity of A3B-CTD is ~15-fold less than that of A3A, although both exhibit similar pH dependence. Interestingly, A3B-CTD with an A3A loop 1 substitution had significantly increased deaminase activity, while a single residue change (H29R) in A3A loop 1 reduced A3A activity to the level seen with A3B-CTD. This establishes that loop 1 plays an important role in A3-catalyzed deamination by precisely positioning the deamination-targeted C into the active site. Overall, our data provide important insights into the determinants for the activities of individual A3 proteins and facilitate understanding of their biological function.
Introduction
In humans, there are seven members of the APOBEC3 (A3) family of proteins, which deaminate deoxycytidine (dC) to deoxyuridine (dU) in single-stranded (ss) DNA substrates and function as DNA mutators1–7 (reviewed in refs.8–11). These proteins play an important role in the innate immune response to human pathogens by restricting exogenous viruses such as human immunodeficiency virus type 1 (HIV-1) (reviewed in refs.8,10–16), human T-lymphotropic leukemia virus type 1 (HTLV-1)17,18, and hepatitis B virus (HBV)19,20, as well as endogenous retroelements (reviewed in refs.10,21). A3 proteins are arranged in tandem arrays on human chromosome 2222 and contain one (A3A, A3C, A3H) or two (A3B, A3D, A3F, A3G) domains that possess characteristic Zn-binding motifs HX1EX23-24CX2-4C (with X being any amino acid) (reviewed in refs.9,23). The histidine and cysteine residues coordinate the Zn2+ ion, whereas the glutamic acid residue acts as a proton shuttle in the deaminase reaction8,24. The Zn-binding motifs have been classified as Z1, Z2, or Z3 type, based on phylogenetic analysis and sequence comparison25,26.
A3B possesses properties in common with the other A3 proteins as well as several features that are unique. Like A3G, its N- and C-terminal Zn-binding motifs belong to the Z2 and Z1 classes, respectively26, but the sequence of the A3B Z1 domain is closer to that of A3A than with A3G Z127 (Table 1). Although an early report suggested that both domains of A3B are catalytically active28, recent data indicate that only the C-terminal domain (CTD) has enzymatic activity, similar to A3G29–32. Furthermore, like all other A3 proteins except A3G, A3B exhibits a preference for substrates containing the TTCA sequence motif (the catalytic C is in bold)27,28,31,33, rather than CCCA, which is the preferred motif for A3G1,4,5,7,34,35.
Table 1.
Sequence comparison of A3B-CTD with other A3 proteins and theoretical isoelectric points (pI)
| Protein | Zinc-binding domain (Z) type | % Sequence identity with A3B-CTD (187–382)a | Theoretical pIb |
|---|---|---|---|
| A3B-CTD (187-382) | Z1 | 100 | 5.1 |
| A3A (10-199) | Z1 | 89 | 6.3 |
| A3G-CTD (191-384) | Z1 | 64 | 6.2 |
| A3C (12-190) | Z2 | 40 | 7.5 |
| A3B-NTD (12-186) | Z2 | 39 | 7.5 |
| A3G-NTD (12-181) | Z2 | 40 | 9.4 |
| A3B-FL (1-382) | - | - | 5.7 |
| A3G-FL (1-384) | - | - | 8.3 |
Sequence comparison was performed with the indicated residues for each protein using LALIGN138.
Theoretical pI values were calculated using the ProtParam online web-based program (http://web.expasy.org/protparam).
As shown for A3A36–45, A3B strongly inhibits retrotransposition of endogenous retroelements such as LINE-1, Alu, and intracytoplasmic A-type particles27,30,36–40,42,46,47. A3B also restricts HBV replication48–50, but there are divergent reports regarding its effect on chronic HBV infection51,52. Potent inhibition of HTLV-1 replication by A3B has been reported18, although restriction was shown by others to be minimal27 or effective for only a very small percentage of genomes53.
At present, it has not been resolved whether A3B mediates HIV-1 restriction or not, and substantially different degrees of restriction in cell-based studies, from high40,54–56 to modest or insignificant have been reported27,29,34,57–60. In addition, in contrast to all other A3 proteins except A3A (61,62; reviewed in refs.8,10–13,15,16,63), A3B restriction is not affected by the presence or absence of HIV-1 Vif34,54,57–59. There is also no clear consensus regarding A3B’s involvement in HIV-1 restriction in humans64,65, although a recent, carefully controlled study60 concluded that HIV-1 risk does not vary with a human polymorphism in which the entire 29.5-kb A3B gene is deleted66.
Strikingly, of all the human A3 proteins, A3B is the only one that has a predominant nuclear localization27,31,38–40,42,46,67. This raises the possibility that A3B might have access to chromosomal DNA and cause mutations or other damage, as was observed for A3A68–70, which is partitioned into the nucleus and the cytoplasm in non-myeloid cells36,38,39,71–73. In fact, in the last few years several groups who are investigating the nucleotide signatures associated with various cancers found indications for A3-induced mutagenesis in tumor genomes74–78. Interestingly, regional clusters of strand-coordinated hypermutation (C>T and/or C>G mutations), termed “kataegis” (Greek word for thunderstorm)75, in the context of TpC dinucleotides were commonly observed in breast75,78–80 and other cancers76–79,81. Although A3A alone is able to induce double-strand DNA breaks69,82–84, both A3A80,81 and A3B31,33,70,81,85,86 (also see review in ref.87) contribute to mutational effects in tumor genomes. Indeed, based on the available genomic data, it appears that A3B plays a major role in carcinogenesis and has limited activity as a restriction factor.
To gain insights into the question of how structure determines the molecular properties of A3B, we solved the solution NMR structure of wild-type (WT) A3B-CTD and investigated its substrate binding and catalytic activity. A3B-CTD has a structure common to all human A3 domains, with the greatest resemblance to the A3A88 and A3G-CTD89,90 structures as well as to the X-ray structures of an A3B-CTD solubility-enhanced mutant with four amino acid substitutions and deletion of loop 3 (i.e., F200S/W228S/L230K/F308K/Δloop3 [QMΔL3])91. Differences between A3B-CTD and A3A are noted for loop 1, which is longer in A3B-CTD and exhibits a larger degree of conformational plasticity than loop 1 in A3A. Under similar pH and buffer conditions, the catalytic activity of A3B-CTD is one order of magnitude less than that of A3A. Interestingly, comparison of the in vitro enzymatic activity of full-length (FL) A3B and A3B-CTD in mammalian cell extracts, shows an only ~2 – 3-fold reduction in activity, suggesting that the catalytic activity resides exclusively in the A3B-CTD, with the N-terminal domain (NTD) possibly enhancing binding affinity. Given A3B’s importance in cancer, elucidation of the A3B-CTD structure and how the details of the structure relate to catalytic activity is an essential first step to develop strategies for combatting the devastating nature of A3B’s DNA mutator activity.
MATERIALS AND METHODS
Construction of clones for expression in mammalian cells and bacteria
The FL WT A3B clone (catalog number 11090), used for 293T cell-based assays, was obtained through the AIDS Research and Reference Reagent Program (Division of AIDS, NIAID, NIH) from Bryan R. Cullen54. It comprises human pA3B-HA in pcDNA3, containing a 1275-bp KpnI/XhoI insert encoding the A3B gene linked to three 3′-terminal HA tags. Sequencing of the clone showed that position 62 is a glutamic acid (GAG codon). FL A3B E255Q (also referred to as E255Q), a catalytically dead mutant, was constructed in the pcDNA3 vector, using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA). A3B-CTD was constructed by using a 699-bp synthetic DNA encoding the 3′ end of A3B, corresponding to amino acid 187 in FL A3B to the end of the third 3′ HA tag, that was flanked by ATG (5′) and TAA (3′) codons. KpnI and XhoI linkers were inserted at the extreme 5′- and 3′-ends, respectively, for cloning into pcDNA3.1(+) (performed by GenScript, Piscataway, NJ). The HiSpeed Plasmid Maxi Prep kit (Qiagen, Inc.) was used for making large-scale plasmid preparations.
For bacterial expression in E. coli Rosetta 2 (DE3), the A3B-CTD coding sequence, comprising residues 187-382 of FL A3B with an added N-terminal methionine and a C-terminal His6-tag (LEHHHHHH), was inserted into the NdeI-XhoI site of the pET21 plasmid (Novagen). The pET21 plasmid, containing an A3B-CTD gene with D205G/ΔP206/ΔL207/ΔV208/L209I/R210G/R212H/Q213K mutations (A3B-CTD L1 mutant containing A3A loop 1) was purchased from GenScript for expression of the mutant protein. WT A3G-CTD (encoding residues 191-384 with an added N-terminal methionine and a C-terminal His6-tag) and WT A3C coding sequences with a C-terminal His6-tag were sub-cloned for expression, using the protocol described above for WT A3B-CTD. The A3A H29R mutation was constructed using the QuikChange Kit and sub-cloned as described above. The A3B sequence in each plasmid used for mammalian cell expression was verified by DNA sequencing performed by ACGT (Wheeling, IL) and verification of the sequences for the bacterially expressed constructs was performed by Genewiz (South Plainfield, NJ).
Preparation of mammalian cell extracts
Propagation of 293T cells, transfection, preparation of cell extracts, and determination of protein concentration were performed as described previously44,92. The relative expression levels of A3B were determined by Western blot analysis of cell extracts (10 μg of total protein for each sample), using the Western Breeze chemiluminescent Western blot kit (Life Technologies, Grand Island, NY). The primary antibodies were anti-HA (Cell Signaling Technologies, Danvers, MA) for detection of all A3B proteins and anti-tubulin (Abcam, Cambridge, MA) for the loading control. The secondary antibodies, AlexaFluor 488 goat anti-rabbit IgG and AlexaFluor 680 goat anti-mouse IgG, were obtained from Life Technologies.
Deaminase assay
Prior to performing the deaminase assay, the 293T cell extract (20 μg of total protein) was incubated at 37 °C for 15 min with RNase A (endonuclease-free, final concentration 1 μg/μl) (Qiagen) in a 20-μl reaction to remove potentially inhibitory cellular RNAs92. Deaminase assays, polyacrylamide gel electrophoresis and data analysis were performed as described previously44,92, except that reactions were incubated for 5 h at 37 °C. The substrates were 40-nt Alexa-Fluor 488-labeled ssDNAs (Integrated DNA Technologies (IDT), Coralville, IA) containing the following sequence: 5′-(Alexa488) ATT ATT ATT ATT ATT ATT ATN NNN TTT ATT TAT TTA TTT A-3′, where NNNN represents the motif for deamination. Note that for ACCCA, the motif consists of five bases: an A was substituted for the T immediately downstream of the targeted C in the 40-nt substrate (see below, JL1095). The motifs that were tested were: TTCA (JL913); TTCT (JL1043); TTCG (JL1094); TGCA (JL1178); and ACCCA (JL1095)92. The deamination activity was expressed as percent deamination product over total signal. To compare the deaminase activities of FL A3B and its variants, the percent deamination product was normalized against protein levels in the cell extracts, as determined by both chemiluminescence and fluorescence scanning of Western blots in the Typhoon 9400 Imager (GE Healthcare) using ImageQuant software (Figure 1A). The data represent the average of two independent determinations (each performed in duplicate) from two different transfections.
Figure 1.

