

ABSTRACT
The realization, that the androgen receptor (AR) is essential for prostate cancer (PC) even after relapse following androgen deprivation therapy motivated the search for novel types of AR inhibitors. We proposed that targeting AR expression versus its function would work in cells having either wild type or mutant AR as well as be independent of androgen synthesis pathways. Previously, using a phenotypic screen in androgen-independent PC cells we identified a small molecule inhibitor of AR, ARTIK-52. Treatment with ARTIK-52 caused the loss of AR protein and death of AR-positive, but not AR-negative, PC cells. Here we present data that ARTIK-52 induces degradation of AR mRNA through a mechanism that we were unable to establish. However, we found that ARTIK-52 is toxic to breast cancer (BC) cells expressing AR, although they were not sensitive to AR knockdown, suggesting an AR-independent mechanism of toxicity. Using different approaches we detected that ARTIK-52 induces replication-dependent double strand DNA breaks exclusively in cancer cells of prostate and breast origin, while not causing DNA damage, or any toxicity, in normal cells, as well as in non-PC and non-BC tumor cells, independent of their proliferation status. This amazing specificity, combined with such a basic mechanism of toxicity, makes ARTIK-52 a potentially useful tool to discover novel attractive targets for the treatment of BC and PC. Thus, phenotypic screening allowed us to identify a compound, whose properties cannot be predicted based on existing knowledge and moreover, uncover a barely known link between AR and DNA damage response in PC and BC epithelial cells.
KEYWORDS: androgen receptor, ARTIK-52, breast cancer, DNA damage, p53, prostate cancer
Introduction
Availability of high content libraries of small molecule opens up the possibility of identifying chemicals with almost any desired biological properties. The choice between target- or phenotype-oriented screenings depends on multiple factors, one of which is the availability of an established or proposed target responsible for specific phenotype. However, even if target is known, a phenotype- or cell-based screening still has certain attractive features.
In phenotypic screening, molecules are selected based on their ability to change a complex phenotype in a model system (e.g. cells). The disadvantage of this approach is that the exact mechanism of compound activity is obscured, since the desired change in cell state may be achieved via multiple paths. Consequently, the exact steps in signaling pathways and biochemical reactions modulated by small molecule remain unknown without special and often laborious investigation. On the other hand this uncertainty may be seen as an advantage, a way to discover unknown and potentially critical druggable nodes of regulation of different cellular processes, which otherwise may not be easily revealed.
The initial stimulus for our phenotypic screening was the realization that androgen receptor continues to be a valid target in prostate cancer (PC) treatment, even at the stage of recurrence of PC after androgen withdrawal therapy.
We confirmed the requirement of AR for relapsed PC cells using RNAi to AR 1 and proposed that complete elimination of AR would be the most effective approach to inhibit AR signaling. We used androgen insensitive PC cells with AR-dependent reporter to identify small molecules that were able to inhibit luciferase activity.2 Some of the identified small molecules inhibiting AR-dependent transcription were able to cause reduction of AR protein level. We noticed that only these compounds induced PC cell death, while molecules that inhibited AR transcription without any effect on AR protein level only suppressed growth of PC cells. The former molecules were named ARTIK or “AR Transcription inhibiting – Killing."2
One of the criteria used for identification of specific compounds was selective toxicity to AR positive PC cells combined with the absence of toxicity to AR-negative prostate or non-prostate cells. To rule out non-specific toxic compounds we used tumor and non-tumor cell lines of different origin.2 A representative set of breast cancer (BC) cell lines was not included in this list because non-specific toxicity toward breast epithelia was of little concern in male patients with PC. Ironically, upon further investigation BC cells were determined to be the only other cell type sensitive to ARTIK compounds found so far.
In this study we focused on ARTIK-52 compound (c52 in ref. 2). We found that ARTIK-52 induces degradation of AR mRNA through yet unidentified mechanism. After observing phenomena caused by ARTIK-52 in cells, we established that this compound possessed a very specific and unusual toxicity profile limited almost exclusively to AR expressing tumor cells of breast and prostate origin. Although this effect explains ARTIK-52 toxicity to PC cells, it is not the case for BC, whose viability does not depend on AR expression. In an attempt to find a mechanism for ARTIK-52 specific toxicity we found that it causes cell type specific replication dependent DNA damage. The idea that DNA damage can be induced in a tissue specific manner, and moreover that this damage can be limited to tumor cells, while leaving non-tumor, even highly proliferating cells, intact, would be very provocative, without identification of such activity of small molecule in almost unrelated phenotypic screening.
Results
ARTIK-52 treatment leads to the reduction of AR and ERBB2 levels
In our previous work we established that ARTIK-52 is toxic to a panel of PC cells expressing AR, independent of their androgen sensitivity status and type of AR mutation.2 To reveal the mechanism of ARTIK-52 activity we assessed its effect on AR protein in several PC cells and observed that it causes a reduction in AR protein levels in a time and dose dependent manner (Fig. 1 A, B). We excluded the effect of ARTIK-52 on AR protein stability, since inhibition of AR translation with cycloheximide, or blockage of its possible degradation with bortezomib, did not abrogate ARTIK-52 induced reduction of AR level (Fig. S1A-C). Measuring AR mRNA levels in the same cells revealed that ARTIK-52 treatment results in a dose and time dependent reduction of AR mRNA (Fig. 1 C, D). We excluded the effect of ARTIK-52 on AR gene expression since we still observed reduction of AR mRNA in ARTIK-52 treated cells when transcription was inhibited with actinomycin D (Fig. S1D, E). However the interpretation of all these experiments was complicated by the fact that all 3 chemicals, cycloheximide, bortezomib and actinomycin D induced cell death faster than ARTIK-52, and the effects of cycloheximide and actinomycin D on AR protein and mRNA levels respectively was also faster than that of ARTIK-52 (Figure S1A, B and data not shown).
Figure 1.
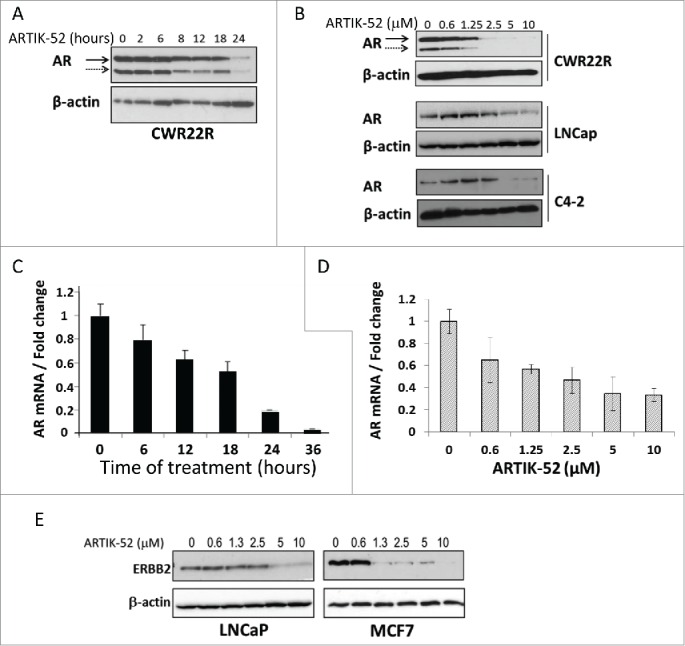
ARTIK-52 treatment leads to the reduction of AR and ERBB2 levels. A, B. Western blotting of protein extracts of CWR22R, LNCaP and C4-2 cells treated with 1 μM of ARTIK-52 for different periods of time (A) or with different concentrations for 24 hours (B) stained with AR and β-actin antibodies. Solid arrow marks full length 110 kDa form of AR, dashed arrow marks 80kDa AR isoform lacking ligand binding domain in CWR22R cells. C, D. Normalized data of qPCR with reversed transcribed total RNA of CWR22R cells treated with 1 μM of ARTIK-52 for different periods of time (C) or with different concentrations for 24 hours (D). Error bars represent standard deviation between 3 replicates within one experiment. E. Western blotting of protein extracts of LNCaP and MCF7 cells treated with ARTIK-52 for 24 hours stained with ERBB2 and β-actin antibodies.
Using a reporter construct with luciferase under the control of AR gene promoter (−424/+884) we did not observe any change in luciferase expression upon ARTIK-52 treatment (Figure S1F), indicating that ARTIK-52 effect is not mediated through AR gene promoter elements or transcriptional factors controlling AR expression.
These data suggested that ARTIK-52 may induce AR mRNA degradation. However, when we tried to inhibit RNA degradation in cells using shRNA to major component of exosome RRP4 (EXOSC2)3 we did not see any reduction of ARTIK-52 effect on AR (Figure S2A, B). Another way of regulation of AR mRNA level is through recently reported miRNA, miR488.4 To test the role of this mechanism we used 2 approaches, (i) siRNA to major endonuclease, responsible for mRNA degradation through miRNA regulated pathway, DICER (Figure S2 C); 5 and (ii) reporter construct with CMV regulated luciferase followed by miR488 binding site from AR mRNA 3'UTR 4 (Fig. S2D). ARTIK-52 neither lost its effect on AR in cells transfected with siRNA to DICER (Figure S2A, C), nor did it have any effect on the activity of miR488 containing luciferase reporter (Fig. S2E).
We also attempted to assess the role of several RNA binding proteins, which may be involved in the regulation of AR mRNA stability, including HuR,6,7 PCBP1, and PCBP2,6 in the mechanism of activity of ARTIK-52 using the same 2 approaches via siRNAs to HuR, PCBP1 and 2, and luciferase reporter constructs with different elements of AR 3'UTR containing the proposed binding sites of these RNA binding proteins.6,7 These experiments also did not reveal any role of these factors in the mechanism of activity of ARTIK-52 (Fig. S2D-F).
In parallel we searched the literature for other mRNAs whose stability is regulated through 3'UTR elements similar to that of AR. We selected ERBB2,8,9 since this gene is expressed in PC cell lines. Additionally, we analyzed ERBB2 expressing BC cells. Surprisingly, we observed that treatment with ARTIK-52 leads to the reduction of ERBB2 levels not only in PC, but also in the BC cell line MCF7 (Fig. 1E). Therefore, although ARTIK-52 interferes with the regulation of AR and ERBB2 mRNA stability, we were unable to elucidate exact mechanism of its effect of these mRNAs.
