

ABSTRACT
Heat stress is one of the best-studied exogenous stress factors; however little is known about its delayed effects. Recently, we have shown that heat stress induces cellular senescence-like G2 arrest exclusively in early S-phase cells. The mechanism of this arrest includes the generation of heat stress-induced single-stranded DNA breaks, the collision of replication forks with these breaks and the formation of difficult-to-repair double-stranded DNA breaks. However, the early S phase-specific effects of heat stress are not limited to the induction of single-stranded DNA breaks. Here, we report that HS induces partial DNA re-replication and centrosome amplification. We suggest that HS-induced alterations in the expression levels of the genes encoding the replication licensing factors are the primary source of such perturbations. Notably, these processes do not contribute to acquisition of a senescence-like phenotype, although they do elicit postponed effects. Specifically, we found that the HeLa cells can escape from the heat stress-induced cellular senescence-like G2 arrest, and the mitosis they enter is multipolar due to the amplified centrosomes.
KEYWORDS: cell cycle, cellular senescence, centrosome amplification, DNA damage, DNA replication, heat stress, licensing
Introduction
In eukaryotes, the cell cycle comprises 4 discrete nonequivalent phases: gap 1 (G1), synthesis (S), gap2 (G2), and mitosis (M). The S and M phases can be further divided into sub-phases. Based on the patterns of the DNA replication foci (i.e., the sites of BrdU/EdU incorporation), the S phase is usually divided into 3 intermingled parts, i.e., early, mid and late S.1,2 The early S phase-specific BrdU/EdU incorporation pattern (i.e., a large number of small foci) reflects the well-documented fact that the majority of replication origins fire during the first few hr of S phase progression.3 However, not only replication origins are licensed in early S phase; it is well known that tightly regulated centrosome duplication also occurs early in the S phase.4 It has been proposed that DNA replication and centrosome duplication may share some controlling/licensing factors due to the similar nature of the limitations on these processes (i.e., the prevention of re-replication and centrosome over-duplication).4,5 It has long been observed that early S-phase cells are more sensitive to particular types of stress. Specifically, at this cell cycle stage, the cells are more susceptible to chemical carcinogens. This effect has been explained primarily through the existence of a DNA replication timing phenomenon – it was assumed that early replicating proto-oncogenes acquire transforming mutations during replication.6,7 Early S-phase cells are also hypersensitive to heat stress (HS). Recently, we described the molecular mechanism that underlies the vulnerability of these cells to HS.8 Specifically, we demonstrated that HS induces cellular senescence-like G2 arrest exclusively in early S-phase cells (Fig. 1 and ref. 8). The mechanism of this arrest includes the generation of HS-induced single-stranded DNA breaks, the collision of replication forks with these breaks and the formation of double-stranded breaks (Fig. 1). Subsequent persistent DNA damage responses lead to a cellular senescence-like proliferation arrest. Notably, the proposed mechanism of the senescence-like growth arrest of early S-phase cells is applicable to different single-stranded DNA break-inducing agents, such as the topoisomerase I inhibitor camptothecin (CPT) and hydrogen peroxide.8 Predictably, the early S phase-specific effects of HS are much more complex than those of CPT or hydrogen peroxide. Here, we report that HS induces partial DNA re-replication and centrosome amplification. We suggest that HS-induced alterations in the expression levels of the genes encoding the replication licensing factors are the primary source of such perturbations. Interestingly, these processes do not contribute to acquisition of a senescence-like phenotype, although affect delayed cell fate decisions.
Figure 1.
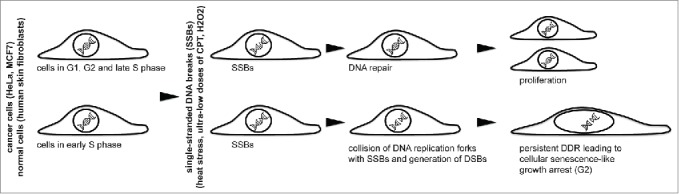
Single-stranded DNA break-inducing agents stimulate cellular senescence-like growth arrest in early S-phase cells. The mechanism of this arrest includes the generation of single-stranded DNA breaks (SSBs), the collision of replication forks with these breaks and the formation of difficult-to-repair double-stranded DNA breaks (DSBs). CPT, camptothecin, H2O2, hydrogen peroxide.