Deaminase activities and sequence specificity of FL A3B and A3B-CTD. (A) Western blot analysis showing FL A3B, FL A3B E255Q, and A3B-CTD protein levels in 293T cell extracts. Transfection of the empty vector and E255Q were used as the negative controls. Note that the lack of A3B expression with the empty vector indicates that no endogenous A3B could be detected in the extracts. The tubulin signal served as a loading control. (B) The percent (%) deamination was calculated as described under Materials and Methods and was plotted against increasing amounts of total protein (μg) added. (C) Deaminase activity measured for cell extracts, visualized by gel electrophoresis. A 40-nt ssDNA containing the TTCA deaminase motif was used as the substrate. The oligonucleotide was incubated in a series of reactions with increasing amounts of each extract (1, 2, 3, and 5 μg of protein). Arrows to the right of the gel indicate the positions of the 40-nt ssDNA substrate and the deamination product. Lanes 1, 6, 11, and 16, no protein control; lanes 2 to 5, FL A3B; lanes 7 to 10, E255Q; lanes 12 to 15, A3B-CTD; lanes 17 to 20, empty vector. (D and E) Deaminase substrate sequence specificity of FL A3B (D) and A3B-CTD (E). Reactions contained 40-nt substrates with one of the following deaminase motifs: TTCA, TTCT, TTCG, TGCA, and ACCCA 92. The data were analyzed and plotted as described in Materials and Methods. Note that the values for deamination of the TTCA substrate with the FL (D) and CTD (E) A3B proteins were taken from the data shown in (B).
Protein expression and purification
Uniform 15N- and 13C-labeling of A3B-CTD was carried out by growth in modified minimal medium at 18 °C, using 15NH4Cl and 13C6-glucose as the sole nitrogen and carbon sources, respectively, and 2H-, 15N-, and 13C-labeling was achieved by growth in D2O, with 15NH4Cl, and 13C6/2H7-glucose as sole nitrogen and carbon sources, respectively. Selective protonation of Tyr/Phe/Ile/Val/Leu side chains was accomplished by adding 40–80 mg of 13C/15N-tyrosine, -phenylalanine, -isoleucine, and -valine to the D2O culture 1 h prior to induction with 0.5 mM isopropyl-1-thio-β-D-galactopyranoside (total induction time: 16 h). Proteins were purified by chromatography on a 5-ml HiTrap His column (GE Healthcare) in 50 mM sodium phosphate buffer (pH 7.5), containing 500 mM NaCl, 2 mM 2-mercaptoethanol, and 0.02% sodium azide, employing a linear gradient of 10–500 mM imidazole. A3B-containing fractions were passed over a Hi-Load Superdex 75 (1.6 cm × 60 cm) column in 25 mM sodium phosphate buffer (pH 7.5), containing 100 mM NaCl, 5 mM dithiothreitol (DTT), and 0.02% sodium azide, followed by ion-exchange chromatography over a 5-ml HiTrap-Q column (GE Healthcare), equilibrated with 25 mM Tris-HCl buffer (pH 8.5), 2 mM DTT, and 0.02% sodium azide, employing a linear gradient of 0–1 M NaCl. The other A3 proteins, i.e., A3B-CTD L1, A3A, A3A H29R, A3G-CTD, and A3C, were uniformly labeled with 15N only and purified according to the protocol described above for WT A3B-CTD. For experiments in which deaminase activity was measured as a function of pH, with the exception of A3C, all samples were buffer exchanged into NMR buffer (25 mM sodium phosphate, pH 5.5–8.1, 10 mM DTT, 0.02% sodium azide, and 7% D2O). The A3C sample was buffer exchanged into 10 mM Hepes buffer, pH 5.5–8.1, 300 mM arginine, 50 mM NaCl, 10 mM DTT, 0.02% sodium azide, and 7% D2O. As judged by electrophoresis in sodium dodecyl sulfate-polyacrylamide gels, all proteins were > 95% pure (data not shown). Importantly, the structural integrity of all proteins was confirmed by 1H-15N HSQC spectra (e.g., see spectra in Supplementary Figure S1).
Multi-angle light scattering
Size-exclusion chromatography/multi-angle light scattering (SEC-MALS) data were obtained at room temperature using an analytical Superdex 200 column with in-line multi-angle light scattering, refractive index (Wyatt Technology, Inc., Santa Barbara, CA), and UV (Agilent Technologies, Santa Clara, CA) detectors. 100 μl of 80 μM A3B-CTD were applied to the column in 25 mM sodium phosphate buffer, pH 6.9, 0.02% sodium azide, and 1 mM DTT at a flow-rate of 0.5 ml/min.
NMR Spectroscopy
All NMR spectra were recorded at 25 °C on Bruker AVANCE900, AVANCE800, AVANCE700, and AVANCE600 spectrometers, equipped with 5-mm triple-resonance, z-axis gradient cryoprobes. Backbone and side chain resonance assignments of A3B-CTD (in NMR buffer, pH 6.9, 25 °C) were performed using NMR data obtained from 2D 1H-15N HSQC, 2D 1H-13C HSQC, 3D HNCACB, HN(CO)CACB, HNCA, HN(CO)CA, HCCH-TOCSY,93 and simultaneous 13C- and 15N-edited NOESY spectra94. Distance restraints were derived from 3D simultaneous 13C- and 15N-edited NOESY and 2D NOESY experiments. All NOESY spectra were acquired at 900 MHz, using a mixing time of 100 ms (non-perdeuterated samples) or 150 ms (perdeuterated samples). Spectra were processed and analyzed with TOPSPIN 2.1 (Bruker), NMRPipe95, and Collaborative Computing Project for NMR (CCPN)96 programs.
NMR structure calculation
Structure calculations used the anneal.py protocol in XPLOR-NIH97. An iterative approach with extensive manual cross-checking of all distance restraints against the NOESY data and the generated structures was employed using CCPN. The final number of the NMR-derived restraints was 3548, with 3071 NOE distances, 168 H-bond distances, identified from NOE patterns for helices and β-sheets, and 309 ϕ and ψ backbone torsion angles from TALOS98. In addition, a Zn2+ ion with tetrahedral coordination to the H253, C284, and C289 side chains88,99,100 was added at a late stage in the calculations, using distances from the X-ray structure of A3G-CTD90,101. 256 structures were generated and the 30 lowest energy structures were selected and analyzed using PROCHECK-NMR102 (Table 2). Pockets in the A3 protein structures were identified by CASTp103–105 and rendered as Connolly surfaces106. All structure figures were generated with MOLMOL107 or PyMOL (the PyMOL Molecular Graphics System, Version 1.7.4 Schrödinger, LLC).
Table 2.
Statistics for the final 30 conformer ensemble of A3B-CTD
| Number of NOE distance constraints | |
| Intra-residue (i−j=0) | 1268 |
| Sequential (|i−j|=1) | 639 |
| Medium range (2≤|i−j|≤4) | 338 |
| Long range (|i−j|≥5) | 826 |
| Total | 3071 |
| Number of hydrogen bond constraints | 168 |
| Number of dihedral angle constraints | |
| ϕ | 155 |
| ψ | 154 |
| Total | 309 |
| Structural Quality | |
| Violationsa | |
| Distances constraints (Å) | 0.031 ± 0.002 |
| Dihedral angles constraints (°) | 0.397 ± 0.050 |
| Deviation from idealized covalent geometry | |
| Bond lengths (Å) | 0.002 ± 0.000 |
| Bond Angles (°) | 0.450 ± 0.007 |
| Improper torsions (°) | 0.255 ± 0.009 |
| Average r.m.s.d. of atomic coordinates (Å)b | |
| Backbone heavy atoms | 0.59 ± 0.04 |
| All heavy atoms | 1.18 ± 0.05 |
| Ramachandran plot analysis (%)c | |
| Most favorable region | 74.5 ± 2.3 |
| Additional allowed regions | 21.5 ± 2.6 |
| Generously allowed regions | 3.0 ± 1.1 |
| Disallowed regions | 1.0 ± 0.6 |
No individual member of the ensemble exhibited distance violations > 0.5 Å or dihedral angle violations >5°.
The average r.m.s. difference in atomic coordinates for residues 191-202, 214-240, and 253-377 was calculated for individual structures with respect to the mean structure. Flexible regions in the A3B-CTD structure (residues 187-190 (N-terminal), 203-213 (loop 1), 241-252 (loop 3), and 378-382 (C-terminus)) were excluded from the statistics.
Statistics were calculated using PROCHECK for the entire A3B-CTD (residues 187-382).
Single-stranded oligonucleotide binding to A3B
HPLC-purified ssDNA oligonucleotide (5′-ATTTUATTT-3′) was obtained from MIDLAND Co. (Midland, TX) or IDT and was prepared as a 21.7 mM stock solution in H2O. Binding was measured by adding aliquots of the stock solution to 42 μM 15N-labeled A3B-CTD in NMR buffer (pH 6.9) and recording binding isotherms from 1HN proton chemical shift changes in 2D 1H-15N HSQC spectra. Dissociation constants were calculated by best fitting of binding curves assuming 1:1 binding for 17 unambiguously traceable amide resonances using CCPN. The final Kd value is the average for the resonances.
A3-catalyzed deamination of ssDNAs
1D 1H NMR spectra of oligodeoxynucleotides were acquired at 25 °C as a function of time after addition of A3B-CTD, A3B-CTD L1, A3A, A3A H29R, A3G-CTD or A3C (~1.6 or ~16 μl of ~20 μM A3 stock solution) to 160 μl of 1 mM oligodeoxynucleotides in NMR buffer at pH ranging from ~5.5 to 8.1. Final concentrations were ~1 mM for oligodeoxynucleotides and ~0.2 or ~2 μM for A3 proteins. Initial rates of the C→U deamination reactions were determined from the intensities of C and U 1H-5 resonances in 1D 1H spectra.
Results
Deaminase activity of A3B-CTD and FL proteins present in mammalian cell extracts
The deaminase activities of FL A3B and A3B-CTD in cell extracts were compared, using proteins expressed in 293T cells (Figure 1A)44,92. The FL A3B E255Q mutant lacking catalytic activity and the empty vector served as negative controls (Figure 1B and C). Using the highest amount of extract (5 μg), FL A3B deaminated ~90% of the 40-nt ssDNA substrate with a TTCA motif in ~5 h, while the equivalent degree of deamination with A3A44 or A3H92 was achieved in ~ 1 h. For A3B-CTD, after 5 h, ~55% product was formed. Using lower protein concentrations, the difference was more pronounced, e.g., for 1 μg A3B-CTD extract, ~4-fold lower product was observed, compared to FL protein (Figure 1B and C). Thus, the NTD is not essential for deaminase activity, although its presence results in a modest increase in activity, presumably by increasing binding affinity to the substrate. Taken together, these results lend further support to earlier studies, concluding that A3B’s catalytic activity is localized to the CTD29–32.
To delineate the substrate specificities for A3B, assays were performed with five substrates that differed in the motif containing the deamination targeted C: TTCA; TTCG; TTCT, TGCA, and ACCCA (Figure 1D and 1E). Interestingly, the FL protein exhibited identical activity with three of these substrates, TTCA, TTCG, and TTCT, and ~2-fold lower activity with ACCCA (Figure 1D), indicating that T at the −1 position is preferred. No deamination was observed for the TGCA substrate, in contrast to A3H, which exhibited a low, but detectable level of deamination of this substrate92. Thus, the A3B substrate requires a T or C immediately 5′ to the targeted C and does not tolerate a G at this position.
Parallel experiments with A3B-CTD (Figure 1E) revealed that substrates that vary 3′ of the targeted C exhibited slight differences, with TTCG > TTCA > TTCT. However, the differences were very small (~1.5-fold) and deamination did not exceed ~70%. As shown for the FL protein, the poorest substrate for A3B-CTD in these assays was ACCCA and no deamination was detected for TGCA.