ARTIK-52 is toxic to AR positive prostate and breast cancer cells
The effect of ARTIK-52 on ERBB2 levels in BC cells prompted us to look at the effect of ARTIK-52 on AR levels in BC, since many of BC cells express AR.10-12 We treated an expanded list of cell lines including BC, and several non-tumor cells with ARTIK-52 and observed reduction of AR protein and mRNA levels in PC and BC cells, but not in non-tumor prostate cells (Fig. 2A-C). We found that ARTIK-52 was also toxic to the same AR-positive BC cell lines, although it did not show any toxicity to many other cells lacking AR expression, including BC (MDA-MB-231, BT549), PC (PC3, Du145), pancreatic adenocarcinoma (PANC1, MiaPaCa2), renal cell carcinoma (RCC45, ACHN) and melanoma (Mel7). (Fig. 2D, E). Moreover, ARTIK-52 was not toxic at all to non-tumor prostate and breast epithelial cells or normal diploid fibroblasts, which also express different levels of AR (Fig. 2E). Thus ARTIK-52 toxicity correlates with its ability to cause reduction of AR in tumor cells.
Figure 2.
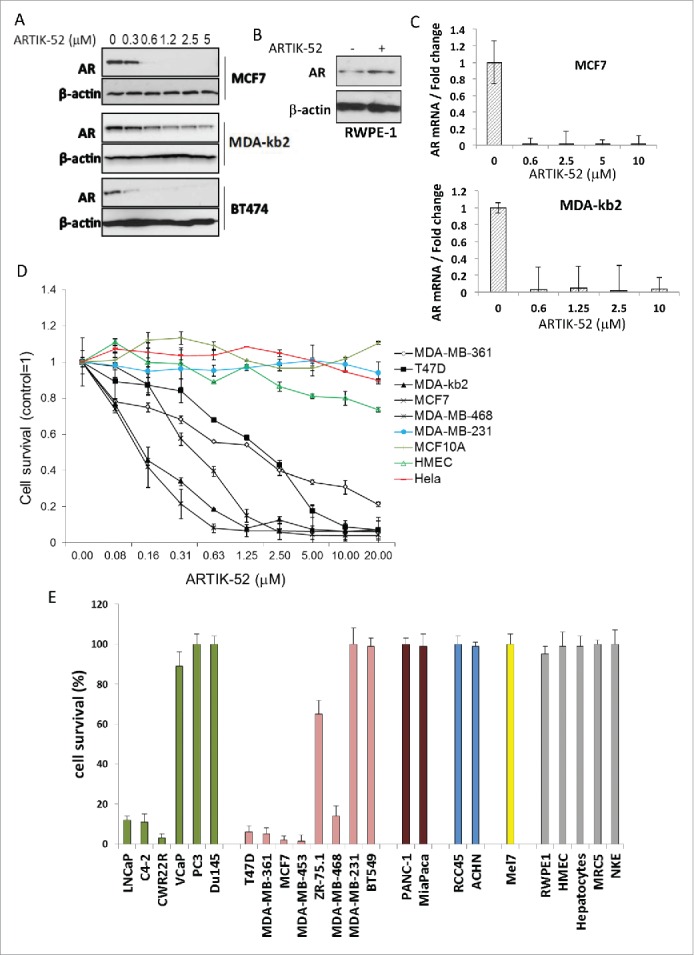
ARTIK-52 treatment causes reduction of AR levels and toxicity in AR expressing tumor cells. A. AR protein detected by western blotting in BC cells treated with indicated concentrations of ARTIK-52 for 24 h. B. Western blotting of extracts of normal prostate cells, RWPE-1, treated with 1 µM of ARTIK-52 for 24 h, stained with the indicated antibodies. C. AR mRNA levels detected using qPCR in BC cells treated with indicated concentrations of ARTIK-52 for 24 h. Error bars represent standard deviation between 3 replicates within one experiment. D. Survival of cells maintained with different concentrations of ARTIK-52 for 72 hours. Black lines correspond to AR positive tumor cell lines, colored lines – to AR negative tumor cell lines and non-tumor cells. Error bars represent standard deviation between 3 replicates in at least 2 different experiments E. Survival of cells treated with 5 µM of ARTIK-52 for 72 h. Untreated cells were taken as 100%. Green bars correspond to prostate cancer cells, pink – breast cancer cells, brown – pancreatic cancer, blue – renal cancer, yellow – melanoma, gray – non-tumor cells: RPWE-1 – prostate epithelial cell line, HMEC – primary mammary epithelia, MRC5 – diploid fibroblasts, NKE – kidney epithelial cell line. Error bars represent standard deviation between 3 replicates in at least 2 different experiments.
To see if ARTIK-52 toxicity is due to AR reduction in BC cells, we tested whether knockdown of AR was toxic to ARTIK-52 sensitive BC cells. Surprisingly we did not see any effect of AR knockdown using siRNAs to AR in BC cells, while the same siRNA reduced viability of PC CWR22R cells (Fig. 3A, B, C).
Figure 3.
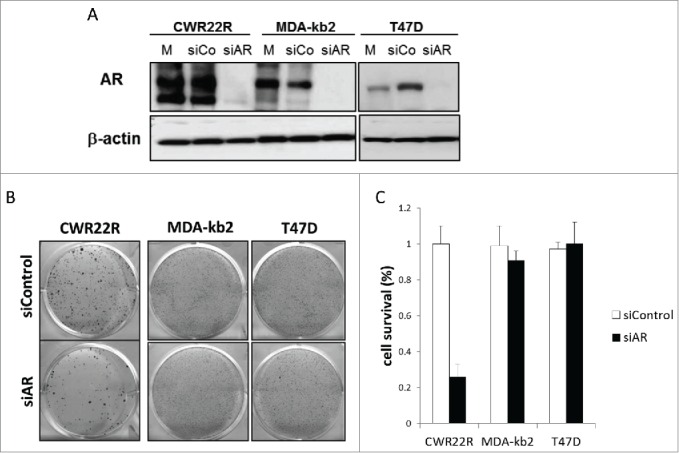
Effect of AR knockdown on survival of ARTIK-52-sensitive BC cells and CWR22R cells. Cells were transfected with siRNA to AR (siAR), scrambled siRNA (siCo) or left untreated (mock control, M). A. Western blotting of total cell extracts 7 d after transfection. B. Methylene-blue staining of colonies of corresponding transfected cells 14 d after transfection. C. Quantitation of methylene blue staining of colonies on panel B.
Thus, although ARTIK-52 has a clear effect on AR levels and its toxicity highly correlates with the AR status of BC cells, the mechanism of its toxicity to these cells appears to be AR independent.
ARTIK-52 toxicity is not specific to any known BC subtype
Trying to understand through which mechanism ARTIK-52 kills BC cells, we tested a panel of 31 well characterized BC cell lines,13 among which we found 16 sensitive and 15 resistant to ARTIK-52 treatment (Table S1). There were more AR positive cell lines among those sensitive to ARTIK-52 than among resistant cell lines, although both AR positive and negative cells were in ARTIK-52 sensitive and resistant categories (Fig. 4A), confirming that AR inhibition cannot explain ARTIK-52 toxicity to BC cells. There was also no correlation between ARTIK-52 sensitivity and expression of any other known markers of BC (ER, PR and ERBB2), or transcriptional subtypes (luminal, basal, claudin-low and ERBB2-amplified) (Fig. 4B). The comparison of toxicity profiles of the panel of BC cells sensitivity to ARTIK-52 and ~70 FDA approved agents 13 previously tested on the same panel did not reveal any similarities between ARTIK-52 and any of these drugs.
Figure 4.
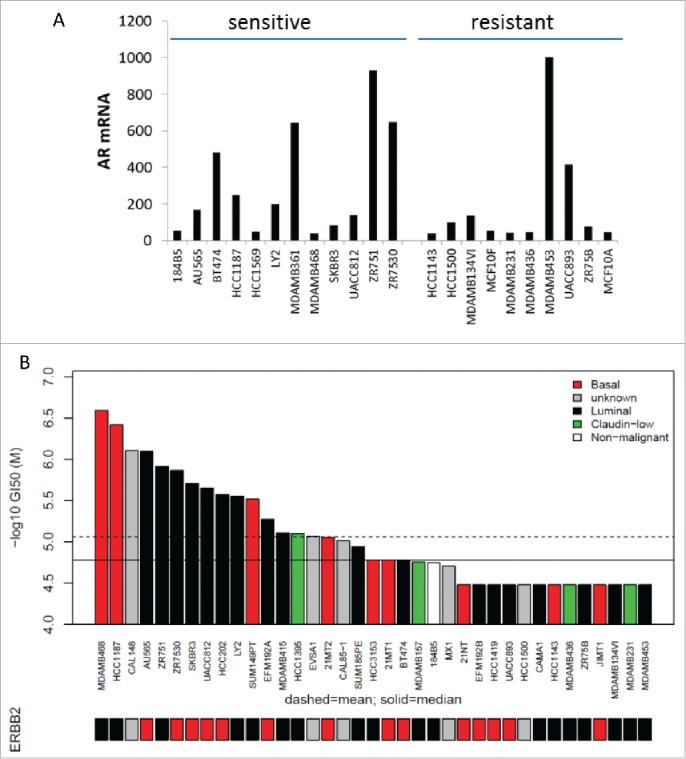
Sensitivity of different subtypes of BC cells to ARTIK-52. A. AR expression in highly ARTIK-52 sensitive (n = 12) and the completely resistant (n = 10) BC cells. Normalized intensity of microarray hybridization signal. B. Concentration of ARTIK-52 causing 50% growth inhibition (GI50) of BC cells of different transcriptional subtypes (luminal, basal, claudin-low and ERBB2-amplified). Bars under the plot show ERBB2 amplification status: red – amplified, black – non-amplified, gray – no data.