Results and discussion
HS induces partial DNA re-replication in early S-phase cells
In the course of studying HS-induced S phase-specific perturbations of cellular processes that can result in the acquisition of a senescence-like phenotype, we investigated whether HS could induce DNA re-replication. Flow cytometry analysis of human HeLa cells that were HS-treated (45°C, 30 min) and recovered in fresh media for 24 hr at 37°C did not reveal any detectable increase in the cellular DNA content (Fig. 2A). In accordance with our previous observations,8 HS-treated and recovered cells underwent cellular senescence-like G2 arrest (4n peak); at the same time, we did not detect any additional cell fractions with DNA content greater than 4n (Fig. 2A). However, DNA re-replication may be partial and restricted to certain genomic loci.9 Thus, we examined whether HS could induce the amplification of specific genomic sites. For this purpose, we used fluorescence in situ hybridization (FISH) with a short (several kb) probe targeted to the unique DNA sequence adjacent to the c-myc replication origin. We chose this locus because it is early replicating in human HeLa cells10 and contains one of the few well-characterized human DNA replication origins.11,12 Early and late S-phase cells that were HS-treated (45°C, 30 min) and recovered for 24 hr at 37°C were hybridized with a biotinylated DNA probe. The biotin was detected using a fluorescent-labeled antibody (Fig. 2B). The proportions of cells with different patterns of hybridization signals were determined. We divided the cells into 3 groups according to the pattern of hybridization signals present: i) the cells containing only single (singlets) or double (doublets; replicated) FISH signals, ii) the cells containing singlets, doublets and at least one triple or quadruple signal, that may represent singlets and/or doublets located close to each other, and, iii) the cells containing at least one amplified FISH signal consisting of more than 4 individual hybridizing spots (Fig. 2B). These amplified FISH signals were assumed to indicate re-replicated c-myc loci. Simple calculations demonstrated that in more than 30% of the early S-phase cells that were HS-treated and recovered for 24 hr, these loci were amplified (33% (n = 242) of HS-treated and recovered cells exhibited amplified FISH signal compared to 5% (n = 173) of control cells with amplified signal; Fig. 2C). Simultaneously, the late S-phase cells did not exhibit amplification of the c-myc loci in response to HS as evidenced by the unchanged numbers of cells that possessed different FISH signals (Fig. 2C). This observation may be related to the fact that DNA replication licensing events primarily occur early in S phase. To ascertain that the re-replication events are limited to early S-phase cells and to early-replicating loci, we hybridized the HS-treated and recovered HeLa cells with the probe for late-replicating chromosome 18 – specific centromeric α-satellite (Figure S1); no amplification of the centromeric FISH probe was observed in these cells. As previously stated, both HS and CPT induce senescence-like growth arrest in early S-phase cells through the identical mechanism.8 DNA re-replication is known to stimulate cellular senescence under certain conditions,13 thus, we investigated whether re-replication contributes to the development of HS-induced senescence-like state. If HS-induced DNA re-replication contributes to development of cellular senescence, it should also occur in CPT-treated early S-phase cells. To examine this, the abovementioned FISH probe was visualized in early S-phase HeLa cells that had been CPT-treated and recovered in fresh media for 24 hr. No increase in the number of cells harboring amplified FISH signals was found (Fig. 2D). Thus, DNA re-replication does not contribute to the development of a senescence-like state in cells treated with either HS or CPT. However, the partial DNA re-replication induced by HS may have other delayed effects.
Figure 2.
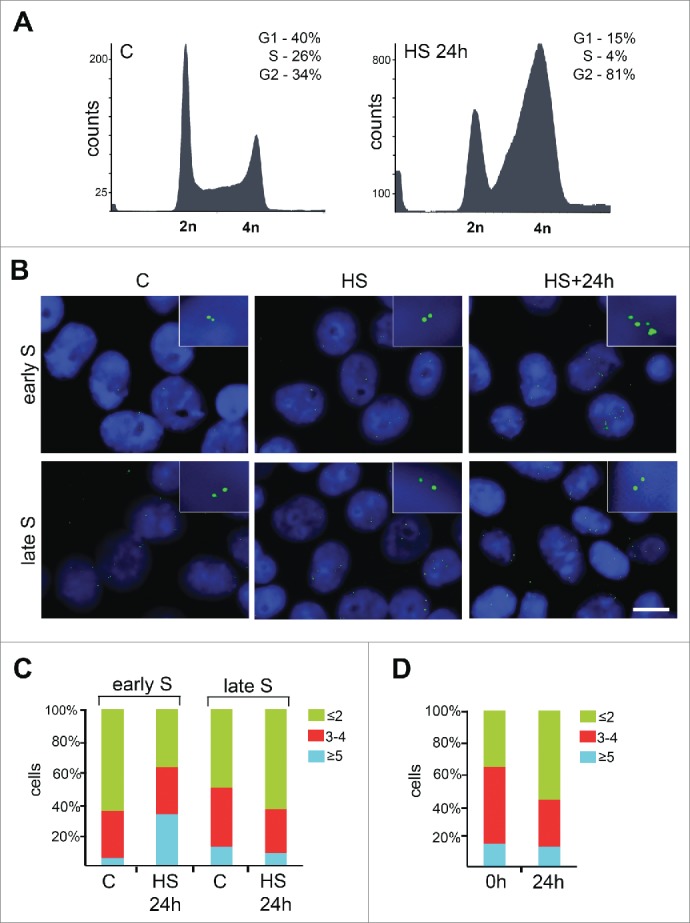
HS induces partial DNA re-replication. (A) Cell cycle profiles of early S-phase HeLa cells that were either mock-treated or HS-treated (45.5°C, 30 min) and recovered for 24 hr. (B) Early and late S-phase HeLa cells that were either untreated (“C”), HS-treated (45.5°C, 30 min; “HS”), or HS-treated and allowed to recover for 24 h (“HS+24h”) were hybridized with a probe specific for c-myc replication origin and stained with DAPI. Scale bar, 15 μm (C) The percentage of cells that were treated and stained as in (B) possessing different hybridization signals. (D) The percentage of early S-phase HeLa cells that were treated with CPT (100 nM, 1 hr), incubated in fresh media for indicated time periods (0 and 24 hr) and hybridized with the abovementioned probe, possessing different hybridization signals. Results of one representative experiment are shown in (C) and (D).