A3B-CTD NMR structure determination
The purified A3B-CTD was monomeric and monodisperse in solution (Figure 2, inset, lower left) and exhibited a well-dispersed 1H-15N HSQC spectrum at a protein concentration of 76 μM and pH values around pH 7 (Figure 2). At concentrations above 100 μM, however, the quality of the 1H-15N HSQC spectrum gradually deteriorated (Supplementary Figure S2), indicating non-specific aggregation. This aggregation can be alleviated by increasing the ionic strength, but only to some extent (Supplementary Figure S3). The final sample conditions used for the NMR structural studies described below were ~80 μM A3B-CTD in the absence of NaCl in 25 mM sodium phosphate buffer, pH 6.9, 10 mM DTT, and 0.02% sodium azide.
Figure 2.

800 MHz 1H-15N HSQC NMR spectrum of 76 μM 13C/15N-labeled A3B-CTD in 25 mM sodium phosphate, pH 6.9, 25° C. Assignments are indicated by residue name and number. An expansion of the boxed region is provided in the inset in the upper left corner. SEC-MALS data are shown in the inset in the lower left corner, with the elution profile indicated with black circles and the estimated molecular mass across the peak with blue triangles.
Nearly complete backbone amide (all, but 12 of 187 observable) (Figure 2) as well as most CαH and side chain (> 70%) resonances of A3B-CTD were assigned using routine multidimensional NMR experiments (see Materials and Methods). Missing backbone HN resonances were associated with loop 1 (D205, V208, L209, R210, and R211) and loop 3 (C239, N240, N244, and L245) residues, most likely due to severe line broadening of these resonances arising from conformational exchange on an intermediate (μ-ms) chemical shift time scale.
The structure of A3B-CTD was calculated on the basis of a total of 3548 NMR-derived experimental restraints (Table 2) and in the final 30-member conformational ensemble, all experimental restraints were satisfied and good covalent geometry was observed. A stereo-view of the 30-member ensemble and a ribbon representation of the lowest energy structure are provided in Figure 3A and 3B, respectively. For the well-ordered parts of the structure, excluding residues 187-190, 203-213, 241-252, and 378-382, the atomic r.m.s differences for the backbone and all heavy atoms with respect to the mean positions are 0.59 ± 0.04 and 1.18 ± 0.05 Å, respectively (Table 2). The overall structure consists of a central five-stranded β-sheet with six surrounding helices, common to all A3 proteins (A3A88,108, A3C109, A3F-CTD110–112, A3G-CTD89,90,99–101,113, and A3G-NTD114), with A3B-CTD most closely resembling the A3A88,108 and A3G-CTD89,90 structures (Supplementary Table S1). The current NMR structure of A3B-CTD is also very similar to the X-ray structure of the A3B-CTD mutant (QMΔL3)91.
Figure 3.

A3B-CTD NMR solution structure. (A) Stereo-view (defocused) of the backbone (N, Cα, C′) atoms of the final 30-conformer ensemble. Regions of helical and beta sheet structures are colored magenta and blue, respectively, and the remainder of the structure is colored grey. The Zn2+ ion is shown as a brown ball. (B) Ribbon representation of the lowest energy structure of the ensemble, using the same color scheme as in (A). Secondary structure elements are labeled. (C) Stereo-view (defocused) of the superposition of the active site regions of the current A3B-CTD NMR and the A3A NMR (PDB: 2M6588) structures. Secondary structure elements of the A3B-CTD structure are colored using the same color scheme as in (A) and (B) and those of A3A are colored in pink (helices), light blue (beta strands), and khaki (loops). The active site residues (H253, E255, C284, and C289 in A3B-CTD and H70, E72, C101, and C106 in A3A) and the Zn2+ ions are shown in ball and stick representation with carbon, nitrogen, oxygen, sulfur, and zinc atoms in green, blue, red, yellow, and brown, respectively, for A3B-CTD, or in pale green, cyan, red, yellow, and orange, respectively, for A3A. The active site residues are labeled only in the A3B-CTD structure. (D) Amino acid sequence alignment of loops 1, 3, 5, and 7 for A3B-CTD and A3A. The entire sequence alignment is given in Supplementary Figure S4. Identical residues are highlighted in yellow; residues in the loop 1 region that were changed to construct the A3B-CTD L1 mutant are enclosed in a black rectangle. Residues R212 (A3B-CTD) and H29 (A3A) are highlighted in magenta.
Interestingly, A3B-CTD possesses an interrupted β2 strand similar to A3A88,108 and A3G-CTD89,90,99,100,113. In addition, the active site residues in the A3B-CTD and A3A NMR structures are located in very similar positions (Figure 3C)88. However, differences between A3A and A3B-CTD were observed in the local conformations of the loops, in particular loop 1 (Figure 3), with the longer A3B-CTD loop 1 exhibiting extensive conformational plasticity (Figure 3A) and the associated amide resonances exhibiting severe broadening. By contrast, in A3A, the most extensive conformational exchange is seen for loop 3 resonances88.
Single-Stranded Oligonucleotide Binding to A3B-CTD
Binding studies were performed with a ssDNA U-containing oligonucleotide (5′-ATTTUATTT-3′), since for enzymatically active protein, using the C-containing substrate results in deamination over the course of the binding experiment and a mixture of substrate and product will be bound in the active site. This is a valid approach, as was previously ascertained for A3A88. Structural mapping of the resonance changes in the 1H-15N HSQC spectra of A3B-CTD after addition of the 5′-ATTTUATTT-3′ oligonucleotide showed that primarily residues near the catalytic site and in the surrounding loops (loops 1, 3, 5, and 7) were involved in binding (Figure 4A). For example, significant chemical shift changes were observed in the titration with the 9-nt oligonucleotide for the amide resonances of H253, E255, C284, and C289 (the Zn+2 coordinating residues in the catalytic site), Q213 and T214 (loop 1), K243, C247, G248, F249, Y250, and R252 (loop 3), W281, S282, S286, W287, and G288 (loop 5), and D314, Y315, and D316 (loop 7) (Figure 4A and 4B). This demonstrates that DNA binding affects an extended surface of A3B-CTD that includes all of these loops. Importantly, the amide resonance of Y315 in loop 7 (Y132 in A3A and D317 in A3G), a residue implicated in determining TC versus CC specificity of A3 deaminases, was also affected115. In addition, a striking resemblance was observed for the magnitude and direction of the (5′-ATTTUATTT-3′)-binding-induced changes in the A3B-CTD (Figure 4B) and A3A (Figure 4C; see Figure 4b and 5a in ref.88) 1H-15N HSQC spectra. This suggests that essentially the same residues in both proteins are involved in the interaction, although binding by A3B-CTD (Kd, 0.119 ± 0.026 mM) is two-fold weaker than by A3A (Kd, 0.058 ± 0.008 mM88).
Figure 4.

Structural mapping of 5′-ATTTUATTT-3′ binding to A3B-CTD. (A) Residues whose resonances experience significant 1H,15N chemical shift changes upon binding are colored red (>0.050 p.p.m.) or orange (0.028–0.050 p.p.m.). 1H,15N-combined chemical shift changes were calculated using , with ΔδHN and ΔδN representing 1HN and 15N chemical shift differences, respectively. (B) and (C) Binding isotherms for representative 1HN resonances of A3B-CTD (B) and A3A (C).
Deamination studies by NMR
A3B-CTD-catalyzed deamination of a 15-nt ssDNA substrate, 5′-ATTATTTCATTTATT-3′, was followed by monitoring the disappearance of the cytosine 1H-5 doublet and appearance of the 1H-5 doublet of uracil in 1D 1H NMR spectra88,100 (Figure 5A, inset). At pH 7.1, ~1 mM substrate was completely converted to product by ~0.2 μM enzyme in ~150 h, with an initial reaction rate of 0.046 mM h−1 (Figure 5A). This rate is one order of magnitude lower (~15-fold) than the rate of the A3A-catalyzed reaction (i.e., 0.75 mM h−1), under similar conditions (pH 6.9) (Figure 5B).
Figure 5.
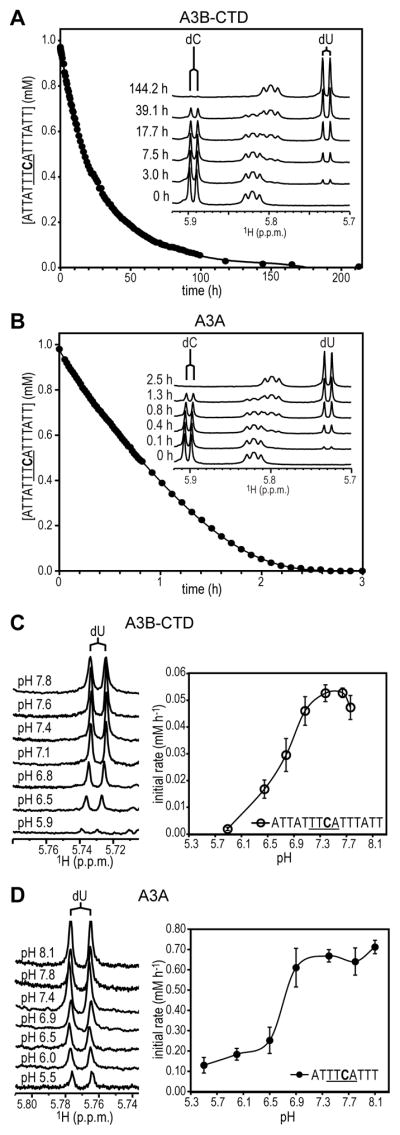
A3B-CTD and A3A catalyzed deamination reactions and pH dependence. A series of 1D 1H NMR spectra of the 15-nt ssDNA (5′-ATTATTTCATTTATT-3′) substrate after addition of A3B-CTD at pH 7.1 (A) or A3A at pH 6.9 (B) were recorded at 25 °C. The time for each 1D measurement was ~1.8 min. The intensities of the 1H-5 resonances in the 1D spectra of the substrate (dC) and product (dU) were measured for calculation of unreacted substrate concentrations, which were plotted as a function of reaction time. Representative 1D 1H NMR spectra recorded at the indicated reaction times are shown in the insets. Spectra at different pH values for A3B-CTD at the 3 h time point (C, left panel) and A3A at the 0.2 h time point (D, left panel) and initial rates of deamination for A3B-CTD (C, right panel) and A3A (D, right panel) with 5′-ATTATTTCATTTATT-3′ and 5′-ATTTCATTT-3′, respectively, as substrates. Using the 9- and 15-nt substrates for activity comparisons is valid, since the dC in these substrates is deaminated at essentially identical rates by A3A 88 as well as by A3B-CTD (data not shown). The initial deamination rates were calculated as in (A) and (B). The error bars represent the SD obtained from two to four independent measurements. Concentrations of ssDNA and enzyme were ~1 mM and ~0.2 μM, respectively.
The pH dependence of the deamination reaction was followed over a pH range from 5.9–7.8 (Figure 5C). Measuring the product (5′-ATTATTTUATTTATT-3′) at the 3 h time point showed the highest activity at basic pH values (Figure 5C, left panel). The initial reaction rate was used to determine the pH optimum, which was ~7.5 (initial rate, 0.053 mM h−1) (Figure 5C, right panel); at lower pH, e.g., pH 5.9, the initial rate was only 0.002 mM h−1, ~30-fold lower than at pH ~7.5. In contrast, A3A-catalyzed deamination of the 15-nt ssDNA substrate 5′-ATTATTTCATTTATT-3′ occurred more rapidly and the reaction was essentially complete at 2 h (Figure 5B, pH 6.9). The pH dependence of the A3A deamination reaction with the 9-nt 5′-ATTTCATTT-3′ substrate was followed over a pH range from 5.5–8.1 and product conversion was measured at the 0.2 h time point (Figure 5D, left panel). The initial reaction rate increased from pH 5.5 to pH 6.9, but did not change significantly for pH values above pH 6.9 (Figure 5D, right panel). Thus, a discrete pH optimum was not observed here for A3A. These results contrast with those of Pham et al.116, who used a different protein construct and different assay conditions.