In the absence of an association between ARTIK-52 sensitivity and the expression of known BC markers we compared gene expression profiles of sensitive and resistant BC cells. We did not find any single gene, which expression clearly discriminated between ARTIK-52 sensitive and resistant cells. The only one gene, which was expressed in almost all resistant but not in sensitive cells, was CAV1 (Fig. S3A), which product is the main component of the caveolae or invaginations of the plasma membrane common in many vertebrate cell types.13,14 Although we confirmed a difference in CAV1 expression between sensitive and resistant BC cell lines, we did not observe any effect of ARTIK-52 treatment on CAV1 level in these cells (Fig. S3B). To test if CAV1 plays any role in ARTIK-52 resistance, we transduced sensitive CWR22R and MCF7 cells with a lentiviral vector containing CAV1 cDNA and treated these cells with ARTIK-52. As shown on Figure S3C and D, overexpression of CAV1 in CWR22R or MCF7 cells did not make sensitive cells resistant to ARTIK-52 treatment.
Assuming that ARTIK-52 sensitivity or resistance may not be associated with any one particular gene, but rather with an expression of a group of genes with similar functions or a pathway, we ran a Gene Set Enrichment Analysis (GSEA) using lists of genes mostly, but not exclusively, expressed either in resistant or sensitive cells. Genes expressed at higher levels in resistant vs. sensitive cells were enriched for those involved in DNA replication, nucleotide excision, and mismatch repair, while genes expressed mostly in sensitive cells showed enrichment for androgen and estrogen response, as well as ABC transporters (Fig. 5).
Figure 5.
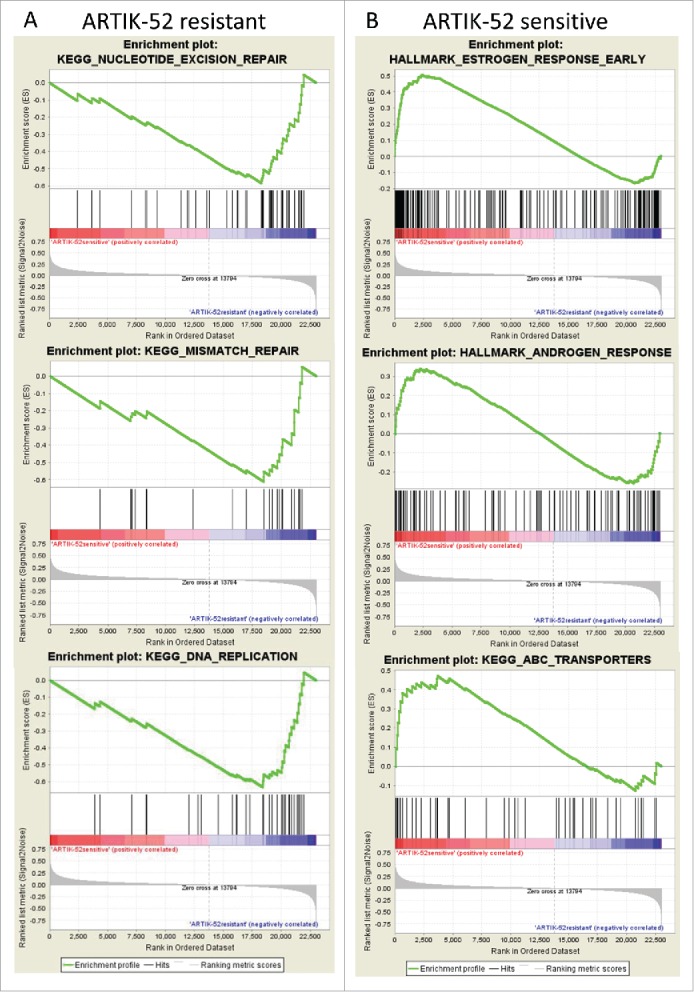
Enrichment of genes belonging to known pathways in BC cells sensitive or resistant to ARTIK-52. Results of GSEA for gene sets predominantly expressed in resistant (A) or sensitive (B) cell lines.
Differences in sensitivity to ARTIK-52 is not explained by differential drug uptake or by intracellular metabolism of the drug
Enrichment of ABC transporters among cell lines sensitive to ARTIK-52 does not easily suggest the mechanism of ARTIK-52 sensitivity, since the major function of ABC transporters is to pump small molecules out of cells 15 and is typically associated with cellular resistance to drugs. We decided however, to compare intracellular drug concentrations in sensitive (CWR22R, MCF7) and resistant (MDA-MB-231, Hela) cells. LC/MS based assay demonstrated that ARTIK-52 was present in all samples independently of sensitivity status, more than that, the highest ARTIK-52 concentration was found in resistant Hela cells, and concentrations, present in resistant BC MDA-MB-231 cells, were higher than that in sensitive, MCF7, cells (Figure S4A), suggesting that ARTIK-52 selectivity is not due to differential uptake/efflux of the drug by cells.
Higher intra-cellular concentration of ARTIK-52 in 2 resistant cell lines versus that in 2 sensitive cell lines suggested that ARTIK-52 undergoes some chemical (metabolic) conversion in sensitive cells, which may be part of its mechanism of activity.
To test this hypothesis we utilized 2 approaches. First, we have co-cultured resistant MDA-MB-231 cells labeled with GFP with sensitive MCF7 cells. Metabolic conversion of ARTIK-52 in any of cells followed by diffusion of metabolite from cells to cell culture medium might change pattern of sensitivity of other cells to ARTIK-52 treatment. We observed that after 72 hour treatment with 1μM ARTIK-52 did not change number of GFP-positive cells compared to untreated control but led to complete disappearance of GFP-negative MCF7 cells demonstrating that co-culturing of 2 cell lines did not trigger sensitivity in MDA-MB-231 or resistance in MCF7 cells (Figure S4B).
As a second approach, we incubated ARTIK-52 sensitive and resistant cells with the drug for 6 and 20 hours and isolated all potential metabolites from them using a methanol extraction method.16 Dried extracted metabolites were reconstituted in cell culture medium and the resulting solutions were tested on ARTIK-52 resistant (MDA-MB-231) or sensitive cells (MCF7, CWR-22R) to detect the presence of cytotoxic activity. Figure S4C demonstrates that cytotoxic activity was still present in extracts from both sensitive and resistant cells, while specificity of this cytotoxic activity was not changed, demonstrating that ARTIK-52 was neither inactivated in resistant MDA-MB-231 cells, nor was it converted in sensitive cells into a compound toxic to MDA-MB-231 cells.
These data ruled out the possibility that ARTIK-52 selectivity is based on differences in drug uptake/efflux or intracellular metabolism between sensitive and resistant cells and suggested that it is rather explained by ARTIK-52 effect on certain molecular pathways in sensitive, but not resistant cells.
ARTIK-52 treatment induces p53 activation in sensitive, but not resistant cells
We performed a genome-scale gene expression analysis using mRNA isolated from sensitive and resistant cell lines treated with ARTIK-52 or control medium for 12 hours (before signs of toxicity were evident) using Illumina HumanHT-12 v4 Expression BeadChip. We used sensitive luminal: MCF7, MDA-kb2, and basal: MDA-MB-468 BC cell lines and resistant luminal ZR-75-1 (these cells are not sensitive to ARTIK-52 at concentration 3 μM, used in this experiment) and basal MDA-MB-231 cells. We also used sensitive PC cells CWR22R. First, we observed significantly less gene expression changes in response to treatment in ARTIK-52 resistant cell lines vs. ARTIK-52 sensitive cells (Fig. 6A). AR or ERBB2 expression was down-regulated in AR positive sensitive cells (CWR22R, MCF7), but not in resistant cells (ZR-75-1) (Figure S5A). There was no overlap between genes significantly changed in sensitive and resistant cells and no common pathway or process was identified among genes which expression was changed upon ARTIK-52 treatment in resistant cells. Gene Ontology analysis of genes, whose expression was significantly upregulated in sensitive cells, unexpectedly revealed signs of DNA damage response (Fig. 6B) including activation of p53 target genes in ARTIK-52 sensitive p53 wild type cells (CWR22R, MCF7, Fig. 6C, D). No activation of p53 response was detected in ARTIK-52 sensitive MDA-kb2, MDA-MB-468 cells in line with the presence of p53 mutations in these cells 17 and in p53 wild type but ARTIK-52 resistant ZR-75-1 cells (Fig. 6C, D). Cell type dependence of p53 response was quite surprising, but explained why it was not noticed before during small molecule primary screening of AR inhibitors 2 that were tested for the absence of activity against other transcriptional factors, including p53: effect of ARTIK-52 on p53 responsive reporter was tested in renal cell carcinoma RCC45 cells, which are resistant to this drug (Fig. 2E).
Figure 6.

Effect of ARTIK-52 treatment on gene expression in sensitive and resistant cells. A. Volcano plots demonstrating all genes which expression was changed in response to treatment with 3 μM of ARTIK-52 for 12 hours. Red dots correspond to genes which expression changed >2 times with p-value <0.05. Data of microarray hybridization analysis. B. Venn diagram showing intersection between lists of genes overexpressed in response to ARTIK-52 in all sensitive cells (>1.5). Genes common for all cells were used for Gene Ontology analysis using MetaCore software. C, D. Changes in p53 targets, CDKN1A (C) and BAX (D) mRNA levels in p53 wild type sensitive, but not resistant cells. Average normalized signal of microarray hybridization. Error bars – deviation between 3 biological replicates. Bars under plots show p53 status of the cell lines, red – wild type, black – mutant.
To confirm cell type specificity of p53 activation, we used several p53 wild type ARTIK-52 sensitive and resistant cells and observed that increase of p53 level occurred only in ARTIK-52 sensitive, but not resistant cells, while curaxins CBL0137, a non-cell type specific p53 activator,18 caused p53 induction in all tested cells (Fig. 7A-C). Thus ARTIK-52 appears to be a first in class small molecule cell-specific p53 activator.
Figure 7.