Reversal of HS-induced cellular senescence–like growth arrest
It is generally thought that cellular senescence is an irreversible growth arrest; however, some experimental data indicate that this arrest can be bypassed in certain circumstances.14 Specifically, cellular senescence can be reversed by the inactivation of the p53/p21 or pRB/p16 pathways.14,15 Cells that escape from senescence usually enter a state of crisis that is characterized by increased genomic instability and cell death that occurs during mitosis or in the subsequent cell cycle.16,17 Little is known about the escape of tumor cells from cellular senescence-like states. To explore this subject, we followed the fate of early S-phase HeLa cells that were treated with acute HS (45°C, 30 min) to induce cellular senescence-like growth arrest for up to 120 hr (Fig. 3A). Expectedly, the cells acquired cellular senescence-like characteristics during the first 24 hr and maintained these characteristics until 72 hr post-HS (Fig. 3A). Surprisingly, the cells started to enter mitosis during the next 24 hr (the 96 hr time point in Fig. 3A). Notably, all of the mitotic spindles were multipolar as demonstrated by α-tubulin immunostaining (100% (n = 73) of multipolar mitotic spindles were formed in HeLa cells that escaped from senescence-like state and underwent mitosis compared to 8% (n = 118) of multipolar mitotic spindles observed in control HeLa cells). This crisis-like event did not lead to extensive cell death; some cells completed the abnormal mitosis and formed seemingly normal progeny (the 120 hr time point in Fig. 3A). However, a relatively large proportion of the cell population became multinucleated, which may have been the consequence of a mitotic catastrophe18 (Fig. 3A). These observations highlight several interesting traits of HS-induced premature senescence in tumor cells. First, the HS-induced senescence-like G2 arrest of HeLa cells can be bypassed. However, the molecular events that underlie this escape are not clear. We know that p21 drives the senescence-like state in heat-stressed HeLa cells,8 thus, the inactivation or downregulation of p21 could be the cause of senescence escape. Another possibility is that G2-arrested HeLa cells accumulate polo-like kinase 1 (plk1) until it reaches a level that is sufficient to enter mitosis regardless of checkpoint signals or DNA damage.19 We tested these assumptions by analyzing expression levels of p21 and plk1 in HS-treated and recovered HeLa cells (Figure S2). Using quantitative reverse transcription-PCR (qRT-PCR) and western blotting, we found that p21 gene expression is upregulated in response to HS and kept constant during subsequent prolonged recovery periods (Figures S2A and B); contrariwise, the expression of plk1 remains unchanged upon cells exposure to HS (Figure S2C). Thus, it seems clear that neither p21 nor plk1 contribute to the escape of HeLa cells from HS-induced senescence-like G2 arrest. Second, HeLa cells that have escaped from a senescence-like state undergo multi-spindle mitosis that is most likely a result of HS-induced centrosome amplification. Third, only a portion of the cell population dies due to abnormal mitosis (presumably due to mitotic catastrophe), and another considerable portion survives. In normal cells, the survivors should be aneuploid and mostly inviable; however, in HeLa cells, the initial abnormal karyotype can compensate for the aneuploidy.
Figure 3.
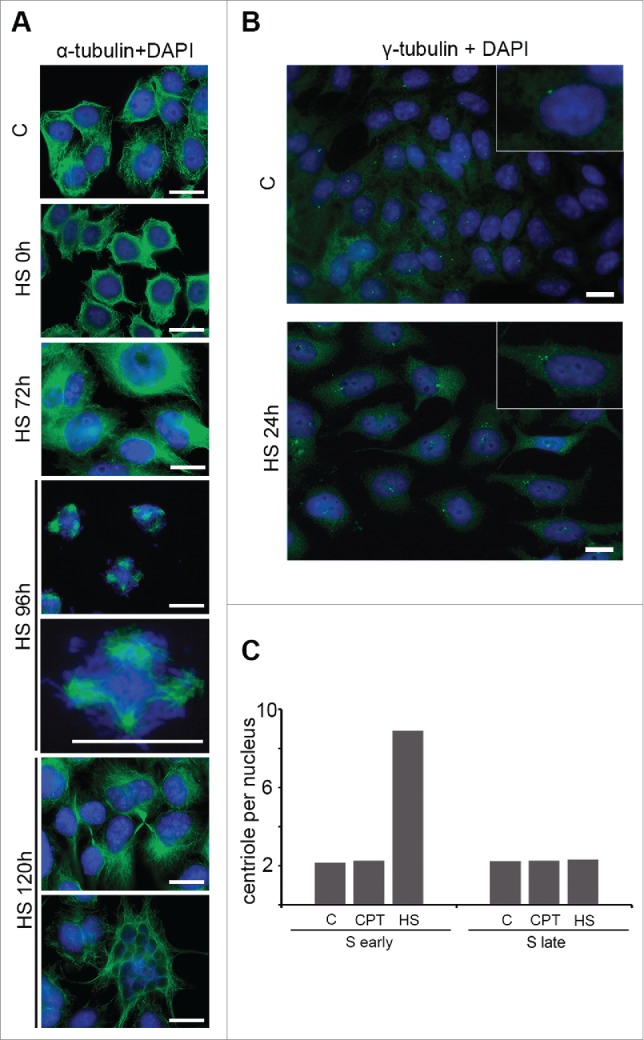
HeLa cells escape from HS-induced cellular senescence-like growth arrest and undergo abnormal mitosis. (A) Early S-phase HeLa cells that were untreated (“C”) or HS-treated (45.5°C, 30 min) and allowed to recover for indicated time periods (0, 72, 96 and 120 hr) were immunostained for α-tubulin (green). The DNA was stained with DAPI (blue). Merged images are shown. Scale bar, 15 μm. Multinucleated cell is shown by white arrow. (B) Early S-phase HeLa cells that were untreated (“C”) or HS-treated (45.5°C, 30 min) and recovered for 24 hr were immunostained for γ-tubulin (green). The DNA was stained with DAPI (blue). Merged images are shown. Scale bar, 15 μm. (C) Early and late S-phase HeLa cells that were untreated (“C”), CPT-treated (300 nM, 1 hr) or HS-treated (45.5°C, 30 min) were incubated in fresh media at 37°C for 24 hr and immunostained for γ-tubulin. The number of centrioles (γ-tubulin dots) was quantified; the average number of centrioles per cell is shown.