Since the largest difference between A3B-CTD and A3A is associated with loop 1 (Figure 3D and Supplementary Figure S4), we prepared a chimaera in which loop 1 from A3A, i.e., 23NNGIGRHKT31, was substituted for loop 1 in A3B-CTD, i.e., 203NNDPLVLRRRQT214. Compared with WT A3B-CTD (Figure 5C, right panel, and Figure 6A), the hybrid protein exhibited one order of magnitude higher activity with TTCA-containing substrates (Figure 6C), i.e., only two-fold lower than the highest activity for A3A (Figure 5D, right panel, and Figure 6B), and a pH dependence similar to that seen for A3A. Changing loop 1 in A3A by replacing a single amino acid, H29R, reduced the activity 10-fold compared to WT, with no change in pH dependence (Figure 6D). This is in contrast to the comparable A3G H216R mutant, which unlike WT A3G, exhibits only a small (1.5-fold) increase in deamination activity when the pH is changed from 7.4 to 5.5117.
Figure 6.

pH dependence of the deamination rates of selected A3 proteins. (A) A3B-CTD WT; (B) A3A WT; (C) A3B-CTD L1 mutant; (D) A3A H29R mutant; (E) A3G-CTD; and (F) A3C. The following substrates were used: 5′-ATCCCATTT-3′, 5′-ATTTCATTT-3′, 5′-ATATCATTT-3′, and 5′-ATTATTTCATTTATT-3′. In the insets in (A), (D), (E), and (F), the y-axes showing the initial rates are expanded. The values for A3B-CTD WT in (A) with the 5′-ATTATTTCATTTATT-3′ substrate (empty circles) and for A3A in (B) with the 5′-ATTTCATTT-3′ substrate (filled circles) were taken from the data shown in Figure 5C and 5D (right panels), respectively. Concentrations of ssDNA and enzymes were ~1 mM and ~0.2 or ~2 μM, respectively. The data were normalized to an enzyme concentration of 0.2 μM and the error bars represent the S.D. from two to four independent measurements.
For comparison, we also investigated the pH dependence of WT A3G-CTD (Figure 6E) and A3C deaminase activities (Figure 6F). For A3G-CTD with 5′-ATCCCATTT-3′, which contains A3G’s preferred motif (CCCA)5,7, activity was higher at low pH values < 6.5; the initial rate was ~0.12 mM h−1. This value is ~6-fold lower than the highest activity of WT A3A (initial rate, ~0.7 mM h−1; Figure 5D, right panel), but ~2-fold higher than the maximum activity of A3B-CTD (initial rate, ~0.05 mM h−1; Figure 5C, right panel). The higher activity of A3G at acidic pH agrees with the results of Harjes et al.117.
Substrate specificities of A3B-CTD, A3A, A3G-CTD, and A3C
Substrate specificities for several A3 proteins were evaluated using identical reaction conditions with several ssDNA substrates containing the following motifs: TTCA; ATCA; and CCCA, ATCC, and TCCC (Figure 6). 5′-ATATCATTT-3′ was the preferred substrate for A3B-CTD, followed by 5′-ATTATTTCATTTATT-3′, 5′-ATCCCATTT-3′, and 5′-ATCCCATTT-3′ (Figure 6A). 5′-ATCCCATTT-3′ was an especially poor substrate and was barely converted to product (initial rate ≤ 0.0001 mM h−1). The ~1.4-fold preference for the ATCA vs. the TTCA motif, while very small, is in agreement with a recent report118 that described A3B’s preference for a purine in the -2 position, based on statistical analyses of deaminase data using long genomic ssDNA substrates in yeast model systems.
For A3A, the substrate preferences were: 5′-ATTTCATTT-3′ (defined as 100%) > 5′-ATATCATTT-3′ ≈ 5′-ATCCCATTT-3′ (~60% and ~50% respectively) > 5′-ATCCCATTT-3′ (~10%) > 5′-ATCCCATTT-3′ (~2%) (Figure 6B). The strong ability of A3A to deaminate the C in substrates containing TTCA or CCCA motifs agrees well with the results of our earlier A3A study88. Although Love et al.119 concluded that A3A lacks a distinct preference for TTC over ATC, GTC, or CTC, our data reveal a small, but reproducible trend, showing that A3A prefers a pyrimidine (TTCA) rather than a purine (ATCA) at the -2 position. These data are in accord with the recent study cited above118, comparing the deaminase motif preferences of A3A and A3B in yeast systems.
Interestingly, although A3A preferred TTCA over ATCA or CCCA under our conditions, the cytosines in the latter two motifs were deaminated fairly effectively, by at least a factor of 6-fold faster than was seen with A3B-CTD and its most preferred substrate, 5′-ATATCATTT-3′. Furthermore, the preference for an A immediately 3′ of the targeted C, also appeared to be important, since the rate was ~5-fold less for a C in this position (compare the rates with 5′-ATATCATTT-3′ and 5′-ATCCCATTT-3′). For deamination by A3A, a distinct pH maximum at pH 6.9 was noted with 5′-ATCCCATTT-3′, whereas for the other substrates, no drop in deamination rates was seen above pH ≥ 7.
Compared with A3B-CTD WT (Figure 6A), the A3B-CTD L1 mutant exhibited substantially higher (5- to 50-fold) deaminase activity towards all of the substrates tested (Figure 6C), essentially transferring A3A characteristic properties to A3B-CTD. Substrate specificity was also affected by the mutation, since the L1 mutant no longer showed a preference for ATCA over TTCA. The L1 mutant also exhibited a shift in the pH optimum from basic pH values (> pH 7) to slightly lower pH values (6.5–6.9), most clearly seen with 5′-ATCCCATTT-3′. Conversely, the A3A H29R mutation converted A3A to A3B-CTD-like and resulted in substantially lower deaminase activity (~10-fold) than A3A WT towards all tested substrates (Figure 6D).
Investigation of the substrate specificities for A3G-CTD confirmed the preference for the CCCA motif in 5′-ATCCCATTT-3′. The rate of deamination of other C bases in this substrate, e.g., TCCC and ATCC, was 100-fold lower or undetectable, respectively (Figure 6E, insert). A3G-CTD deamination of the ATCA motif in 5′-ATATCATTT-3′ and the TTCA motif in 5′-ATTTCATTT-3′ was at the limit of detection, i.e., 200- to 400-fold lower than was seen with the preferred CCCA motif.
In contrast to the other A3 proteins, none of the C bases in the 5′-ATCCCATTT-3′ substrate was deaminated by A3C (pH 5.5–8.0) and deamination of 5′-ATTTCATTT-3′ by A3C was very poor under the experimental conditions used here (pH 6.1–7.5) (Figure 6F), even after 1 day of incubation with 0.2 μM of the enzyme. For higher concentrations of enzyme, 2 μM A3C, and 1 mM substrate, 1–2% conversion to product was observed within 10 h and the initial rate was 0.002–0.004 mM h−1. Thus, A3C deaminase activity is ~2000-fold lower than that of A3A. These data are consistent with the low levels observed in assays for A3C DNA mutator activity57,120 and the finding that A3C-induced mutations were observed only infrequently (even at preferred 5′ TC sites) in the DNA of A3C-expressing cells infected with several patient-derived HIV-1 molecular clones121.
In summary, A3A is the most powerful deaminase among all of the A3 proteins tested in this study and is able to deaminate cytidines in all substrates tested. The pH dependence of A3A, the A3A H29R mutant, A3B-CTD, and the A3B-CTD L1 mutant was strikingly different from that of A3G-CTD (Figure 6). Finally, the rank order of deaminase activity for the A3 proteins examined here is: A3A ≫ A3G-CTD ≥ A3B-CTD ⋙ A3C.
Structural basis for different deamination activities of A3 proteins
Since A3B and A3A exhibited dramatic differences in enzyme activities, despite similar global binding sites and affinities for ss DNA oligonucleotides, a detailed comparison of their active site pockets and surrounding loops was performed. The binding site pocket in the A3B-CTD NMR structure (Figure 7A) (230 Å3, this study) is much smaller than the pocket found in the A3A NMR structure (Figure 7B) (843 Å3, PDB: 2M6588). In addition, loop conformations and dynamics in or adjacent to the binding sites exhibit distinct features in the two structures, which clearly modulate activity. For example, the A3A loop 1 is short, while A3B-CTD, A3G-CTD, and A3C possess longer loops, which may partially occlude the active site. If conformational selection is important in the formation of the productive catalytic complex, the binding competent conformations have to be selected from the flexible loop 1, loop 3, loop 5, and loop 7 ensemble for accurately accommodating the cytosine in the active site. Thus, for A3 proteins, it seems likely that in addition to the size of the active site pocket, the details of the surrounding loops are the most critical determinants for enzyme activity.
Figure 7.

Comparison of the active sites in the NMR solution structures of A3B-CTD and A3A. Pockets in (A) A3B-CTD WT (this study) and (B) A3A WT (PDB: 2M6588) are shown as grey meshes.
Discussion
In the present work, we report the NMR solution structure of the catalytic CTD of WT A3B and provide a detailed analysis of substrate binding and deaminase activity for several members of the A3 family. The A3B CTD structure exhibits the same architecture as other A3 proteins, consisting of a five-stranded β-sheet core that is surrounded by six α-helices. It is very similar to the recently determined A3B-CTD X-ray structure91, exhibiting a pairwise r.m.s. difference of 1.56 Å (N, Cα, C′ backbone atoms), and is most similar to A3A, compared to other A3 proteins (Supplementary Table S1). However, distinct differences are seen in the loop conformations, most notably for loop 1, which is longer and more flexible in A3B-CTD than the corresponding loop in A3A.
Despite numerous reports describing A3 protein oligomerization and its relation to deaminase activity10,90,122–131, there is still no consensus regarding the oligomerization states of some of these proteins (see for example, A3A refs.88,108,116,119,130,132). Interestingly, FL A3B has been reported to form multimers in cells130. However, here, we show unequivocally by SEC-MALS and NMR that WT A3B-CTD (Figure 2) exists solely as a monomer in solution, in agreement with Shi et al., who reported that the A3B-CTD QMΔL3 mutant is monomeric91. In contrast, Siriwardena et al.133 reported that A3B-CTD (residues 193-382, a construct with a six-residue truncation at the N-terminus, compared to the construct used in this study and by Shi et al.91), exists as a monomer-dimer equilibrium. Importantly, both the NMR (this study) and X-ray91 A3B-CTD structures show that the missing residues in the truncated protein (e.g., Y191 and L192) are an integral part of the folded protein structure, suggesting that truncation most likely destabilizes the protein and that the shorter A3B-CTD may undergo non-specific association.
Although all A3 structures exhibit the same global fold (Supplementary Table S1), with very similar catalytic sites, there are differences in the structural and motional properties of the loops, particularly loops 1, 3, 5 and 7, surrounding the catalytic site. For example, for A3B-CTD, the most significant conformational variability is seen for loop 1 and loop 3 (Figure 3). This conformational flexibility is important for substrate binding and several resonances of loop 1 are very broad and undetectable in the free enzyme. Loop 1 (Q213 and T214), loop 3 (K243, C247, G248, F249, Y250, and R252), loop 5 (W281, S282, S286, W287, and G288), and loop 7 (D314, Y315, and D316) residues experience large amide chemical shift changes upon ssDNA binding, suggesting that the conformation of these loops is rearranged upon substrate binding (Figure 4A and 4B). The striking similarity around the catalytic site in both A3B-CTD and A3A, exploited for oligonucleotide binding, can be seen by the DNA binding-induced chemical shift changes for residues in or close to the active site (Figure 4B and 4C). For example, T214/T31, E255/E72, W287/W104, and C289/C106 exhibit remarkably similar changes upon 5′-ATTTUATTT-3′ oligonucleotide binding. Since the catalytic activities of A3B-CTD and A3A are very different, this demonstrates that binding alone is only necessary, but not sufficient for catalysis.