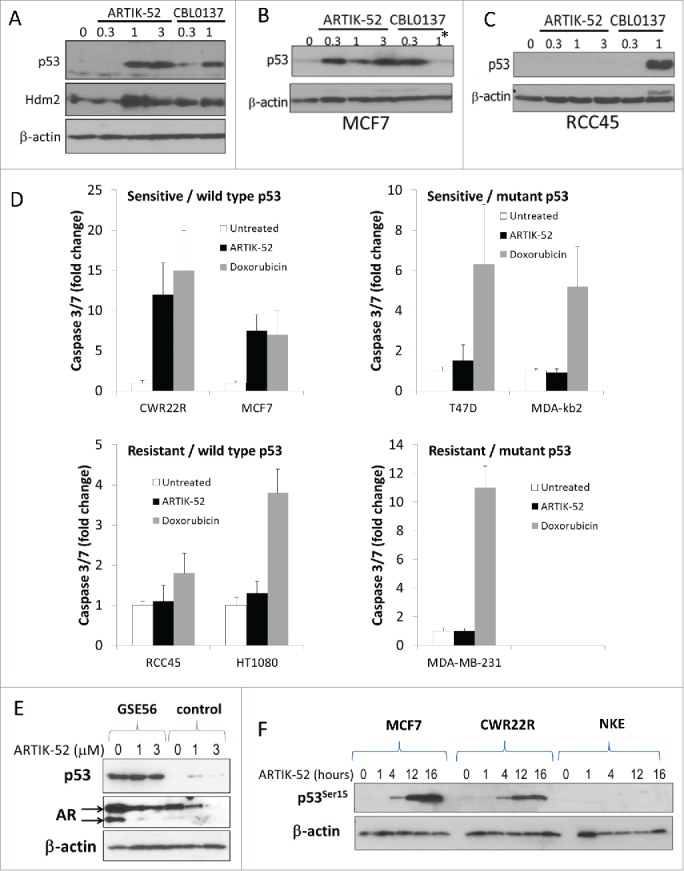
Activation of p53 and induction of p53-dependent apoptosis by ARTIK-52 in sensitive but not resistant cells. A-C. Western blotting of total cell extracts of ARTIK-52 sensitive, CWR22R (A) and MCF7 (B) cells, or resistant, RCC45 cells (C), treated with ARTIK-52 or CBL0137 (in μM) for 16 hours. * - reduction of p53 level in MCF7 cells in response to 1μM of CBL0137 is due to high toxicity of CBL0137 for these cells. D. Caspase 3/7 activation in p53 wild type and mutant cells in response to treatment with ARTIK-52 (1 μM) or doxorubicin (1 μM) for 16 hours. Bars correspond to fold changed compared with untreated control. Error bars – standard deviation between 3 replicates within one experiment. E. Effect of p53 inactivation by dominant negative mutant GSE56 on ARTIK-52 induced reduction of AR level in CWR22R cells. Western blotting of extracts of cells transduced with either GSE56 or empty vector and treated with indicated concentrations of ARTIK-52 for 16 hours. F. ARTIK-52 treatment causes phosphorylation of serine 15 of p53 in sensitive cells. Western blotting of extracts of ARTIK-52 sensitive (MCF7 and CWR22R) or resistant (NKE) cells treated with 1 μM of ARTIK-52 during indicated periods of time.
At the same moment, we could not explain toxicity of ARTIK-52s exclusively by p53 activation, as there is a set of p53-mutant BC cells sensitive to ARTIK-52 (all sensitive PC cells have wild type p53). However, though both p53-wild type and p53-mutant sensitive cells died upon treatment, only those with wild type p53 died by conventional apoptosis, which was revealed by activation of caspases-3,-7 upon treatment (Fig. 7D). In parallel with ARTIK-52 in these experiments we used doxorubicin, a non cell-specific DNA damaging agent, which induced caspase activation in all cells independent of p53 status or sensitivity to ARTIK-52 (Fig. 7D).
Activation of p53 by ARTIK-52 opens up the possibility to explain the reduction of AR in treated cells through upregulation of the p53 target, Hdm2 (Fig. 7A), since it was shown that AR is a substrate of the E3 ubiquitin ligase activity of Hdm2.19,20 Although the observed reduction of AR mRNA could not be explained by ubiquitin ligase activity of Hdm2, we decided to test if inactivation of p53 in ARTIK-52 sensitive cells would abrogate the reduction of AR protein level and the sensitivity of cells to ARTIK-52 treatment. We inactivated p53 in CWR22R cells with p53 dominant negative mutant GSE56.21 Basal level of AR protein was elevated upon p53 inactivation as expected,22 however, we did not notice any difference in the response of original and GSE56 expressing cells to ARTIK-52 (Fig. 7E).
p53 activation did not explain toxicity of ARTIK-52 to sensitive cells, nevertheless, we hypothesized that if we would find why p53 is activated, we will understand the mechanism of activity of ARTIK-52. First, we looked at the presence of phosphorylation of N-terminal p53 serine 15, a sign of activation of DNA-damage sensitive kinases, in ARTIK-52 treated cells. Treatment of CWR22R and MCF7 sensitive, but not resistant NKE cells, clearly induced phosphorylation of serine 15 (Fig. 7F). Thus, there is a possibility that ARTIK-52 induces DNA damage in sensitive cells.
ARTIK-52 induces DNA damage in sensitive, but not resistant cells
We conducted a panel of assays to detect the presence of DNA damage after ARTIK-52 treatment. We observed that in sensitive, but not resistant cells, an increase in γH2AX staining, the appearance of “comets” under alkali conditions in a Comet Assay, and the appearance of damaged DNA upon pulse field gel electrophoresis (PFGE) of total cellular DNA lysed in gel plugs (Fig. 8A–D). Thus, ARTIK-52 treatment induces DNA damage in sensitive, but not in resistant cells, although, as we showed before, the drug is present inside both types of cells (Fig. S4A).
Figure 8.
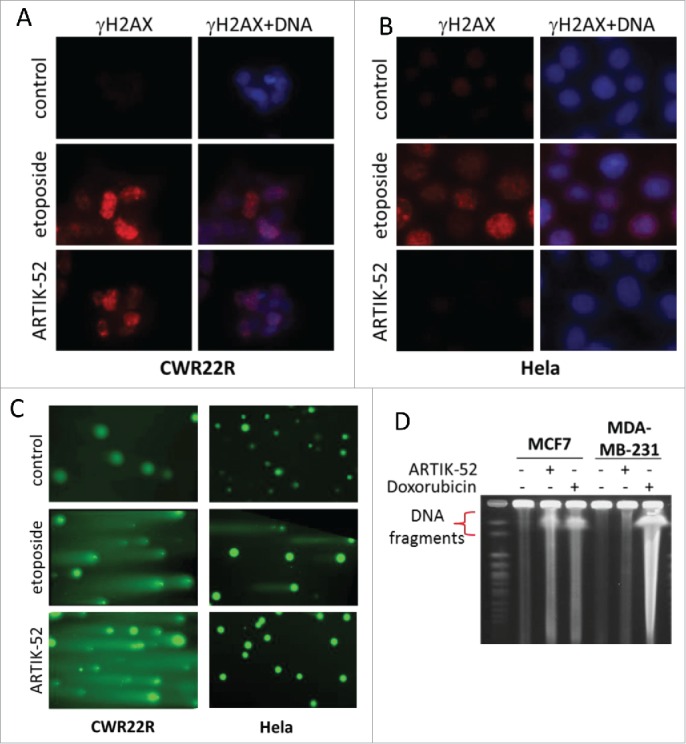
ARTIK-52 treatment induces DNA damage in sensitive, but not resistant cells. A, B. Immunofluorescent staining of ARTIK-52 sensitive, CWR22R (A) or resistant, Hela (B) cells treated with 1 μM of ARTIK-52 or 1 μg/ml of etoposide for 6 hours with γH2AX antibody and Hoechst 33342 DNA stain. C. Comet assay in alkali conditions with cells treated with 1 μM of ARTIK-52 or 1 μg/ml of etoposide for 16 hours. D. PFGE of DNA from cells treated with 1 μM of ARTIK-52 or 1 μg/ml of doxorubicin for 16 hours.
Next, we tested what kind of DNA damage ARTIK-52 induces. The results of PFGE and positive γH2AX staining suggested the presence of double strand breaks; however they may be result of single strand breaks converted into double strand breaks in the process of DNA replication.23 The presence of single strand breaks slows down replication due to replication fork stalling followed by collapse at these sites and therefore accumulation of cells in S phase of cell cycle.24 In line with this we observed that treatment with ARTIK-52 inhibits EdU incorporation in sensitive, but not resistant cells, and leads to accumulation of sensitive cells in S phase (Fig. 9A-D and S6).
Figure 9.
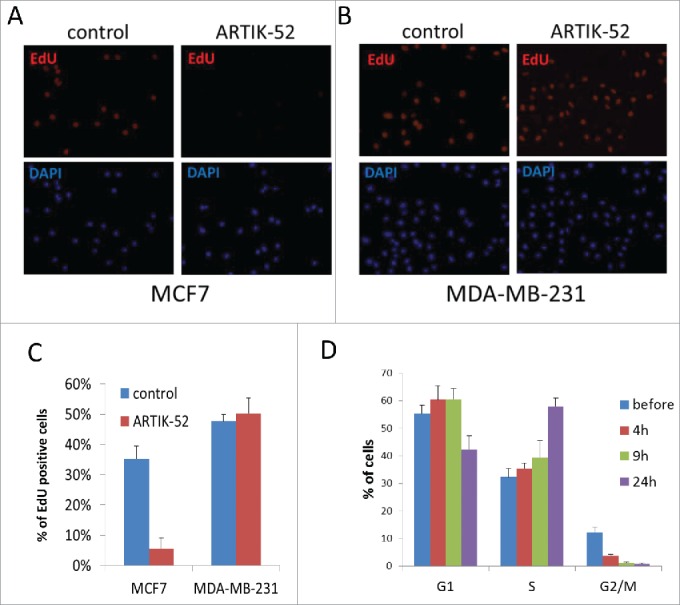
ARTIK-52 inhibits replication and causes accumulation of cells in S-phase of cell cycle. A, B. EdU incorporation in ARTIK-52 sensitive MCF7 (A) or resistant MDA-MB-231 (B) cells treated with 1 μM of ARTIK-52 for 9 hours. EdU was added for the last 2 hours of incubation. C. Quantitation of EdU staining shown on panels A and B done by counting of EdU positive and DAPI positive cells in 10 fields of view (10X magnification). Error bars – standard deviation between % of EdU positive cells in 10 fields. D. FACS analysis of cell cycle distribution of CWR22R cells before and after treatment with 1 µM of ARTIK-52 quantitated ModFit LT (Verity Software House). Error bars are deviations between 2 replicates within one experiment.