It is well known that HS can induce centrosome amplification in mammalian cells.20-22 To determine whether this phenomenon occurs in our model system, we immunostained the centrosomal protein γ-tubulin in HeLa cells that were HS-treated (45°C, 30 min) in early S-phase and recovered for 24 hr. Inspection of immunostained cells clearly demonstrated that the HS induced an amplification of the centrosomes (Fig. 3B). To ascertain whether the induction of centrosome amplification by HS was temporarily restricted to the early S phase, we counted the numbers of centrosomes in the HS-treated (45°C, 30 min) and recovered (24 hr) early and late S-phase cells. We found that the numbers of centrosomes in the late S-phase cells that were subjected to HS remained unchanged (Fig. 3C). This cell cycle specificity of HS-induced centrosome perturbations is not surprising; it has previously been shown that centrosomes are G1-specific targets of HS.20 Collectively, these results indicate that HS not only disrupts pericentriolar material (ref. 20 and our data) but also affects a centrosome duplication licensing system that operates in G1/S. To ensure that the observed centrosome overduplication was not linked to the HS-induced cellular senescence of early S-phase HeLa cells, we counted the centrosomes in early S-phase HeLa cells that had been treated with CPT and recovered in fresh media for 24 hr (Fig. 3C). As expected, the CPT did not induce centrosome amplification, although it is known to trigger the development of a senescence-like phenotype.8 This observation demonstrated the lack of a causal relationship between centrosome overduplication and stress-induced cellular senescence.
Deregulation of the expression of replication licensing factor genes may be the cause of HS-induced DNA re-replication and centrosome amplification
One of the most tightly regulated steps of the DNA replication process is initiation. Cells should avoid firing particular DNA replication origins more than once during the cell cycle. The replication licensing system controls origin firing, and geminin, Cdt1 and cell division cycle 6 (Cdc6) appear to be the most critical components of this control process.23 Cdt1 and Cdc6 license DNA replication by forming a pre-replication complex, and geminin negatively regulates the initiation of DNA replication by inhibiting Cdt1.23 It is well known that the deregulation of the expression of replication licensing factors, e.g., the upregulation of Cdt1 or Cdc6 and the downregulation/depletion of geminin, leads to genome over-replication.24-26 Interestingly, increases in the Cdt1-to-geminin ratio can also cause centrosome amplification in addition to DNA re-replication.27,28 Moreover, Cdc6 and geminin have been reported to partially reside in the centrosomes in interphase cells, which suggests the existence of previously unnoticed centrosome-related functions of Cdc6 and geminin.28-30 To investigate the bases of HS-induced DNA re-replication and centrosome overduplication, we measured the levels of the geminin, Cdt1 and Cdc6 mRNAs in the early S-phase HeLa cells that had been heat-stressed (45°C, 30 min) and recovered at 37°C for different time intervals (0, 3, 6 and 24 hr). The control cells were not heat-stressed but were otherwise subjected to the same manipulations. qRT-PCR analysis revealed that the expressions of Cdt1 and geminin were not altered in the HS-treated cells either immediately after the treatment or the during recovery period (Fig. 4), which indicates that the Cdt1-to-geminin ratio was generally preserved upon HS. At the same time, expression of Cdc6 increased in response to HS (Fig. 4). The upregulation of Cdc6 expression was gradual and reached the maximum level 6 hr post-HS (Fig. 4). The subsequent downregulation of Cdc6 expression at 24 hr post-HS apparently reflected the fact that the cells had entered senescence-associated G2 arrest. Notably, expression of Cdc6 was upregulated even by mild HS (41°C, 30 min), as previously demonstrated in human lymphoma U937 cells.31 Hence, the HS-induced upregulation of Cdc6 expression could be the cause of the partial DNA re-replication and centrosome amplification that occurs in early S-phase cells that are subjected to HS. We further checked whether expression levels of several other factors necessary for initiation of DNA replication (Cdc45, PSF2) and centrosome duplication (SAS6) were altered in response to HS. Using qRT-PCR we found that expression of these genes remained unchanged in HeLa cells treated with acute HS (45°C, 30 min) and recovered at 37°C for different time intervals (0, 3, 6 and 24 hr; Figure S3).