The tryptophan and glycine residues between the two Zn+2 coordinating cysteines (C101 and C106 in A3A; C284 and C289 in A3B-CTD), present as a two-residue insertion only in A3A and A3B, disrupt a helical conformation, but keep the catalytic site structure intact (Figure 7). This region protrudes out from the body of the protein in the NMR structures of WT A3A and A3B-CTD (Supplementary Figure S5), while it is folded back onto the protein in the X-ray structures of the inactive A3A E72A mutant108 and the A3B-CTD QMΔL3 mutant91 (Supplementary Figure S5). This difference may simply reflect the possibility that both conformations can exist, given the flexibility of this region in solution (Figure 3A).
Despite the 89% sequence identity (Table 1), A3A and A3B-CTD exhibit substantially different activities: A3B-CTD is a much less active enzyme compared to A3A (Figure 5 and 6), even in the context of the FL enzyme (Figure 1)88. Comparison of the amino acid sequences of A3B-CTD and A3A (Supplementary Figure S4) reveals that most differences reside in the first ~30 amino acids of the two proteins (15 out of 25). They cluster in α1 and loop 1, with the latter being three residues shorter in A3A, compared to all other A3 proteins (Figure 3D)92. Since grafting the shorter A3A loop 1 onto A3B-CTD (A3B-CTD L1 mutant) resulted in an order of magnitude increase in deaminase activity, it appears that a shorter loop 1 enhances enzyme function. Within loop 1 of A3A, H29 plays a pivotal role, since changing the histidine to arginine, the corresponding residue in A3B-CTD (Figure 3D), dramatically decreased enzymatic activity in the A3A H29R mutant to the level observed with A3B-CTD (Figure 6). Inspecting the pH dependence of the activity reveals that the A3A and A3B-CTD L1 mutant activities are highest above pH 7, clearly implicating this histidine in modulating activity. Among the other A3 proteins, A3G-CTD also possesses a histidine (H216) in loop 1, but this loop is longer than the A3A loop 1. In this case, A3G-CTD activity is higher at the low pH side of the histidine pKa (Figure 6E) and a protonated H216 imidazole ring may be necessary for A3G-CTD activity, as discussed by Harjes et al.117, again highlighting the importance of the detailed conformation of loop1 residues for substrate recognition and positioning.
Although the correlation between deaminase activity and the size of the catalytic pocket in A3A and A3B-CTD seems intriguing (Figure 7), it is likely that the flexible nature of the surrounding loops is a more critical feature governing the enzymatic functions of A3 proteins. Indeed, it appears that catalytic activity is compatible with conformational rearrangements of the structure for optimally positioning substrates into the active site. The C (A3G) or T (all other A3 proteins) specificity at the −1 position appears to be governed by loop 7115,134–137 and a single residue changes in loop 7 (D314R in A3B) switches the −1 specificity from T to C91. Similarly, our data show that A3B-CTD and A3A, for which differences in the amino acid sequences are found mainly in the N-terminal 30 amino acids, particularly loop 1, also differ in the −2 position specificity (TTCA vs. ATCA, Figure 6A and B), suggesting that the −2 position specificity is affected by the N-terminal region of the proteins, including loop 1. Interestingly, interactions between loop1 and loop 7 residues have been noted in several A3 structures, and it may be the loss or gain of interactions between loop 1 and loop 7 that governs the selectivity of bases around the deamination-targeted base, permitting precise positioning of the proteins’ catalytic residues, necessary for performing the chemical reaction.
In summary, analysis of the human A3B-CTD NMR solution structure reported in the present study in conjunction with a detailed evaluation of DNA binding and catalytic activity for this protein explains why, among the A3 proteins, A3B-CTD is a weak deaminase and A3A is the most active enzyme. Yet, despite its poor activity, A3B’s genomic DNA mutator activity causes a wide variety of tumors. Knowledge of which determinants in the three-dimensional structure of the A3B catalytic domain influence enzymatic activity should greatly strengthen efforts to develop novel agents that prevent the devastating consequences of human cancer caused by the A3B enzyme.
Supplementary Material
Figure S1. 1H-15N HSQC NMR spectra of A3B-CTD WT, A3B-CTD L1 mutant, A3A WT, and A3A H29R mutant in NMR buffer (PDF)
Figure S2. 1H-15N HSQC NMR spectra of A3B-CTD WT in NMR buffer at protein concentrations of 43 μM, 76 μM, and 170 μM (PDF)
Figure S3. 1H-15N HSQC NMR spectra of A3B-CTD WT in NMR buffer containing 100 mM NaCl at protein concentrations of 50 μM, 143 μM, and 290 μM (PDF)
Figure S4. Amino acid sequence alignment of A3B-CTD and A3A (PDF)
Figure S5 Stereo-view of the superposition of the active site regions of the A3B-CTD NMR and the A3B-CTD X-ray structures (PDF)
Table S1. Structural comparison of APOBEC3 protein (PDF)
Acknowledgments
The authors thank Dr. Jinwoo Ahn for helpful advice on protein purification, Doug Bevan for computer technical support, Michael J. Delk for NMR instrumental support, Dr. Charles D. Schwieters for the Zn2+ coordination parameter and topology files, and the NIH AIDS Research and Reference Reagent Program for the A3B clone, as detailed in the text.
FUNDING
This work was supported by National Institutes of Health Grant P50GM082251 (to A.M.G.) and the Intramural Research Program at the National Institutes of Health, Eunice Kennedy Shriver National Institute of Child Health and Human Development (to J.G.L.).
Abbreviations
- A3
APOBEC3
- A3B
APOBEC3B
- CTD
C-terminal domain
- dC
deoxycytidine
- dU
deoxyuridine
- ss
single-stranded
- HIV-1
human immunodeficiency virus type 1
- HTLV-1
human T-lymphotropic leukemia virus type 1
- HBV
hepatitis B virus
- FL
full-length
- NTD
N-terminal domain
- WT
wild-type
Footnotes
Accession Codes
The A3B-CTD atomic coordinates and NMR constraints have been deposited in the PDB with the accession code 2nbq and the 1H, 13C, and 15N NMR chemical shift data of A3B-CTD have been deposited in the BMRB under entry code 25985.
Notes
The authors declare no competing financial interest.
The supporting information is available free of charge on the ACS Publications website at DOI: XXXX.
References
- 1.Harris RS, Petersen-Mahrt SK, Neuberger MS. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol Cell. 2002;10:1247–1253. doi: 10.1016/s1097-2765(02)00742-6. [DOI] [PubMed] [Google Scholar]
- 2.Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH. DNA deamination mediates innate immunity to retroviral infection. Cell. 2003;113:803–809. doi: 10.1016/s0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- 3.Lecossier D, Bouchonnet F, Clavel F, Hance AJ. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science. 2003;300:1112. doi: 10.1126/science.1083338. [DOI] [PubMed] [Google Scholar]
- 4.Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature. 2003;424:94–98. doi: 10.1038/nature01707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu Q, König R, Pillai S, Chiles K, Kearney M, Palmer S, Richman D, Coffin JM, Landau NR. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat Struct Mol Biol. 2004;11:435–442. doi: 10.1038/nsmb758. [DOI] [PubMed] [Google Scholar]
- 6.Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424:99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- 7.Chelico L, Pham P, Calabrese P, Goodman MF. APOBEC3G DNA deaminase acts processively 3′ --> 5′ on single-stranded DNA. Nat Struct Mol Biol. 2006;13:392–399. doi: 10.1038/nsmb1086. [DOI] [PubMed] [Google Scholar]
- 8.Harris RS, Liddament MT. Retroviral restriction by APOBEC proteins. Nat Rev Immunol. 2004;4:868–877. doi: 10.1038/nri1489. [DOI] [PubMed] [Google Scholar]
- 9.Bransteitter R, Prochnow C, Chen XS. The current structural and functional understanding of APOBEC deaminases. Cell Mol Life Sci. 2009;66:3137–3147. doi: 10.1007/s00018-009-0070-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiu YL, Greene WC. The APOBEC3 cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu Rev Immunol. 2008;26:317–353. doi: 10.1146/annurev.immunol.26.021607.090350. [DOI] [PubMed] [Google Scholar]
- 11.Malim MH. APOBEC proteins and intrinsic resistance to HIV-1 infection. Phil Trans R Soc Lond B Biol Sci. 2009;364:675–687. doi: 10.1098/rstb.2008.0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goila-Gaur R, Strebel K. HIV-1 Vif, APOBEC, and intrinsic immunity. Retrovirology. 2008;5:51. doi: 10.1186/1742-4690-5-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Imahashi M, Nakashima M, Iwatani Y. Antiviral mechanism and biochemical basis of the human APOBEC3 family. Front Microbiol. 2012;3:250. doi: 10.3389/fmicb.2012.00250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duggal NK, Emerman M. Evolutionary conflicts between viruses and restriction factors shape immunity. Nat Rev Immunol. 2012;12:687–695. doi: 10.1038/nri3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Desimmie BA, Delviks-Frankenberrry KA, Burdick RC, Qi D, Izumi T, Pathak VK. Multiple APOBEC3 restriction factors for HIV-1 and one Vif to rule them all. J Mol Biol. 2014;426:1220–1245. doi: 10.1016/j.jmb.2013.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng Y, Baig TT, Love RP, Chelico L. Suppression of APOBEC3-mediated restriction of HIV-1 by Vif. Front Microbiol. 2014;5:450. doi: 10.3389/fmicb.2014.00450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sasada A, Takaori-Kondo A, Shirakawa K, Kobayashi M, Abudu A, Hishizawa M, Imada K, Tanaka Y, Uchiyama T. APOBEC3G targets human T-cell leukemia virus type 1. Retrovirology. 2005;2:32. doi: 10.1186/1742-4690-2-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ooms M, Krikoni A, Kress AK, Simon V, Münk C. APOBEC3A, APOBEC3B, and APOBEC3H haplotype 2 restrict human T-lymphotropic virus type 1. J Virol. 2012;86:6097–6108. doi: 10.1128/JVI.06570-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turelli P, Mangeat B, Jost S, Vianin S, Trono D. Inhibition of hepatitis B virus replication by APOBEC3G. Science. 2004;303:1829. doi: 10.1126/science.1092066. [DOI] [PubMed] [Google Scholar]
- 20.Köck J, Blum HE. Hypermutation of hepatitis B virus genomes by APOBEC3G, APOBEC3C and APOBEC3H. J Gen Virol. 2008;89:1184–1191. doi: 10.1099/vir.0.83507-0. [DOI] [PubMed] [Google Scholar]
- 21.Koito A, Ikeda T. Intrinsic immunity against retrotransposons by APOBEC cytidine deaminases. Front Microbiol. 2013;4:28. doi: 10.3389/fmicb.2013.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jarmuz A, Chester A, Bayliss J, Gisbourne J, Dunham I, Scott J, Navaratnam N. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics. 2002;79:285–296. doi: 10.1006/geno.2002.6718. [DOI] [PubMed] [Google Scholar]