We also compared the presence of single stranded DNA in nuclei of sensitive and resistant cells treated with ARTIK-52 using antibodies to 2 proteins able to bind single stranded DNA, RPA1 and XRCC1.25-27 We observed an enhancement of staining in sensitive MCF7 cells, while there was no change in ARTIK-52 resistant MDA-MB-231 cells (Fig. 10A). To confirm that double strand DNA breaks in ARTIK-52 treated cells are associated with replication, we labeled sites of replication in MCF7 cells with EdU and co-stained the same cells with antibody to γH2AX. We observed that in cells treated with ARTIK-52 all γH2AX foci were co-localized with EdU (Fig. 10B, C).
Figure 10.
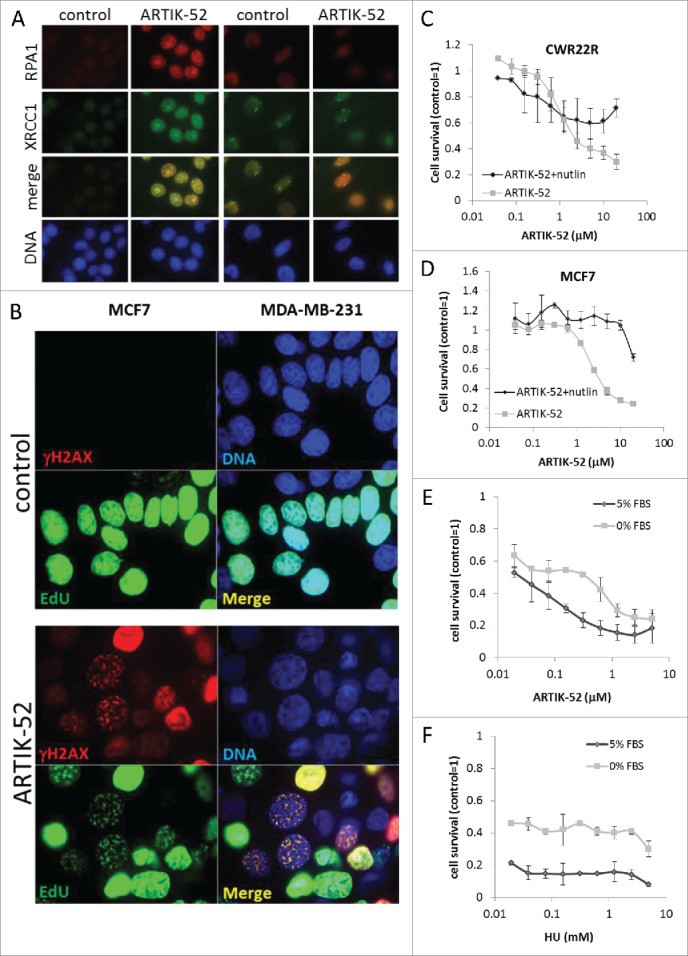
DNA damage and toxicity caused by ARTIK-52 treatment are replication dependent. A. Immunofluorescent staining of cells treated with 1 μM of ARTIK-52 for 6 hours with antibodies to RPA1, XRCC1 and DAPI. B. Immunofluorescent images of MCF7 cells incubated with ARTIK-52 (1 µM) and EdU for 16 hours followed by Click-IT assay, and staining with γH2AX antibody and DAPI. C, D. E. Cytotoxicity assay on CWR22R (C) and MCF7 (D) cells and either pre-treated with nutlin (CWR22R −10 µ M, MCF7 −5 µM) or not for 24 hours before addition of ARTIK-52. Nutlin was left with cells untill the end of experiment, but diluted twice due to addition of ARTIK-52 solutions. E, F. Cytotoxicity assays on arrested or growing CWR22R cells with ARTIK-52 or HU. Error bars on C-F are standard deviation between 3 replicates in one experiment.
Well-known inhibitor of replication, ribonucleotide reductase inhibitor, hydroxyurea (HU),28 is toxic to cells only if they undergo proliferation. Thus, we compared ARTIK-52 toxicity in cycling and quiescent cells. We used 2 approaches to induce quiescence in CWR22R and MCF7 cells, serum withdrawal for 48 hours and treatment with nutlin, an inhibitor of p53/mdm2 interaction, which causes growth arrest in p53 wild type cells at certain concentration.29 Both approaches demonstrated that ARTIK-52 is more toxic to cycling than growth arrested cells (Fig. 10C-F).
Thus, ARTIK-52 is toxic to a subset of tumor cells of prostate and breast origin due to its cell-type specific ability to induce replication-associated DNA damage in sensitive cells. Why only a part of cancer cells from these tissues are sensitive to ARTIK-52 and why normal cells of the same origin are not sensitive, as well as what role AR plays in ARTIK-52 induced DNA damage – are questions which require further investigation.
Discussion
There are still debates in the drug discovery field about the benefits and deficits of target- vs phenotype-based drug discovery approaches, and most importantly, what screening strategy is the most fruitful. Although target-oriented drug discovery seems more straightforward, several recent analyses revealed that during last decade phenotype-based drug screenings were the major source of FDA approved anti-cancer drugs.30-33 The most probable reason of this is our still quite limited understanding of the complex behavior of living cells. In this situation forecasting the consequences of our manipulations of potential targets is very inaccurate.
Using phenotype based approaches, we avoid these predictions, but instead observe a correlation between our random manipulations and phenotype changes. In the event where we establish a link between certain manipulation and a desirable phenotype change, this same manipulation has a much higher chance of causing similar phenotypic changes in clinical settings. In this way we also learn how to cause the most effective change in phenotype, although there may be almost no clue as to the underlying mechanism. It means that we have a model to study this novel mechanism, which in many cases we were not aware of at the beginning of the screening. This is in fact an unbiased way to find new and critical mechanisms of regulation of multiple processes in cells. In some cases going for the strongest hits we find the most critical node to affect the whole network, and occasionally some unexpected additional links between different functional systems.
Our current study presents a typical example of this. We identified a chemical inhibiting AR reporter activity through induction of AR mRNA degradation; however, eventually we found that this chemical induces cell type dependent DNA damage, which was not explicitly described before. In recent years, multiple links between AR transcriptional activity and DNA damage response and repair were established.34 Functional and physical interactions of AR with several checkpoint and DNA damage detection factors were observed, such as RAD9, a component of the 9-1-1 complex, 35CHK1,36 BRCA1,37,38 BRCA2 39 and several others (for review see ref. 36). Moreover, 2 recent reports demonstrated that inhibition of AR activity through ligand depletion, or with novel AR inhibitors (ARN-509 or MDV3100) increases DNA damage induced by gamma-radiation and even basal level of DNA damage in PC cells.34,40 Thus, it was at first tempting to speculate that ARTIK-52 induces DNA damage in PC cells through inhibition of AR function. However, we were unable to establish this link. ARTIK-52 induces DNA damage in multiple BC cell lines, some of which are AR negative, suggesting that it can cause DNA damage through an AR-independent mechanism. Moreover, inhibition of AR in these BC cells with siRNA was not toxic for these cells, which would be expected if inhibition of AR is responsible for the induction of DNA damage. Thus, we have to conclude that 2 phenotypic changes induced by ARTIK-52 in PC and BC cells: inhibition of AR and induction of DNA damage, are not directly related. To the best of our knowledge, the effect of specific AR inhibition (androgen deprivation therapy, direct AR inhibitors or RNAi to AR) on DNA damage induction in BC cells expressing AR has not been tested so far in any published study.
Broad testing of ARTIK-52 toxicity on a panel of >50 cell lines revealed an intriguing observation: very specific toxicity to cancer cells of prostate and breast origin. We did not observe any effect of ARTIK-52 on all tested cells of different origin, including normal epithelia of prostate and breast at concentrations up to 2 orders of magnitude higher than in PC and BC cells. Although DNA damage and toxicity of ARTIK-52 depends on replication, this does not explain absence of toxicity to non-tumor mammary and prostate epithelial cells, which actively proliferate in culture and which are completely resistant to ARTIK-52. This exclusively specific toxicity conflicts with the DNA damage inducing property of ARTIK-52. While the toxic effect of DNA damage on cell viability is well known, there are no examples of cell specific DNA damaging compounds. Previously established ways to explain different sensitivity of cells to DNA damaging stimuli were limited to variability in proliferative rates, ability of cells to pump out or metabolize DNA damaging agents, or expression of enzymes involved in DNA metabolism or repair. We ruled out practically 3 out of 4 possibilities since we did not observe significant differences among proliferating rates of sensitive and resistant to ARTIK-52 cells. We did not observe higher concentrations of ARTIK-52 in sensitive versus resistant cells. We were unable to detect toxic metabolites of ARTIK-52 in sensitive cells, or metabolic inactivation of ARTIK-52 in resistant cells. The fourth possibility may be the case since genes expressed at higher level in ARTIK-52 resistant cells are enriched in pathways involved in nucleotide excision and mismatch repair pathway. However, expression of no single gene which may be responsible for ARTIK-52 mechanism of action was different between the 2 categories of cells.
We did not find examples of cell-type specific expression of genes involved in DNA metabolism, repair, or damage detection in literature except reduction of expression of these sorts of factors upon terminal differentiation of some normal cells.23 However, this mystery of ARTIK-52 specificity of DNA damage is reminiscent of the long existing mystery of why hereditary mutations of DNA damage detection or repair genes in most cases cause not a general cancer susceptibility, but appearance of very tissue specific types of cancer. Few examples of this include: germ line mutations in 2 components of mismatch repair machinery, MSH2 and MLH1 predispose bearers almost exclusively to nonpolyposis colorectal cancer,41,42 while mutation in MLH2 increases risk of endometrial cancer,43 MSH6 – colon and gastric carcinoma.44 BRCA1, a protein involved in different aspects of DNA damage detection and repair, significantly increases the chances of developing basal breast and ovarian cancers; 45,46 while BRCA2, an important player of the homologous recombination pathway, prompts early onset breast cancer of luminal type and prostate adenocarcinomas.47,48
This list of tissue specific predisposition to cancer upon loss of function of BRCA2 is quite similar to the list of tissues sensitive to DNA damaging effect of ARTIK-52, which suggests a potential link between ARTIK-52 mechanism of action and homologous recombination. Unfortunately, it does not aid us in the understanding of why defects in homologous recombination cause cancers almost exclusively in luminal epithelia of breast and prostate. However, the cell-type specific activity of ARTIK-52 may serve as a model and tool to approach this intriguing question.