Figure 4.
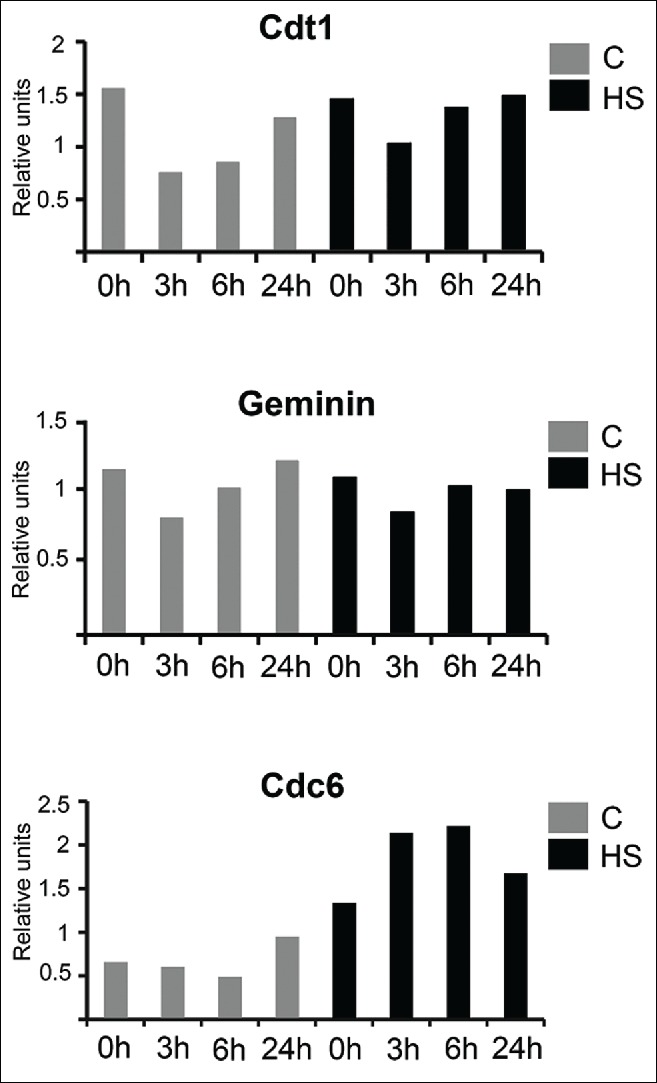
Early S-phase HeLa cells were either mock-treated or HS-treated (45.5°C, 30 min) and recovered for indicated time periods (0, 3, 6 and 24 hr). Quantitative reverse-transcription PCR (qRT-PCR) analysis of cdt1, geminin and cdc6 mRNA levels was performed. RNA extracted from treated and non-treated cells was reverse transcribed, and the cDNA obtained was analyzed using a SYBR Green-based quantitative PCR. The amplification levels of the analyzed cDNAs were normalized to the amplification level of GAPDH cDNA. The results of one representative experiment are shown.
Conclusions
Recently, we described the molecular mechanism that underlies early S-phase cell hypersensitivity in response to HS and single-stranded DNA break-inducing agents.8 We demonstrated that HS and very low concentrations of DNA-damaging agents induce the development of a cellular senescence-like phenotype specifically in early S-phase cells (Fig. 1). This hypersensitivity of early S-phase cells depends on the large number of DNA replication forks that operate simultaneously in this stage and may encounter single-stranded DNA breaks generated via inhibition of topoisomerase I. However, the early S phase-specific effects of HS are much more complex. Here, we show that HS induces partial DNA re-replication and centrosome overduplication in early S-phase cells. The upregulation of Cdc6 expression induced by HS is the most probable cause of these phenomena. Notably, DNA re-replication and centrosome amplification are not directly linked to the development of HS-induced cellular senescence, although they do elicit postponed effects. Specifically, when HeLa cells escape from the HS-induced cellular senescence-like state, the mitosis they enter is multipolar due to the amplified centrosomes. In this respect, the most important future goal is uncovering the molecular mechanisms that release tumor cells from senescence-like states.
Materials and methods
Antibodies
The following primary antibodies were used for immunofluorescence: α-tubulin (mouse; Sigma-Aldrich, T9026; 1:500 dilution), and γ-tubulin (a gift from Dr. F. Gioeva, Institute of Protein Research, Pushchino, Russia; 1:200 dilution). The secondary antibodies conjugated to either Alexa Fluor 488 or Alexa Fluor 555 were purchased from Molecular Probes/Invitrogen (1:200 dilution). The mouse monoclonal antibodies conjugated to Alexa Fluor 488 against biotin were purchased from Molecular Probes/Invitrogen (A11242, 1:200 dilution).
Cell culture and synchronization
Human HeLa cells were cultured in DMEM (C415, PanEco) supplemented with 10% fetal bovine serum (SV30160.03, Hyclone). The cells were cultured at 37ºC in a conventional humidified CO2 incubator. For synchronization via double thymidine block, the cells were treated with 2 mM thymidine (194754, MP Biomedicals) for 16 h, released from the block for 9 h, and then treated with thymidine for an additional 16 h. To release the cells from double thymidine, they were washed twice with phosphate-buffered saline (PBS, 7 mM Na2HPO4, 1.5 mM KH2PO4, (pH 7.4), 137 NaCl, 2.7 mM KCl) and released in drug-free medium. The cells were released in drug-free medium for 2 hrs to obtain early S-phase cells and for 6 hrs to obtain late S-phase cells according to the protocol adapted in ref. 8.