- 23.Holmes RK, Malim MH, Bishop KN. APOBEC-mediated viral restriction: not simply editing? Trends Biochem Sci. 2007;32:118–128. doi: 10.1016/j.tibs.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 24.Betts L, Xiang S, Short SA, Wolfenden R, Carter CW., Jr Cytidine deaminase. The 2.3 Å crystal structure of an enzyme: transition-state analog complex. J Mol Biol. 1994;235:635–656. doi: 10.1006/jmbi.1994.1018. [DOI] [PubMed] [Google Scholar]
- 25.LaRue RS, Jónsson SR, Silverstein KA, Lajoie M, Bertrand D, El-Mabrouk N, Hotzel I, Andrésdóttir V, Smith TPL, Harris RS. The artiodactyl APOBEC3 innate immune repertoire shows evidence for a multi-functional domain organization that existed in the ancestor of placental mammals. BMC Mol Biol. 2008;9:104. doi: 10.1186/1471-2199-9-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.LaRue RS, Andrésdóttir V, Blanchard Y, Conticello SG, Derse D, Emerman M, Greene WC, Jónsson SR, Landau NR, Löchelt M, Malik HS, Malim MH, Münk C, O’Brien SJ, Pathak VK, Strebel K, Wain-Hobson S, Yu X-F, Yuhki N, Harris RS. Guidelines for naming nonprimate APOBEC3 genes and proteins. J Virol. 2009;83:494–497. doi: 10.1128/JVI.01976-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pak V, Heidecker G, Pathak VK, Derse D. The role of amino-terminal sequences in cellular localization and antiviral activity of APOBEC3B. J Virol. 2011;85:8538–8547. doi: 10.1128/JVI.02645-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bogerd HP, Wiegand HL, Doehle BP, Cullen BR. The intrinsic antiretroviral factor APOBEC3B contains two enzymatically active cytidine deaminase domains. Virology. 2007;364:486–493. doi: 10.1016/j.virol.2007.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hakata Y, Landau NR. Reversed functional organization of mouse and human APOBEC3 cytidine deaminase domains. J Biol Chem. 2006;281:36624–36631. doi: 10.1074/jbc.M604980200. [DOI] [PubMed] [Google Scholar]
- 30.Wissing S, Montano M, Garcia-Perez JL, Moran JV, Greene WC. Endogenous APOBEC3B restricts LINE-1 retrotransposition in transformed cells and human embryonic stem cells. J Biol Chem. 2011;286:36427–36437. doi: 10.1074/jbc.M111.251058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, Refsland EW, Kotandeniya D, Tretyakova N, Nikas JB, Yee D, Temiz NA, Donohue DE, McDougle RM, Brown WL, Law EK, Harris RS. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013;494:366–370. doi: 10.1038/nature11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fu Y, Ito F, Zhang G, Fernandez B, Yang H, Chen XS. DNA cytosine and methylcytosine deamination by APOBEC3B: enhancing methylcytosine deamination by engineering APOBEC3B. Biochem J. 2015;471:25–35. doi: 10.1042/BJ20150382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leonard B, Hart SN, Burns MB, Carpenter MA, Temiz NA, Rathore A, Vogel RI, Nikas JB, Law EK, Brown WL, Li Y, Zhang Y, Maurer MJ, Oberg AL, Cunningham JM, Shridhar V, Bell DA, April C, Bentley D, Bibikova M, Cheetham RK, Fan JB, Grocock R, Humphray S, Kingsbury Z, Peden J, Chien J, Swisher EM, Hartmann LC, Kalli KR, Goode EL, Sicotte H, Kaufmann SH, Harris RS. APOBEC3B upregulation and genomic mutation patterns in serous ovarian carcinoma. Cancer Res. 2013;73:7222–7231. doi: 10.1158/0008-5472.CAN-13-1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho SJ, Malim MH. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr Biol. 2004;14:1392–1396. doi: 10.1016/j.cub.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 35.Suspène R, Sommer P, Henry M, Ferris S, Guétard D, Pochet S, Chester A, Navaratnam N, Wain-Hobson S, Vartanian J-P. APOBEC3G is a single-stranded DNA cytidine deaminase and functions independently of HIV reverse transcriptase. Nucleic Acids Res. 2004;32:2421–2429. doi: 10.1093/nar/gkh554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen H, Lilley CE, Yu Q, Lee DV, Chou J, Narvaiza I, Landau NR, Weitzman MD. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr Biol. 2006;16:480–485. doi: 10.1016/j.cub.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 37.Bogerd HP, Wiegand HL, Doehle BP, Lueders KK, Cullen BR. APOBEC3A and APOBEC3B are potent inhibitors of LTR-retrotransposon function in human cells. Nucleic Acids Res. 2006;34:89–95. doi: 10.1093/nar/gkj416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bogerd HP, Wiegand HL, Hulme AE, Garcia-Perez JL, O’Shea KS, Moran JV, Cullen BR. Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc Natl Acad Sci USA. 2006;103:8780–8785. doi: 10.1073/pnas.0603313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muckenfuss H, Hamdorf M, Held U, Perkovic M, Löwer J, Cichutek K, Flory E, Schumann GG, Münk C. APOBEC3 proteins inhibit human LINE-1 retrotransposition. J Biol Chem. 2006;281:22161–22172. doi: 10.1074/jbc.M601716200. [DOI] [PubMed] [Google Scholar]
- 40.Kinomoto M, Kanno T, Shimura M, Ishizaka Y, Kojima A, Kurata T, Sata T, Tokunaga K. All APOBEC3 family proteins differentially inhibit LINE-1 retrotransposition. Nucleic Acids Res. 2007;35:2955–2964. doi: 10.1093/nar/gkm181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Niewiadomska AM, Tian C, Tan L, Wang T, Sarkis PTN, Yu X-F. Differential inhibition of long interspersed element 1 by APOBEC3 does not correlate with high-molecular-mass-complex formation or P-body association. J Virol. 2007;81:9577–9583. doi: 10.1128/JVI.02800-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lovšin N, Peterlin BM. APOBEC3 proteins inhibit LINE-1 retrotransposition in the absence of ORF1p binding. Ann N Y Acad Sci. 2009;1178:268–275. doi: 10.1111/j.1749-6632.2009.05006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bulliard Y, Narvaiza I, Bertero A, Peddi S, Röhrig UF, Ortiz M, Zoete V, Castro-Díaz N, Turelli P, Telenti A, Michielin O, Weitzman MD, Trono D. Structure-function analyses point to a polynucleotide-accommodating groove essential for APOBEC3A restriction activities. J Virol. 2011;85:1765–1776. doi: 10.1128/JVI.01651-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mitra M, Hercík K, Byeon I-JL, Ahn J, Hill S, Hinchee-Rodriguez K, Singer D, Byeon C-H, Charlton LM, Nam G, Heidecker G, Gronenborn AM, Levin JG. Structural determinants of human APOBEC3A enzymatic and nucleic acid binding properties. Nucleic Acids Res. 2014;42:1095–1110. doi: 10.1093/nar/gkt945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Richardson SR, Narvaiza I, Planegger RA, Weitzman MD, Moran JV. APOBEC3A deaminates transiently exposed single-strand DNA during LINE-1 retrotransposition. eLife. 2014;3:e02008. doi: 10.7554/eLife.02008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stenglein MD, Harris RS. APOBEC3B and APOBEC3F inhibit L1 retrotransposition by a DNA deamination-independent mechanism. J Biol Chem. 2006;281:16837–16841. doi: 10.1074/jbc.M602367200. [DOI] [PubMed] [Google Scholar]
- 47.Marchetto MCN, Narvaiza I, Denli AM, Benner C, Lazzarini TA, Nathanson JL, Paquola ACM, Desai KN, Herai RH, Weitzman MD, Yeo GW, Muotri AR, Gage FH. Differential L1 regulation in pluripotent stem cells of humans and apes. Nature. 2013;503:525–529. doi: 10.1038/nature12686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Suspène R, Guétard D, Henry M, Sommer P, Wain-Hobson S, Vartanian JP. Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in vivo. Proc Natl Acad Sci USA. 2005;102:8321–8326. doi: 10.1073/pnas.0408223102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bonvin M, Greeve J. Effects of point mutations in the cytidine deaminase domains of APOBEC3B on replication and hypermutation of hepatitis B virus in vitro. J Gen Virol. 2007;88:3270–3274. doi: 10.1099/vir.0.83149-0. [DOI] [PubMed] [Google Scholar]
- 50.Lucifora J, Xia Y, Reisinger F, Zhang K, Stadler D, Cheng X, Sprinzl MF, Koppensteiner H, Makowska Z, Volz T, Remouchamps C, Chou WM, Thasler WE, Huser N, Durantel D, Liang TJ, Munk C, Heim MH, Browning JL, Dejardin E, Dandri M, Schindler M, Heikenwalder M, Protzer U. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science. 2014;343:1221–1228. doi: 10.1126/science.1243462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu R, Zhang X, Zhang W, Fang Y, Zheng S, Yu X-F. Association of human APOBEC3 cytidine deaminases with the generation of hepatitis virus B x antigen mutants and hepatocellular carcinoma. Hepatology. 2007;46:1810–1820. doi: 10.1002/hep.21893. [DOI] [PubMed] [Google Scholar]
- 52.Abe H, Ochi H, Maekawa T, Hatakeyama T, Tsuge M, Kitamura S, Kimura T, Miki D, Mitsui F, Hiraga N, Imamura M, Fujimoto Y, Takahashi S, Nakamura Y, Kumada H, Chayama K. Effects of structural variations of APOBEC3A and APOBEC3B genes in chronic hepatitis B virus infection. Hepatol Res. 2009;39:1159–1168. doi: 10.1111/j.1872-034X.2009.00566.x. [DOI] [PubMed] [Google Scholar]
- 53.Mahieux R, Suspène R, Delebecque F, Henry M, Schwartz O, Wain-Hobson S, Vartanian JP. Extensive editing of a small fraction of human T-cell leukemia virus type 1 genomes by four APOBEC3 cytidine deaminases. J Gen Virol. 2005;86:2489–2494. doi: 10.1099/vir.0.80973-0. [DOI] [PubMed] [Google Scholar]
- 54.Doehle BP, Schäfer A, Cullen BR. Human APOBEC3B is a potent inhibitor of HIV-1 infectivity and is resistant to HIV-1 Vif. Virology. 2005;339:281–288. doi: 10.1016/j.virol.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 55.Dang Y, Wang X, Esselman WJ, Zheng Y-H. Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J Virol. 2006;80:10522–10533. doi: 10.1128/JVI.01123-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bogerd HP, Kornepati AV, Marshall JB, Kennedy EM, Cullen BR. Specific induction of endogenous viral restriction factors using CRISPR/Cas-derived transcriptional activators. Proc Natl Acad Sci USA. 2015;112:E7249–7256. doi: 10.1073/pnas.1516305112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu Q, Chen D, König R, Mariani R, Unutmaz D, Landau NR. APOBEC3B and APOBEC3C are potent inhibitors of simian immunodeficiency virus replication. J Biol Chem. 2004;279:53379–53386. doi: 10.1074/jbc.M408802200. [DOI] [PubMed] [Google Scholar]
- 58.Rose KM, Marin M, Kozak SL, Kabat D. Regulated production and anti-HIV type 1 activities of cytidine deaminases APOBEC3B, 3F, and 3G. AIDS Res Hum Retroviruses. 2005;21:611–619. doi: 10.1089/aid.2005.21.611. [DOI] [PubMed] [Google Scholar]