Materials and methods
Chemicals
The following reagents were purchased from Sigma-Aldrich: Actinomycin D (A9415), doxorubicin (D1515), cycloheximide (C4859) and hydroxyurea (H8627). ARTIK-52 (1-(3-chlorophenyl)-N-(3-morpholin-4-ylpropyl)-9H-pyrido[3,4-b]indole-3-carboxamide) was purchased from Chembridge Chemical Corporation, bortezomib (sc-217785) – from SantaCruz Technologies. Etoposide (45-E2600000) and Nutlin (45-N8287) were purchased from Krackler Scientific. CBL0137 was provided by Incuron, LLC (Buffalo, NY).
Cells
CWR22R cells were obtained from Warren Heston (Cleveland Clinic), ACHN, BT474, BT549, C4-2, Du145, Hela, HT1080, LNCaP, MCF7, MCF10a, MDA-kb2, MDA-MB-231, MDA-MB-468, MiaPaCa2, PANC1, PC3, RWPE1, T47D, VCaP, ZR-75-1, Mel7 and 293T were obtained from ATCC, frozen at early passages (<5) and used for no more than 3 months after thawing. All cells were propagated as suggested by ATCC, except all tumor cells were kept at 5% Fetal Bovine Serum (FBS), instead of recommended 10%. NKE-hTert and RCC45 cells were described in.49 Human Mammary Epithelial Cells (HMECs) and primary human hepatocytes were purchased from Lonza and propagated as suggested. The following media and supplements were used: RPMI 1640 (Gibco, 11875119), DMEM-F12 (Cellgro, 40-101-CV), DMEM (Gibco, 11995073), FBS (Gibco, 10437028), 1M HEPES Buffer (Corning, 25-060-CI), 100 mM Sodium Pyruvate (Corning, 25-000-CI), 2Mercaptoethanol (Gibco, 21985-023), MEM Nonessential Amino Acids (Corning, 25-030-CI), Penicillin(10000 U/ml)-Streptomycin(10000 ug/ml) mix (Corning, 30-002-CI). Transfections were done using Lipofectamine 2000 Reagent (Invitrogen, 52887).
Constructs
pLGSE56SP expressing construct was described in; 49 ARE-luc construct, expressing firefly luciferase from AR-responsive element was described in ref. 1 Gaussia luciferase driven by AR-promoter was purchased from Genecopoeia (HPRM11148). Lentiviral construct expressing CAV1 was generated through cloning RT-PCR amplified 605 bp fragment of CAV1 (primers used: Fw: CAGAACAAACCTTTGGCGGG; Rv: GGGCTTGTAGATGTTGCCCT) from MDA-MB-231 cells into pLV-CMV-SV40-puro vector through XbaI and BamHI restriction sites. Lentiviral constructs expressing CMV-driven firefly luciferase fused to AR 3'UTR fragments were generated through cloning of the following annealed oligonucleotides into the MicroRNA Target Selection System Vector (SystemBio):
AR1234: AATTCGAATTCTATTTGCTGGGCTTTTTTT-TTCTCTTTCTCTCCTTTCTTTTTCTTCTTCCCTCCCTA-TCTAACCCTCCCATGGCAG + GATCCTGCCATGGG-A-GGGTTAGATAGGGAGGGAAGAAGAAAAAGAAAGGAG-AGAAAGAGAAAAAAAAAGCCCAGCAAATAGAATTC-G
AR 123: AATTCCTGGGCTTTTTTTTTCTCTTTCTCTC-CTTTCTTTTTCTTCTTCCCTCCCTAG + GATCCTAGGGAGGGAAGAAGAAAAAGAAAGGAGAGAAAGAGAAA-AAAAAAGCCCAGG
AR 12: AATTCGAATTCTATTTGCTGGGCTTTTTT-TTTCTCTTTCTCTCCTTTCTTTTTCTTCG + GATCCGA-AGAAAAAGAAAGGAGAGAAAGAGAAAAAAAAAG-CCCAGCAAATAGAATTCG
AR 34: AATTCCTTCCCTCCCTAUCTAACCCTCCCAT-GGCAG + GATCCTGCCATGGGAGGGTTAGATAGGGA-GGG-AAGG
AR 234: AATTCCTCTCCTTTCTTTTTCTTCTTCCCTC-CCTATCTAACCCTCCCATGGCAG + GATCCTGCCATG-GGAGGGTTAGATAGGGAGGGAAGAAGAAAAAGAAAG-GAGAGG
ARmiR: AATTCGGAAGTTTAGAGAGCTAAGATTATC-TGGGGAAATCAAAACG + GATCCGTTTTGATTTCCCC-AGATAATCTTAGCTCTCTAAACTTCCG
All oligonucleotides were synthesized by Integrated DNA Technologies (Coralville IA).
shRNA
The pLPCPw-shAR lentiviral vector directing expression of shRNA against human AR was generated as described in.1 The shRNA sequence corresponding to the 3'UTR of human AR was TGATCCTCATATGGCCCAG; sequence corresponding to the 5'UTR of human AR was GTCAGGTCTTCAGTAGCCA.
siRNA to Dicer (EHU051411), HuR (EHU063491), PCBP1 (SASI_Hs01_00034329) and PCBP2 (SASI_Hs02_00319506) were purchased from Sigma-Aldrich. Transfections were done utilizing Lipofectamine reagent (Invitrogen, 52887) with siRNAs at concentration of 20 nM. siRNA to ExoSC2 (L-016975-01-0005) and to AR were purchased from Dharmacon, transfections were done wth DharmaFECT reagent (Dharmacon, T2001-01). Each company provided a corresponding scrambled siRNA control.
Lentiviral transduction was done as already described in ref. 49 Transduced cells were selected using 2μg/ml of Puromycin
Cytotoxicity assay
Cells were plated on 96 well plates at concentration of 5000 cells/well. Next day cells were treated with drug at different concentrations. 9-aminoacridine (50μM) was used as a positive control, while 0.1% DMSO was used as a negative control. 72 hours after start of treatment cell viability was assessed using CellTiter-Blue™ Cell Viability Assay (Promega, G8080).
Colony assay
Cells were plated in 6 well plates at 10(5)/well in duplicates. siAR and siControl was transfected into cells at 100 nM using DharmaFECT reagent from Dharmacon (T2001-01). Next day all cells were plated in p100 plates in regular media. At 7 d cells were collected for western blotting and at 14 d cells fixed and stained with Methylene Blue. Quantitation of methylene blue extracted from plates with 1% SDS solution was done using spectrophotometer at λ = 600 nm.
Comparison of ARTIK-52 toxicity to a panel of 31 BC cell lines was done as described in ref. 13 In brief, cells were treated in triplicates for 48 h with ARTIK-52 in 1:5 serial dilutions. Cell proliferation was estimated using the Cell Titer-Glo assay (Promega). GI50 (50% growth inhibition), TGI (concentration for complete growth inhibition) and LC50 (concentration that reduced population by 50%) were calculated as described in ref. 13
Quantitative RT-PCR
Total RNA was isolated using TRIzol reagent (Life Technologies, 15596018) according to the manufacturer's protocol. First-strand cDNA was synthesized from 1 µg of total RNA using an iScript cDNA Synthesis kit (BioRad, 170-8891) according to the manufacturer's protocol. Quantitative real-time PCR (qPCR) was performed with primers purchased from Applied Biosystems: AR - Hs00171172_m1 (FAM), 72bp and 18S – Hs99999901_s1 (FAM), 187 bp, using TaqMan gene Expression Master Mix (Applied Biosystems, 4369016) and the default parameters of the 7900HT sequence detection system (ABI PRISM; Applied Biosystems). To compare gene expressions among samples, the threshold cycle (CT) value was normalized using the mean CT for the reference gene, 18S rRNA (rRNA). The normalized mRNA level was defined as ΔCT = CT (test gene) − CT (mean for the reference gene). The final data were expressed as the fold difference between the test sample and the control sample, which was defined as 2(ΔCT test − ΔCT control). All reactions were performed in triplicates, the results are presented as the mean of their values.
Northern blot hybridization
Total RNA was extracted from cells using TRIzol (Invitrogen, 15596018). 5 μg of RNA was separated by electrophoresis on a 1% agarose-formaldehyde gel and blotted onto a Hybond N+ membrane (Amersham Biosciences, RPN 303N). Dried and UV cross-linked membranes were hybridized for 16 h with specific [32P]-labeled probes: products of reverse-transcription PCR from CWR22R RNA with primers specific to AR (Fw: cggaagctgaagaaacttgg; Rv: atggcttccaggacattcag) or GAPDH (Fw: ggcttccgtgtccccactgc and Rv: ggctggtggtccaggggtct-3'). Primers were synthesized by Integrated DNA Technologies. PCR was performed using Taq-polymerase PCR Master mix (Affymetrix, 71162) at Tm 57°C and 58°C for AR and GAPDH, respectfully, 32 cycles of PCR were held in order to produce enough of product material. The DNA oligonucleotides were labeled with Random Primed DNA Labeling Kit (Roche, 11004760001) in the presence of [α-32P]dCTP according to manufacturer's protocol. Membranes were exposed to X-ray film for 16h at −80°C.
Western blotting
Protein lysates were obtained by lysing cell pellets with Passive Lysis Buffer (Promega, 15593-031) supplied with protease inhibitor cocktail (Roche, 4693116001). The following antibodies were used for western blotting or immune-fluorescence: Sigma-Aldrich: β-Actin (AC-15); BD Pharminogen: AR (554225); Santa Cruz: p53 (DO1) (sc126), HDM2: SMP14 (sc965), Goat-Anti-mouse (Sc2005), Goat-Anti-rabbit (Sc2004); Cell Signaling: p53-Ser15 - 16G8 (9286), phospho-histone H2A.X (9718S), ErbB2 - 29D8 (2165S); AbCam: XRCC1 (ab1838), RPA70 (ab79398). Invitrogen: Alexa Fluor→ 488 Donkey Anti-Mouse IgG (H+L) (A-21202); Alexa Fluor→ 488 Donkey Anti-Rabbit IgG (H+L) (A-21206), Alexa Fluor→ 594 Donkey Anti-Mouse IgG (H+L) (A-21203), Alexa Fluor→ 594 Donkey Anti-Rabbit IgG (H+L) (A-21207).