Hyperthermia and drug treatment
The cells were immersed in precision controlled water-bath at 45.5°C (±0.05°C) for 30 min. No marked changes in pH were detected in the medium during the treatment in these experimental conditions.
For the topoisomerase I inhibition experiments, the cells were treated with 100 nM camptothecin (CPT; E1383, Sigma-Aldrich) for 1 h.
Flow cytometry
The cells were trypsinized with 0.25% trypsin for several minutes at 37°C. The trypsin was inactivated with a 4-fold volume of culture medium. Next, the cells were washed in Versen solution and fixed with 70% ice-cold ethanol for 20 min. After fixation, the cells were washed 3 times with PBS and then incubated for 30 min in staining solution (0.1% sodium citrate, 0.3% NP-40, 50 µg/ml propidium iodide, 50 µg/ml RNaseA). After washing, the samples were analyzed using a MACSQuant flow cytometer.
Immunofluorescence
The cells were grown on microscope slides. All samples were fixed and permeabilized in CSK buffer (10 mM PIPES (pH 7.0), 100 mM NaCl, 1.5 mM MgCl2, 300 mM sucrose) supplemented with 1% paraformaldehyde (PFA; P6148, Sigma-Aldrich) and 2.5% Triton X-100 (T8787, Sigma-Aldrich) for 15 min at room temperature or 100% cold methanol (−20˚C) for 10 min. The fixed cells were washed in PBS, pre-incubated with 1% bovine serum albumin (BSA; A7906, Sigma-Aldrich) and 0.05% Tween-20 (P9416, Sigma-Aldrich) in PBS for 30 min and then incubated with antibodies in PBS supplemented with 1% BSA and 0.05% Tween-20 for 1 h at room temperature. After incubation, the cells were washed 3 times (5 min each) with PBS supplemented with 0.2% BSA and 0.05% Tween 20. The primary antibodies were visualized using Alexa Fluor 488- or Alexa Fluor 555-conjugated secondary antibodies (1:200 dilution). The DNA was stained with a DAPI fluorescent dye (D9542, Sigma-Aldrich) for 10 min at room temperature. The results of immunostaining were analyzed using a Zeiss AxioScope A.1 fluorescence microscope.
Fluorescent in situ hybridization (FISH)
The sequence spanning the c-myc replication origin was PCR-amplified from human genomic DNA and cloned into the pGL3-basic plasmid vector (Promega). The following primers were used for the PCR: ACTTTCGCAAACCTGAACGC, and TAAGGGGAAGGGATGGGAGG. Plasmids were used for the production of the FISH probe using a Biotin Nick Translation Kit (11745824910, Roche Applied Science) according to the manufacturer's instructions. FISH for chromosome 18 centromere was performed using biotinylated probe specific for the human chromosome 18 α-satellite (gift of Dr. I. Iourov; ref 32).
After hyperthermia or CPT treatment, the cells were trypsinized with 0.25% trypsin for a few minutes at 37˚C. After trypsin inactivation, the cells were incubated in 75 mM KCl at 37°C for 20 min, fixed in a methanol/acetic acid fixative (3:1) for 10 min at 4°C and dropped onto prefrozen slides (S4651-72EA, Sigma-Aldrich). The fixed cells were washed in PBS and treated with RNase A at 200 µg/ml (EN0531, Thermo Fisher Scientific) in 2x SSC buffer (0.3 M NaCl, 30 mM sodium citrate, pH 7.0) for 30 min at 37°C. Next, the samples were washed in 2x SSC and treated with 0.1% pepsin (P7012, Sigma-Aldrich) in 0.01 N HCl for 10 min at 37°C. The slides were washed in PBS and stabilized in 1% PFA for 10 min at room temperature. The samples were washed in 2x SSC buffer and dehydrated in a 70%, 80%, and 95% ethanol series (5 min each) at 4°C. The DNA was denatured for 5 min at 72°C in hybridization solution (70% deionized formamide (F9037, Sigma-Aldrich) in 2x SSC), dehydrated in an ice-cold 70, 80 and 95% ethanol series for 5 min and air-dried. The biotinylated probe was mixed with hybridization solution (50% deionized formamide, 10% dextran sulfate (67578, Sigma-Aldrich), 1% Tween-20 in 2x SSC supplemented with 500 ng/µl salmon sperm DNA (D1626, Sigma-Aldrich), 100 ng/µl COT1 DNA (11581074001, Roche Applied Science), 500 ng/µl tRNA (R8508, Sigma-Aldrich). The probes were denatured for 10 min at 72°C and placed on the slides. The samples were covered and hybridized with the probe at 37°C overnight in a humidified chamber. After hybridization, the samples were washed with 2x SSC (10 min, RT), 2x SSC supplemented with 50% deionized formamide (twice, 10 min, 42°C), 0.1x SSC (5 min, 42°C) and 2x SSC (10 min, RT). The preparation was blocked in PBS supplemented with 1% BSA and 0.05% Tween-20 for 30 minutes at room temperature and immunostained with Alexa 488-conjugated mouse anti-biotin antibody (1:200 dilution) and Alexa 488-conjugated gout anti-mouse antibody (1:200 dilution). The samples were counterstained with DAPI. The images were analyzed using a Zeiss AxioScope A.1 fluorescence microscope.