- 59.Hultquist JF, Lengyel JA, Refsland EW, LaRue RS, Lackey L, Brown WL, Harris RS. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J Virol. 2011;85:11220–11234. doi: 10.1128/JVI.05238-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Imahashi M, Izumi T, Watanabe D, Imamura J, Matsuoka K, Ode H, Masaoka T, Sato K, Kaneko N, Ichikawa S, Koyanagi Y, Takaori-Kondo A, Utsumi M, Yokomaku Y, Shirasaka T, Sugiura W, Iwatani Y, Naoe T. Lack of association between intact/deletion polymorphisms of the APOBEC3B gene and HIV-1 risk. PloS One. 2014;9:e92861. doi: 10.1371/journal.pone.0092861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 62.Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu X-F. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302:1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- 63.Shandilya SMD, Bohn M-F, Schiffer CA. A computational analysis of the structural determinants of APOBEC3’s catalytic activity and vulnerability to HIV-1 Vif. Virology. 2014;471–473:105–116. doi: 10.1016/j.virol.2014.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.An P, Johnson R, Phair J, Kirk GD, Yu XF, Donfield S, Buchbinder S, Goedert JJ, Winkler CA. APOBEC3B deletion and risk of HIV-1 acquisition. J Infect Dis. 2009;200:1054–1058. doi: 10.1086/605644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Itaya S, Nakajima T, Kaur G, Terunuma H, Ohtani H, Mehra N, Kimura A. No evidence of an association between the APOBEC3B deletion polymorphism and susceptibility to HIV infection and AIDS in Japanese and Indian populations. J Infect Dis. 2010;202:815–816. doi: 10.1086/655227. author reply 816–817. [DOI] [PubMed] [Google Scholar]
- 66.Kidd JM, Newman TL, Tuzun E, Kaul R, Eichler EE. Population stratification of a common APOBEC gene deletion polymorphism. PLoS Genet. 2007;3:e63. doi: 10.1371/journal.pgen.0030063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lackey L, Demorest ZL, Land AM, Hultquist JF, Brown WL, Harris RS. APOBEC3B and AID have similar nuclear import mechanisms. J Mol Biol. 2012;419:301–314. doi: 10.1016/j.jmb.2012.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Suspène R, Aynaud MM, Guétard D, Henry M, Eckhoff G, Marchio A, Pineau P, Dejean A, Vartanian JP, Wain-Hobson S. Somatic hypermutation of human mitochondrial and nuclear DNA by APOBEC3 cytidine deaminases, a pathway for DNA catabolism. Proc Natl Acad Sci USA. 2011;108:4858–4863. doi: 10.1073/pnas.1009687108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Landry S, Narvaiza I, Linfesty DC, Weitzman MD. APOBEC3A can activate the DNA damage response and cause cell-cycle arrest. EMBO Rep. 2011;12:444–450. doi: 10.1038/embor.2011.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shinohara M, Io K, Shindo K, Matsui M, Sakamoto T, Tada K, Kobayashi M, Kadowaki N, Takaori-Kondo A. APOBEC3B can impair genomic stability by inducing base substitutions in genomic DNA in human cells. Sci Rep. 2012;2:806. doi: 10.1038/srep00806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goila-Gaur R, Khan MA, Miyagi E, Kao S, Strebel K. Targeting APOBEC3A to the viral nucleoprotein complex confers antiviral activity. Retrovirology. 2007;4:61. doi: 10.1186/1742-4690-4-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lackey L, Law EK, Brown WL, Harris RS. Subcellular localization of the APOBEC3 proteins during mitosis and implications for genomic DNA deamination. Cell Cycle. 2013;12:762–772. doi: 10.4161/cc.23713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Land AM, Law EK, Carpenter MA, Lackey L, Brown WL, Harris RS. Endogenous APOBEC3A DNA cytosine deaminase is cytoplasmic and nongenotoxic. J Biol Chem. 2013;288:17253–17260. doi: 10.1074/jbc.M113.458661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stephens P, Edkins S, Davies H, Greenman C, Cox C, Hunter C, Bignell G, Teague J, Smith R, Stevens C, O’Meara S, Parker A, Tarpey P, Avis T, Barthorpe A, Brackenbury L, Buck G, Butler A, Clements J, Cole J, Dicks E, Edwards K, Forbes S, Gorton M, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Jones D, Kosmidou V, Laman R, Lugg R, Menzies A, Perry J, Petty R, Raine K, Shepherd R, Small A, Solomon H, Stephens Y, Tofts C, Varian J, Webb A, West S, Widaa S, Yates A, Brasseur F, Cooper CS, Flanagan AM, Green A, Knowles M, Leung SY, Looijenga LH, Malkowicz B, Pierotti MA, Teh B, Yuen ST, Nicholson AG, Lakhani S, Easton DF, Weber BL, Stratton MR, Futreal PA, Wooster R. A screen of the complete protein kinase gene family identifies diverse patterns of somatic mutations in human breast cancer. Nat Genet. 2005;37:590–592. doi: 10.1038/ng1571. [DOI] [PubMed] [Google Scholar]
- 75.Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, Menzies A, Martin S, Leung K, Chen L, Leroy C, Ramakrishna M, Rance R, Lau KW, Mudie LJ, Varela I, McBride DJ, Bignell GR, Cooke SL, Shlien A, Gamble J, Whitmore I, Maddison M, Tarpey PS, Davies HR, Papaemmanuil E, Stephens PJ, McLaren S, Butler AP, Teague JW, Jonsson G, Garber JE, Silver D, Miron P, Fatima A, Boyault S, Langerod A, Tutt A, Martens JW, Aparicio SA, Borg A, Salomon AV, Thomas G, Borresen-Dale AL, Richardson AL, Neuberger MS, Futreal PA, Campbell PJ, Stratton MR Breast Cancer Working Group of the International Cancer Genome C. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–993. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Roberts SA, Sterling J, Thompson C, Harris S, Mav D, Shah R, Klimczak LJ, Kryukov GV, Malc E, Mieczkowski PA, Resnick MA, Gordenin DA. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol Cell. 2012;46:424–435. doi: 10.1016/j.molcel.2012.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Drier Y, Lawrence MS, Carter SL, Stewart C, Gabriel SB, Lander ES, Meyerson M, Beroukhim R, Getz G. Somatic rearrangements across cancer reveal classes of samples with distinct patterns of DNA breakage and rearrangement-induced hypermutability. Genome Res. 2013;23:228–235. doi: 10.1101/gr.141382.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL, Saksena G, Harris S, Shah RR, Resnick MA, Getz G, Gordenin DA. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet. 2013;45:970–976. doi: 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjord JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Jager N, Jones DT, Jones D, Knappskog S, Kool M, Lakhani SR, Lopez-Otin C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt AN, Valdes-Mas R, van Buuren MM, van ‘t Veer L, Vincent-Salomon A, Waddell N, Yates LR, PedBrain I, Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Taylor BJM, Nik-Zainal S, Wu YL, Stebbings LA, Raine K, Campbell PJ, Rada C, Stratton MR, Neuberger MS. DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. eLife. 2013;2:e00534. doi: 10.7554/eLife.00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Walker BA, Wardell CP, Murison A, Boyle EM, Begum DB, Dahir NM, Proszek PZ, Melchor L, Pawlyn C, Kaiser MF, Johnson DC, Qiang YW, Jones JR, Cairns DA, Gregory WM, Owen RG, Cook G, Drayson MT, Jackson GH, Davies FE, Morgan GJ Australian Pancreatic Cancer Genome I Consortium IBC, Consortium IM-S. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat Commun. 2015;6:6997. doi: 10.1038/ncomms7997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mussil B, Suspène R, Aynaud M-M, Gauvrit A, Vartanian J-P, Wain-Hobson S. Human APOBEC3A isoforms translocate to the nucleus and induce DNA double strand breaks leading to cell stress and death. PloS One. 2013;8:e73641. doi: 10.1371/journal.pone.0073641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Caval V, Suspène R, Shapira M, Vartanian JP, Wain-Hobson S. A prevalent cancer susceptibility APOBEC3A hybrid allele bearing APOBEC3B 3′UTR enhances chromosomal DNA damage. Nat Commun. 2014;5:5129. doi: 10.1038/ncomms6129. [DOI] [PubMed] [Google Scholar]
- 84.Caval V, Bouzidi MS, Suspène R, Laude H, Dumargne MC, Bashamboo A, Krey T, Vartanian J-P, Wain-Hobson S. Molecular basis of the attenuated phenotype of human APOBEC3B DNA mutator enzyme. Nucleic Acids Res. 2015;43:9340–9349. doi: 10.1093/nar/gkv935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Burns MB, Temiz NA, Harris RS. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat Genet. 2013;45:977–983. doi: 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cescon DW, Haibe-Kains B, Mak TW. APOBEC3B expression in breast cancer reflects cellular proliferation, while a deletion polymorphism is associated with immune activation. Proc Natl Acad Sci USA. 2015;112:2841–2846. doi: 10.1073/pnas.1424869112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Harris RS. Molecular mechanism and clinical impact of APOBEC3B-catalyzed mutagenesis in breast cancer. Breast Cancer Res. 2015;17:8. doi: 10.1186/s13058-014-0498-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Byeon IJL, Ahn J, Mitra M, Byeon CH, Hercík K, Hritz J, Charlton LM, Levin JG, Gronenborn AM. NMR structure of human restriction factor APOBEC3A reveals substrate binding and enzyme specificity. Nat Commun. 2013;4:1890. doi: 10.1038/ncomms2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li M, Shandilya SMD, Carpenter MA, Rathore A, Brown WL, Perkins AL, Harki DA, Solberg J, Hook DJ, Pandey KK, Parniak MA, Johnson JR, Krogan NJ, Somasundaran M, Ali A, Schiffer CA, Harris RS. First-in-class small molecule inhibitors of the single-strand DNA cytosine deaminase APOBEC3G. ACS Chem Biol. 2012;7:506–517. doi: 10.1021/cb200440y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shandilya SMD, Nalam MNL, Nalivaika EA, Gross PJ, Valesano JC, Shindo K, Li M, Munson M, Royer WE, Harjes E, Kono T, Matsuo H, Harris RS, Somasundaran M, Schiffer CA. Crystal structure of the APOBEC3G catalytic domain reveals potential oligomerization interfaces. Structure. 2010;18:28–38. doi: 10.1016/j.str.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shi K, Carpenter MA, Kurahashi K, Harris RS, Aihara H. Crystal Structure of the DNA Deaminase APOBEC3B Catalytic Domain. J Biol Chem. 2015;290:28120–28130. doi: 10.1074/jbc.M115.679951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mitra M, Singer D, Mano Y, Hritz J, Nam G, Gorelick RJ, Byeon I-JL, Gronenborn AM, Iwatani Y, Levin JG. Sequence and structural determinants of human APOBEC3H deaminase and anti-HIV-1 activities. Retrovirology. 2015;12:3. doi: 10.1186/s12977-014-0130-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Clore GM, Gronenborn AM. Determining the structures of large proteins and protein complexes by NMR. Trends Biotechnol. 1998;16:22–34. doi: 10.1016/S0167-7799(97)01135-9. [DOI] [PubMed] [Google Scholar]
- 94.Sattler M, Maurer M, Schleucher J, Griesinger C. A simultaneous 15N,1H- and 13C,1H-HSQC with sensitivity enhancement and a heteronuclear gradient echo. J Biomol NMR. 1995;5:97–102. doi: 10.1007/BF00227475. [DOI] [PubMed] [Google Scholar]
- 95.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 96.Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 97.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 98.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 99.Chen K-M, Harjes E, Gross PJ, Fahmy A, Lu Y, Shindo K, Harris RS, Matsuo H. Structure of the DNA deaminase domain of the HIV-1 restriction factor APOBEC3G. Nature. 2008;452:116–119. doi: 10.1038/nature06638. [DOI] [PubMed] [Google Scholar]