Luciferase activity of AR-promoter carrying construct was measured by Secrete-Pair Dual Luminescence Assay Kit (Genecopoeia, SPDA-D010), for the rest of the constructs luciferase activity was measured by utilizing Bright Glo kit (Promega, E2620). In both cases measurements were performed on VICTOR3 spectrophotometer/luminometer (PerkinElmer).
Caspases 3 and 7 activity was detected in cell lysates incubated with Ac-DEVD-AMC (Enzo Life Sciences) - a synthetic tetrapeptide fluorogenic substrate containing the amino acid sequence of the PARP cleavage site at Asp-216. Fluorescence was read at Ex 355 and Em 485.
Immunofluorescence staining
Cells were plated on 35 mm glass-bottom plates (MatTek, p35G-0-140C) and upon treatment fixed for 10 minutes with 3%PFA in PBS with 0.3% Triton X-100, then rinsed with 0.3% Triton x100 in PBS twice and blocked for 20 minutes with 3% BSA solution at room temperature. Cells were incubated with primary antibodies (1:500 dilution in 3%BSA with 0.1% Triton X-100 in PBS) for 2 hours at room temperature or overnight at +4°C, prior to addition of secondary antibody. Secondary antibodies were diluted 1:500 in 3%BSA with 0.1% Triton X-100 and staining lasted 1 hour at room temperature. After washings cell were stained with 10 μg/ml Hoechst (Invitrogen, H3570). Images were obtained using fluorescence microscope Axio Observer A1 and AxioVision Rel.4.8 software (Zeiss).
Comet assay
CWR22R or Hela cells treated either with 1 μM of ARTIK-52 or 1μg/ml of etoposide for 6 hours. Comet assay was performed under alkali conditions using the Comet Assay kit (Trevigen, 4250-050) according to the manufacturer's protocol.
Measurement of ARTIK-52 intracellular concentration
5*10(6) cells were plated in a regular growth media. Next day cells were treated with 0, 1 or 5 μM ARTIK-52. For each condition 3 ml of corresponding solution of growth media with drug were saved for future analysis. Six h upon treatment initiation media with drug was collected from cell plates and 3 ml were stored for analysis. Cells were washed 3 times with PBS, trypsinized and collected in fresh drug-free media. Amount of collected cells was calculated for each condition and cell line. Cells were pelleted, pellet was once washed with PBS and than frozen. LC-MS detection of ARTIK-52 metabolites corresponding pick in cells extracts was preformed at Buffalo BioLabs, Inc.
Extraction of ARTIK-52 metabolites from cells
Methanol extraction of ARTIK-52 from cells was done according to.16 Briefly, 1.5 *10(6) of cells were treated with 0 or 5 μM ARTIK-52 for 6 or 20 h in regular growth media. During treatment cells were incubated as usual at 37°C and 5%CO2. Upon the end of the treatment cells were intensively washed with sterile PBS, scratched from plates and collected in 1 ml of methanol and incubated for overnight at −20°C. Then cells were spin down for 5 min at 6000 rpm at 4°C. Supertnatant was transfered into new tubes and evaporated using SpeedVac (30°C, 1 h). Each sample was reconstituted in 80 μL DMSO. 520 μl of regular growth media was added to dilute extract and 10 serial 1:2 dilutions of this solution were used in cytotoxicity assay.
Microarray hybridization
Cells were plated and treated in triplicates. Total RNA was isolated using TRIzol reagent (Invitrogen, 15596018). cRNA was synthesized and labeled using TargetAmp™-Nano Labeling Kit (Illumina) and hybridized with HumanHT-12 v4 Expression BeadChip Kit (Illumina). Data was processed and analyzed using GeneSpring GX Software (Agilent Technologies).
Fluorescence activated cell sorting analysis of cell cycle
For cell cycle analysis CWR22R cells were incubated with 1μM of ARTIK-52. Cells were collected prior to the treatment and at several time points upon treatment initiation in duplicates. Cells were collected by trypsinization and fixed in ice-cold methanol fot 20 minutes at −20°C. Cells were then pelleted, methanol – aspirated and cell pellet resuspended in 50 μg/ml propidium iodide (Invitrogen, P3566) and 100 μg/ml Ribonuclease A (Sigma- Aldrich, R6513). Samples were incubated at 37°C for 2 hours. Propidium iodide incorporation per cell was assessed using FacsCalibur (Becton Dickinson) and cell cycle distribution of cells was analyzed using ModFit LT™ (Verity Software House).
Pulse-field gel electrophoresis
Pulse-field gel electrophoresis was done as previously described. Briefly, control or treated cells were collected by trypsinisation, resuspended in 1% InCert-agarose (Lonza, 50121) in 37°C PBS to a final concentration of 1.5 million cells/100 μl. Solubilized agarose inserts were incubated in 0.5 M EDTA-1% N-laurylsarcosyl-proteinase K (1 mg/ml) at 50°C for 48h and thereafter washed 4 times in TE buffer prior to loading onto a 1% agarose (chromosomal grade) gel and separation by pulsed-field gel electrophoresis for 24 h (Bio-Rad; 120° angle, 60 to 240 s switch time, 4 V/cm). The gel was subsequently stained with ethidium bromide and analyzed with Image Gauge software (FLA-3000; Fujifilm).
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This study was supported by research grant from Panacela, Inc.
References
- 1.Tararova ND, Narizhneva N, Krivokrisenko V, Gudkov AV, Gurova KV. Prostate cancer cells tolerate a narrow range of androgen receptor expression and activity. Prostate 2007; 67:1801-15; PMID:17935158; http://dx.doi.org/ 10.1002/pros.20662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Narizhneva NV, Tararova ND, Ryabokon P, Shyshynova I, Prokvolit A, Komarov PG, Purmal AA, Gudkov AV, Gurova KV. Small molecule screening reveals a transcription-independent pro-survival function of androgen receptor in castration-resistant prostate cancer. Cell Cycle 2009; 8:4155-67; PMID:19946220; http://dx.doi.org/ 10.4161/cc.8.24.10316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen CY, Gherzi R, Ong SE, Chan EL, Raijmakers R, Pruijn GJ, Stoecklin G, Moroni C, Mann M, Karin M. AU binding proteins recruit the exosome to degrade ARE-containing mRNAs. Cell 2001; 107:451-64; PMID:11719186; http://dx.doi.org/ 10.1016/S0092-8674(01)00578-5 [DOI] [PubMed] [Google Scholar]
- 4.Sikand K, Slaibi JE, Singh R, Slane SD, Shukla GC. miR 488* inhibits androgen receptor expression in prostate carcinoma cells. Inter J Cancer 2011; 129:810-9; PMID:21710544; http://dx.doi.org/ 10.1002/ijc.25753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Provost P, Dishart D, Doucet J, Frendewey D, Samuelsson B, Radmark O. Ribonuclease activity and RNA binding of recombinant human Dicer. EMBO J 2002; 21:5864-74; PMID:12411504; http://dx.doi.org/ 10.1093/emboj/cdf578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yeap BB, Wilce JA, Leedman PJ. The androgen receptor mRNA. Bio Essays 2004; 26:672-82; PMID:15170865; http://dx.doi.org/ 10.1002/bies.20051 [DOI] [PubMed] [Google Scholar]
- 7.Yeap BB, Voon DC, Vivian JP, McCulloch RK, Thomson AM, Giles KM, Czyzyk-Krzeska MF, Furneaux H, Wilce MC, Wilce JA, et al. . Novel binding of HuR and poly(C)-binding protein to a conserved UC-rich motif within the 3'-untranslated region of the androgen receptor messenger RNA. J Biol Chem 2002; 277:27183-92; PMID:12011088; http://dx.doi.org/ 10.1074/jbc.M202883200 [DOI] [PubMed] [Google Scholar]
- 8.Epis MR, Giles KM, Barker A, Kendrick TS, Leedman PJ. miR-331-3p regulates ERBB-2 expression and androgen receptor signaling in prostate cancer. J Biol Chem 2009; 284:24696-704; PMID:19584056; http://dx.doi.org/ 10.1074/jbc.M109.030098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Epis MR, Barker A, Giles KM, Beveridge DJ, Leedman PJ. The RNA-binding protein HuR opposes the repression of ERBB-2 gene expression by microRNA miR-331-3p in prostate cancer cells. J Biol Chem 2011; 286:41442-54; PMID:21971048; http://dx.doi.org/ 10.1074/jbc.M111.301481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yeap BB, Krueger RG, Leedman PJ. Differential posttranscriptional regulation of androgen receptor gene expression by androgen in prostate and breast cancer cells. Endocrinology 1999; 140:3282-91; PMID:10385425 [DOI] [PubMed] [Google Scholar]
- 11.Safarpour D, Tavassoli FA. A Targetable Androgen Receptor-Positive Breast Cancer Subtype Hidden Among the Triple-Negative Cancers. Arch Pathol Laborat Med 2014; PMID:25310144 [DOI] [PubMed] [Google Scholar]
- 12.McNamara KM, Moore NL, Hickey TE, Sasano H, Tilley WD. Complexities of androgen receptor signalling in breast cancer. Endocr Relat Cancer 2014; 21:T161-81; PMID:24951107; http://dx.doi.org/ 10.1530/ERC-14-0243 [DOI] [PubMed] [Google Scholar]
- 13.Heiser LM, Sadanandam A, Kuo WL, Benz SC, Goldstein TC, Ng S, Gibb WJ, Wang NJ, Ziyad S, Tong F, et al. . Subtype and pathway specific responses to anticancer compounds in breast cancer. Proc Natl Acad Sci U S A 2012; 109:2724-9; PMID:22003129; http://dx.doi.org/ 10.1073/pnas.1018854108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bennett N, Hooper JD, Lee CS, Gobe GC. Androgen receptor and caveolin-1 in prostate cancer. IUBMB Life 2009; 61:961-70; PMID:19787702; http://dx.doi.org/ 10.1002/iub.244 [DOI] [PubMed] [Google Scholar]