Supplementary Material
Abbreviations
- HS
heat stress
- CPT
camptothecin
- SSB
single-stranded DNA break
- DSB
double-stranded DNA break
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This study was supported by the Russian Science Foundation grant #14-24-00022
References
- 1.Wei X, Samarabandu J, Devdhar RS, Siegel AJ, Acharya R, Berezney R. Segregation of transcription and replication sites into higher order domains. Science 1998; 281:1502-6; PMID:9727975; http://dx.doi.org/ 10.1126/science.281.5382.1502 [DOI] [PubMed] [Google Scholar]
- 2.Jackson D, Wang X, Rudner DZ. Spatio-temporal organization of replication in bacteria and eukaryotes (nucleoids and nuclei). Cold Spring Harb Perspect Biol 2012; 4:a010389; PMID:22855726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Picard F, Cadoret JC, Audit B, Arneodo A, Alberti A, Battail C, Duret L, Prioleau MN. The spatiotemporal program of DNA replication is associated with specific combinations of chromatin marks in human cells. PLoS Genet 2014; 10:e1004282; PMID:24785686; http://dx.doi.org/ 10.1371/journal.pgen.1004282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bettencourt-Dias M, Glover DM. Centrosome biogenesis and function: centrosomics brings new understanding. Nat Rev Mol Cell Biol 2007; 8:451-63; PMID:17505520; http://dx.doi.org/ 10.1038/nrm2180 [DOI] [PubMed] [Google Scholar]
- 5.Knockleby J, Lee H. Same partners, different dance: involvement of DNA replication proteins in centrosome regulation. Cell Cycle 2010; 9:4487-91; PMID:21088489; http://dx.doi.org/ 10.4161/cc.9.22.14047 [DOI] [PubMed] [Google Scholar]
- 6.Doggett NA, Cordeiro-Stone M, Chae CB, Kaufman DG. Timing of proto-oncogene replication: a possible determinant of early S phase sensitivity of C3H 10T1/2 cells to transformation by chemical carcinogens. Mol Carcinog 1988; 1:41-9; PMID:3255390; http://dx.doi.org/ 10.1002/mc.2940010110 [DOI] [PubMed] [Google Scholar]
- 7.Kaufman DG, Cordeiro-Stone M, Brylawski BP, Cohen SM, Chastain PD. Early S phase DNA replication: a search for targets of carcinogenesis. Adv Enzyme Regul 2007; 47:127-38; PMID:17337290; http://dx.doi.org/ 10.1016/j.advenzreg.2006.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Velichko AK, Petrova NV, Razin SV, Kantidze OL. Mechanism of heat stress-induced cellular senescence elucidates the exclusive vulnerability of early S-phase cells to mild genotoxic stress. Nucleic Acids Res 2015; 43:6309-20; PMID:26032771; http://dx.doi.org/ 10.1093/nar/gkv573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dorn ES, Chastain PD 2nd, Hall JR, Cook JG. Analysis of re-replication from deregulated origin licensing by DNA fiber spreading. Nucleic Acids Res 2009; 37:60-9; PMID:19010964; http://dx.doi.org/ 10.1093/nar/gkn912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hansen RS, Thomas S, Sandstrom R, Canfield TK, Thurman RE, Weaver M, Dorschner MO, Gartler SM, Stamatoyannopoulos JA. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc Natl Acad Sci U S A 2010; 107:139-44; PMID:19966280; http://dx.doi.org/ 10.1073/pnas.0912402107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Waltz SE, Trivedi AA, Leffak M. DNA replication initiates non-randomly at multiple sites near the c-myc gene in HeLa cells. Nucleic Acids Res 1996; 24:1887-94; PMID:8657570; http://dx.doi.org/ 10.1093/nar/24.10.1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tao L, Dong Z, Leffak M, Zannis-Hadjopoulos M, Price G. Major DNA replication initiation sites in the c-myc locus in human cells. J Cell Biochem 2000; 78:442-57; PMID:10861842; http://dx.doi.org/ 10.1002/1097-4644(20000901)78:3%3c442::AID-JCB9%3e3.0.CO;2-1 [DOI] [PubMed] [Google Scholar]
- 13.Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre' M, Nuciforo PG, Bensimon A, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006; 444:638-42; PMID:17136094; http://dx.doi.org/ 10.1038/nature05327 [DOI] [PubMed] [Google Scholar]
- 14.Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 2005; 120:513-22; PMID:15734683; http://dx.doi.org/ 10.1016/j.cell.2005.02.003 [DOI] [PubMed] [Google Scholar]
- 15.Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 2003; 22:4212-22; PMID:12912919; http://dx.doi.org/ 10.1093/emboj/cdg417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayashi MT, Cesare AJ, Rivera T, Karlseder J. Cell death during crisis is mediated by mitotic telomere deprotection. Nature 2015; 522:492-6; PMID:26108857; http://dx.doi.org/ 10.1038/nature14513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei W, Sedivy JM. Differentiation between senescence (M1) and crisis (M2) in human fibroblast cultures. Exp Cell Res 1999; 253:519-22; PMID:10585275; http://dx.doi.org/ 10.1006/excr.1999.4665 [DOI] [PubMed] [Google Scholar]