- 100.Furukawa A, Nagata T, Matsugami A, Habu Y, Sugiyama R, Hayashi F, Kobayashi N, Yokoyama S, Takaku H, Katahira M. Structure, interaction and real-time monitoring of the enzymatic reaction of wild-type APOBEC3G. EMBO J. 2009;28:440–451. doi: 10.1038/emboj.2008.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Holden LG, Prochnow C, Chang YP, Bransteitter R, Chelico L, Sen U, Stevens RC, Goodman MF, Chen XS. Crystal structure of the anti-viral APOBEC3G catalytic domain and functional implications. Nature. 2008;456:121–124. doi: 10.1038/nature07357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 103.Liang J, Edelsbrunner H, Woodward C. Anatomy of protein pockets and cavities: measurement of binding site geometry and implications for ligand design. Protein Sci. 1998;7:1884–1897. doi: 10.1002/pro.5560070905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liang J, Edelsbrunner H, Fu P, Sudhakar PV, Subramaniam S. Analytical shape computation of macromolecules: I. Molecular area and volume through alpha shape. Proteins. 1998;33:1–17. [PubMed] [Google Scholar]
- 105.Liang J, Edelsbrunner H, Fu P, Sudhakar PV, Subramaniam S. Analytical shape computation of macromolecules: II. Inaccessible cavities in proteins. Proteins. 1998;33:18–29. [PubMed] [Google Scholar]
- 106.Connolly ML. Analytical molecular surface calculation. J Appl Cryst. 1983;16:548–558. [Google Scholar]
- 107.Koradi R, Billeter M, Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55. 29–32. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 108.Bohn M-F, Shandilya SMD, Silvas TV, Nalivaika EA, Kouno T, Kelch BA, Ryder SP, Kurt-Yilmaz N, Somasundaran M, Schiffer CA. The ssDNA mutator APOBEC3A is regulated by cooperative dimerization. Structure. 2015;23:903–911. doi: 10.1016/j.str.2015.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kitamura S, Ode H, Nakashima M, Imahashi M, Naganawa Y, Kurosawa T, Yokomaku Y, Yamane T, Watanabe N, Suzuki A, Sugiura W, Iwatani Y. The APOBEC3C crystal structure and the interface for HIV-1 Vif binding. Nat Struct Mol Biol. 2012;19:1005–1010. doi: 10.1038/nsmb.2378. [DOI] [PubMed] [Google Scholar]
- 110.Bohn M-F, Shandilya SMD, Albin JS, Kouno T, Anderson BD, McDougle RM, Carpenter MA, Rathore A, Evans L, Davis AN, Zhang J, Lu Y, Somasundaran M, Matsuo H, Harris RS, Schiffer CA. Crystal structure of the DNA cytosine deaminase APOBEC3F: the catalytically active and HIV-1 Vif-binding domain. Structure. 2013;21:1042–1050. doi: 10.1016/j.str.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Siu KK, Sultana A, Azimi FC, Lee JE. Structural determinants of HIV-1 Vif susceptibility and DNA binding in APOBEC3F. Nat Commun. 2013;4:2593. doi: 10.1038/ncomms3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nakashima M, Ode H, Kawamura T, Kitamura S, Naganawa Y, Awazu H, Tsuzuki S, Matsuoka K, Nemoto M, Hachiya A, Sugiura W, Yokomaku Y, Watanabe N, Iwatani Y. Structural Insights into HIV-1 Vif-APOBEC3F Interaction. J Virol. 2016;90:1034–1047. doi: 10.1128/JVI.02369-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Harjes E, Gross PJ, Chen KM, Lu Y, Shindo K, Nowarski R, Gross JD, Kotler M, Harris RS, Matsuo H. An extended structure of the APOBEC3G catalytic domain suggests a unique holoenzyme model. J Mol Biol. 2009;389:819–832. doi: 10.1016/j.jmb.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kouno T, Luengas EM, Shigematsu M, Shandilya SMD, Zhang J, Chen L, Hara M, Schiffer CA, Harris RS, Matsuo H. Structure of the Vif-binding domain of the antiviral enzyme APOBEC3G. Nat Struct Mol Biol. 2015;22:485–491. doi: 10.1038/nsmb.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rathore A, Carpenter MA, Demir O, Ikeda T, Li M, Shaban NM, Law EK, Anokhin D, Brown WL, Amaro RE, Harris RS. The local dinucleotide preference of APOBEC3G can be altered from 5′-CC to 5′-TC by a single amino acid substitution. J Mol Biol. 2013;425:4442–4454. doi: 10.1016/j.jmb.2013.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pham P, Landolph A, Mendez C, Li N, Goodman MF. A biochemical analysis linking APOBEC3A to disparate HIV-1 restriction and skin cancer. J Biol Chem. 2013;288:29294–29304. doi: 10.1074/jbc.M113.504175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Harjes S, Solomon WC, Li M, Chen K-M, Harjes E, Harris RS, Matsuo H. Impact of H216 on the DNA binding and catalytic activities of the HIV restriction factor APOBEC3G. J Virol. 2013;87:7008–7014. doi: 10.1128/JVI.03173-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chan K, Roberts SA, Klimczak LJ, Sterling JF, Saini N, Malc EP, Kim J, Kwiatkowski DJ, Fargo DC, Mieczkowski PA, Getz G, Gordenin DA. An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat Genet. 2015;47:1067–1072. doi: 10.1038/ng.3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Love RP, Xu H, Chelico L. Biochemical analysis of hypermutation by the deoxycytidine deaminase APOBEC3A. J Biol Chem. 2012;287:30812–30822. doi: 10.1074/jbc.M112.393181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Langlois MA, Beale RCL, Conticello SG, Neuberger MS. Mutational comparison of the single-domained APOBEC3C and double-domained APOBEC3F/G anti-retroviral cytidine deaminases provides insight into their DNA target site specificities. Nucleic Acids Res. 2005;33:1913–1923. doi: 10.1093/nar/gki343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bourara K, Liegler TJ, Grant RM. Target cell APOBEC3C can induce limited G-to-A mutation in HIV-1. PLoS Pathog. 2007;3:e153. doi: 10.1371/journal.ppat.0030153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chelico L, Sacho EJ, Erie DA, Goodman MF. A model for oligomeric regulation of APOBEC3G cytosine deaminase-dependent restriction of HIV. J Biol Chem. 2008;283:13780–13791. doi: 10.1074/jbc.M801004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Huthoff H, Autore F, Gallois-Montbrun S, Fraternali F, Malim MH. RNA-dependent oligomerization of APOBEC3G is required for restriction of HIV-1. PLoS Pathog. 2009;5:e1000330. doi: 10.1371/journal.ppat.1000330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Salter JD, Krucinska J, Raina J, Smith HC, Wedekind JE. A hydrodynamic analysis of APOBEC3G reveals a monomer-dimer-tetramer self-association that has implications for anti-HIV function. Biochemistry. 2009;48:10685–10687. doi: 10.1021/bi901642c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chelico L, Prochnow C, Erie DA, Chen XS, Goodman MF. Structural model for deoxycytidine deamination mechanisms of the HIV-1 inactivation enzyme APOBEC3G. J Biol Chem. 2010;285:16195–16205. doi: 10.1074/jbc.M110.107987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.McDougall WM, Okany C, Smith HC. Deaminase activity on single-stranded DNA (ssDNA) occurs in vitro when APOBEC3G cytidine deaminase forms homotetramers and higher-order complexes. J Biol Chem. 2011;286:30655–30661. doi: 10.1074/jbc.M111.269506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bélanger K, Savoie M, Rosales Gerpe MC, Couture JF, Langlois M-A. Binding of RNA by APOBEC3G controls deamination-independent restriction of retroviruses. Nucleic Acids Res. 2013;41:7438–7452. doi: 10.1093/nar/gkt527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chaurasiya KR, McCauley MJ, Wang W, Qualley DF, Wu T, Kitamura S, Geertsema H, Chan DSB, Hertz A, Iwatani Y, Levin JG, Musier-Forsyth K, Rouzina I, Williams MC. Oligomerization transforms human APOBEC3G from an efficient enzyme to a slowly dissociating nucleic acid-binding protein. Nat Chem. 2014;6:28–33. doi: 10.1038/nchem.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Feng Y, Chelico L. Intensity of deoxycytidine deamination of HIV-1 proviral DNA by the retroviral restriction factor APOBEC3G is mediated by the noncatalytic domain. J Biol Chem. 2011;286:11415–11426. doi: 10.1074/jbc.M110.199604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Li J, Chen Y, Li M, Carpenter MA, McDougle RM, Luengas EM, Macdonald PJ, Harris RS, Mueller JD. APOBEC3 multimerization correlates with HIV-1 packaging and restriction activity in living cells. J Mol Biol. 2014;426:1296–1307. doi: 10.1016/j.jmb.2013.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Feng Y, Love RP, Ara A, Baig TT, Adolph MB, Chelico L. Natural polymorphisms and oligomerization of human APOBEC3H contribute to single-stranded DNA scanning ability. J Biol Chem. 2015;290:27188–27203. doi: 10.1074/jbc.M115.666065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Logue EC, Bloch N, Dhuey E, Zhang R, Cao P, Herate C, Chauveau L, Hubbard SR, Landau NR. A DNA sequence recognition loop on APOBEC3A controls substrate specificity. PloS One. 2014;9:e97062. doi: 10.1371/journal.pone.0097062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Siriwardena SU, Guruge TA, Bhagwat AS. Characterization of the catalytic domain of human APOBEC3B and the critical structural role for a conserved methionine. J Mol Biol. 2015;427:3042–3055. doi: 10.1016/j.jmb.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Carpenter MA, Rajagurubandara E, Wijesinghe P, Bhagwat AS. Determinants of sequence-specificity within human AID and APOBEC3G. DNA Repair (Amst) 2010;9:579–587. doi: 10.1016/j.dnarep.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang M, Rada C, Neuberger MS. Altering the spectrum of immunoglobulin V gene somatic hypermutation by modifying the active site of AID. J Exp Med. 2010;207:141–153. doi: 10.1084/jem.20092238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kohli RM, Abrams SR, Gajula KS, Maul RW, Gearhart PJ, Stivers JT. A portable hot spot recognition loop transfers sequence preferences from APOBEC family members to activation-induced cytidine deaminase. J Biol Chem. 2009;284:22898–22904. doi: 10.1074/jbc.M109.025536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kohli RM, Maul RW, Guminski AF, McClure RL, Gajula KS, Saribasak H, McMahon MA, Siliciano RF, Gearhart PJ, Stivers JT. Local sequence targeting in the AID/APOBEC family differentially impacts retroviral restriction and antibody diversification. J Biol Chem. 2010;285:40956–40964. doi: 10.1074/jbc.M110.177402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Huang XQ, Miller W. A time-efficient, linear-space local similarity algorithm. Adv Appl Math. 1991;12:337–357. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. 1H-15N HSQC NMR spectra of A3B-CTD WT, A3B-CTD L1 mutant, A3A WT, and A3A H29R mutant in NMR buffer (PDF)
Figure S2. 1H-15N HSQC NMR spectra of A3B-CTD WT in NMR buffer at protein concentrations of 43 μM, 76 μM, and 170 μM (PDF)
Figure S3. 1H-15N HSQC NMR spectra of A3B-CTD WT in NMR buffer containing 100 mM NaCl at protein concentrations of 50 μM, 143 μM, and 290 μM (PDF)
Figure S4. Amino acid sequence alignment of A3B-CTD and A3A (PDF)
Figure S5 Stereo-view of the superposition of the active site regions of the A3B-CTD NMR and the A3B-CTD X-ray structures (PDF)
Table S1. Structural comparison of APOBEC3 protein (PDF)