- 18.Vakifahmetoglu H, Olsson M, Zhivotovsky B. Death through a tragedy: mitotic catastrophe. Cell Death Differ 2008; 15:1153-62; PMID:18404154; http://dx.doi.org/ 10.1038/cdd.2008.47 [DOI] [PubMed] [Google Scholar]
- 19.Liang H, Esposito A, De S, Ber S, Collin P, Surana U, Venkitaraman AR. Homeostatic control of polo-like kinase-1 engenders non-genetic heterogeneity in G2 checkpoint fidelity and timing. Nat Commun 2014; 5:4048; PMID:24893992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vidair CA, Doxsey SJ, Dewey WC. Heat shock alters centrosome organization leading to mitotic dysfunction and cell death. J Cell Physiol 1993; 154:443-55; PMID:8436595; http://dx.doi.org/ 10.1002/jcp.1041-540302 [DOI] [PubMed] [Google Scholar]
- 21.Nakahata K, Miyakoda M, Suzuki K, Kodama S, Watanabe M. Heat shock induces centrosomal dysfunction, and causes non-apoptotic mitotic catastrophe in human tumour cells. Int J Hyperthermia 2002; 18:332-43; PMID:12079588; http://dx.doi.org/ 10.1080/026567302101-29736 [DOI] [PubMed] [Google Scholar]
- 22.Debec A, Courgeon AM, Maingourd M, Maisonhaute C. The response of the centrosome to heat shock and related stresses in a Drosophila cell line. J Cell Sci 1990; 96:403-12; PMID:2121747 [DOI] [PubMed] [Google Scholar]
- 23.Fragkos M, Ganier O, Coulombe P, Mechali M. DNA replication origin activation in space and time. Nat Rev Mol Cell Biol 2015; 16:360-74; PMID:25999062; http://dx.doi.org/ 10.1038/nrm4002 [DOI] [PubMed] [Google Scholar]
- 24.Rakotomalala L, Studach L, Wang WH, Gregori G, Hullinger RL, Andrisani O. Hepatitis B virus X protein increases the Cdt1-to-geminin ratio inducing DNA re-replication and polyploidy. J Biol Chem 2008; 283:28729-40; PMID:18693245; http://dx.doi.org/ 10.1074/jbc.M802751200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klotz-Noack K, McIntosh D, Schurch N, Pratt N, Blow JJ. Re-replication induced by geminin depletion occurs from G2 and is enhanced by checkpoint activation. J Cell Sci 2012; 125:2436-45; PMID:22366459; http://dx.doi.org/ 10.1242/jcs.100883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iwahori S, Kohmon D, Kobayashi J, Tani Y, Yugawa T, Komatsu K, Kiyono T, Sugimoto N, Fujita M. ATM regulates Cdt1 stability during the unperturbed S phase to prevent re-replication. Cell Cycle 2014; 13:471-81; PMID:24280901; http://dx.doi.org/ 10.4161/cc.27274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tachibana KE, Gonzalez MA, Guarguaglini G, Nigg EA, Laskey RA. Depletion of licensing inhibitor geminin causes centrosome overduplication and mitotic defects. EMBO Rep 2005; 6:1052-7; PMID:16179947; http://dx.doi.org/ 10.1038/sj.embor.7400527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu F, Lan R, Zhang H, Jiang Q, Zhang C. Geminin is partially localized to the centrosome and plays a role in proper centrosome duplication. Biol Cell 2009; 101:273-85; PMID:18798731; http://dx.doi.org/ 10.1042/BC20080109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim GS, Kang J, Bang SW, Hwang DS. Cdc6 localizes to S- and G2-phase centrosomes in a cell cycle-dependent manner. Biochem Biophys Res Commun 2015; 456:763-7; PMID:25498505; http://dx.doi.org/ 10.1016/j.bbrc.2014.12.018 [DOI] [PubMed] [Google Scholar]
- 30.Kalfalah FM, Berg E, Christensen MO, Linka RM, Dirks WG, Boege F, Mielke C. Spatio-temporal regulation of the human licensing factor Cdc6 in replication and mitosis. Cell Cycle 2015; 14:1704-15; PMID:25875233; http://dx.doi.org/ 10.1080/15384101.2014.1000182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tabuchi Y, Takasaki I, Wada S, Zhao QL, Hori T, Nomura T, Ohtsuka K, Kondo T. Genes and genetic networks responsive to mild hyperthermia in human lymphoma U937 cells. Int J Hyperthermia 2008; 24:613-22; PMID:18608577; http://dx.doi.org/ 10.1080/02656730802140777 [DOI] [PubMed] [Google Scholar]
- 32.Yurov YB, Iourov IY, Monakhov VV, Soloviev IV, Vostrikov VM, Vorsanova SG. The variation of aneuploidy frequency in the developing and adult human brain revealed by an interphase FISH study. J Histochem Cytochem 2005; 53:385-90; PMID:15750026; http://dx.doi.org/ 10.1369/jhc.4A6430.2005 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.