- 15.Marquez B, Van Bambeke F. ABC multidrug transporters: target for modulation of drug pharmacokinetics and drug-drug interactions. Curr Drug Targets 2011; 12:600-20; PMID:21039335; http://dx.doi.org/ 10.2174/138945011795378504 [DOI] [PubMed] [Google Scholar]
- 16.Sheikh KD, Khanna S, Byers SW, Fornace A Jr., Cheema AK. Small molecule metabolite extraction strategy for improving LC/MS detection of cancer cell metabolome. J Biomol Tech 2011; 22:1-4; PMID:21455475 [PMC free article] [PubMed] [Google Scholar]
- 17.Lacroix M, Toillon RA, Leclercq G. p53 and breast cancer, an update. Endocr Relat Cancer 2006; 13:293-325; PMID:16728565; http://dx.doi.org/ 10.1677/erc.1.01172 [DOI] [PubMed] [Google Scholar]
- 18.Gasparian AV, Burkhart CA, Purmal AA, Brodsky L, Pal M, Saranadasa M, Bosykh DA, Commane M, Guryanova OA, Pal S, et al. . Curaxins: anticancer compounds that simultaneously suppress NF-kappaB and activate p53 by targeting FACT. Sci Trans Med 2011; 3:95ra74; PMID:21832239; http://dx.doi.org/ 10.1126/scitranslmed.3002530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu T, Li Y, Gu H, Zhu G, Li J, Cao L, Li F. p21-Activated kinase 6 (PAK6) inhibits prostate cancer growth via phosphorylation of androgen receptor and tumorigenic E3 ligase murine double minute-2 (Mdm2). J Biol Chem 2013; 288:3359-69; PMID:23132866; http://dx.doi.org/ 10.1074/jbc.M112.384289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gaughan L, Logan IR, Neal DE, Robson CN. Regulation of androgen receptor and histone deacetylase 1 by Mdm2-mediated ubiquitylation. Nucl Acids Res 2005; 33:13-26; PMID:15640443; http://dx.doi.org/ 10.1093/nar/gki141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ossovskaya VS, Mazo IA, Chernov MV, Chernova OB, Strezoska Z, Kondratov R, Stark GR, Chumakov PM, Gudkov AV. Use of genetic suppressor elements to dissect distinct biological effects of separate p53 domains. Proc Natl Acad Sci U S A 1996; 93:10309-14; PMID:8816796; http://dx.doi.org/ 10.1073/pnas.93.19.10309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alimirah F, Panchanathan R, Chen J, Zhang X, Ho SM, Choubey D. Expression of androgen receptor is negatively regulated by p53. Neoplasia 2007; 9:1152-9; PMID:18084622; http://dx.doi.org/ 10.1593/neo.07769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iyama T, Wilson DM. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair 2013; 12:620-36; PMID:23684800; http://dx.doi.org/ 10.1016/j.dnarep.2013.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhuyan BK, Fraser TJ, Gray LG, Kuentzel SL, Neil GL. Cell-kill kinetics of several S-phase-specific drugs. Cancer Res 1973; 33:888-94; PMID:4735241 [PubMed] [Google Scholar]
- 25.Caldecott KW. XRCC1 and DNA strand break repair. DNA Repair 2003; 2:955-69; PMID:12967653; http://dx.doi.org/ 10.1016/S1568-7864(03)00118-6 [DOI] [PubMed] [Google Scholar]
- 26.Missura M, Buterin T, Hindges R, Hubscher U, Kasparkova J, Brabec V, Naegeli H. Double-check probing of DNA bending and unwinding by XPA-RPA: an architectural function in DNA repair. EMBO J 2001; 20:3554-64; PMID:11432842; http://dx.doi.org/ 10.1093/emboj/20.13.3554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wold MS. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem 1997; 66:61-92; PMID:9242902; http://dx.doi.org/ 10.1146/annurev.biochem.66.1.61 [DOI] [PubMed] [Google Scholar]
- 28.Feng W, Di Rienzi SC, Raghuraman MK, Brewer BJ. Replication stress-induced chromosome breakage is correlated with replication fork progression and is preceded by single-stranded DNA formation. G3 2011; 1:327-35; PMID:22384343; http://dx.doi.org/full_text [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rigatti MJ, Verma R, Belinsky GS, Rosenberg DW, Giardina C. Pharmacological inhibition of Mdm2 triggers growth arrest and promotes DNA breakage in mouse colon tumors and human colon cancer cells. Mol Carcinogen 2012; 51:363-78; PMID:21557332; http://dx.doi.org/ 10.1002/mc.20795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng W, Thorne N, McKew JC. Phenotypic screens as a renewed approach for drug discovery. Drug Dis Today 2013; 18:1067-73; PMID:23850704; http://dx.doi.org/ 10.1016/j.drudis.2013.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee JA, Shinn P, Jaken S, Oliver S, Willard FS, Heidler S, Peery RB, Oler J, Chu S, Southall N, et al. . Novel Phenotypic Outcomes Identified for a Public Collection of Approved Drugs from a Publicly Accessible Panel of Assays. PloS One 2015; 10:e0130796; PMID:26177200; http://dx.doi.org/ 10.1371/journal.pone.0130796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eder J, Sedrani R, Wiesmann C. The discovery of first-in-class drugs: origins and evolution. Nat Rev Drug Dis 2014; 13:577-87; PMID:25033734; http://dx.doi.org/ 10.1038/nrd4336 [DOI] [PubMed] [Google Scholar]
- 33.Moffat JG, Rudolph J, Bailey D. Phenotypic screening in cancer drug discovery - past, present and future. Nat Rev Drug Dis 2014; 13:588-602; PMID:25033736; http://dx.doi.org/ 10.1038/nrd4366 [DOI] [PubMed] [Google Scholar]
- 34.Polkinghorn WR, Parker JS, Lee MX, Kass EM, Spratt DE, Iaquinta PJ, Arora VK, Yen WF, Cai L, Zheng D, et al. . Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Dis 2013; 3:1245-53; PMID:24027196; http://dx.doi.org/ 10.1158/2159-8290.CD-13-0172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang L, Hsu CL, Ni J, Wang PH, Yeh S, Keng P, Chang C. Human checkpoint protein hRad9 functions as a negative coregulator to repress androgen receptor transactivation in prostate cancer cells. Mol Cell Biol 2004; 24:2202-13; PMID:14966297; http://dx.doi.org/ 10.1128/MCB.24.5.2202-2213.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ta HQ, Gioeli D. The convergence of DNA damage checkpoint pathways and androgen receptor signaling in prostate cancer. Endocr Related Cancer 2014; 21:R395-407; PMID:25096064; http://dx.doi.org/ 10.1530/ERC-14-0217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yeh S, Hu YC, Rahman M, Lin HK, Hsu CL, Ting HJ, Kang HY, Chang C. Increase of androgen-induced cell death and androgen receptor transactivation by BRCA1 in prostate cancer cells. Proc Natl Acad Sci U A 2000; 97:11256-61; PMID:11016951; http://dx.doi.org/ 10.1073/pnas.190353897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park JJ, Irvine RA, Buchanan G, Koh SS, Park JM, Tilley WD, Stallcup MR, Press MF, Coetzee GA. Breast cancer susceptibility gene 1 (BRCAI) is a coactivator of the androgen receptor. Cancer Res 2000; 60:5946-9; PMID:11085509 [PubMed] [Google Scholar]
- 39.Shin S, Verma IM. BRCA2 cooperates with histone acetyltransferases in androgen receptor-mediated transcription. Proc Natl Acad Sci U S A 2003; 100:7201-6; PMID:12756300; http://dx.doi.org/ 10.1073/pnas.1132020100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wo JY, Zietman AL. Why does androgen deprivation enhance the results of radiation therapy? Urologic Oncol 2008; 26:522-9; PMID:18774467; http://dx.doi.org/ 10.1016/j.urolonc.2008.03.008 [DOI] [PubMed] [Google Scholar]
- 41.Bronner CE, Baker SM, Morrison PT, Warren G, Smith LG, Lescoe MK, Kane M, Earabino C, Lipford J, Lindblom A, et al. . Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature 1994; 368:258-61; PMID:8145827; http://dx.doi.org/ 10.1038/368258a0 [DOI] [PubMed] [Google Scholar]
- 42.Fishel R, Lescoe MK, Rao MR, Copeland NG, Jenkins NA, Garber J, Kane M, Kolodner R. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1994; 77:1 p following 166; PMID:8156592; http://dx.doi.org/ 10.1016/0092-8674(94)90226-7 [DOI] [PubMed] [Google Scholar]
- 43.Beiner ME, Rosen B, Fyles A, Harley I, Pal T, Siminovitch K, Zhang S, Sun P, Narod SA. Endometrial cancer risk is associated with variants of the mismatch repair genes MLH1 and MSH2. Cancer Epidemiol Biomarkers Prev 2006; 15:1636-40; PMID:16985024; http://dx.doi.org/ 10.1158/1055-9965.EPI-06-0257 [DOI] [PubMed] [Google Scholar]
- 44.Jasperson KW, Tuohy TM, Neklason DW, Burt RW. Hereditary and familial colon cancer. Gastroenterology 2010; 138:2044-58; PMID:20420945; http://dx.doi.org/ 10.1053/j.gastro.2010.01.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, et al. . A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994; 266:66-71; PMID:7545954; http://dx.doi.org/ 10.1126/science.7545954 [DOI] [PubMed] [Google Scholar]
- 46.Hall JM, Lee MK, Newman B, Morrow JE, Anderson LA, Huey B, King MC. Linkage of early-onset familial breast cancer to chromosome 17q21. Science 1990; 250:1684-9; PMID:2270482; http://dx.doi.org/ 10.1126/science.2270482 [DOI] [PubMed] [Google Scholar]
- 47.Maia S, Cardoso M, Paulo P, Pinheiro M, Pinto P, Santos C, Pinto C, Peixoto A, Henrique R, Teixeira MR. The role of germline mutations in the BRCA1/2 and mismatch repair genes in men ascertained for early-onset and/or familial prostate cancer. Familial Cancer 2015; PMID:26289772 [DOI] [PubMed] [Google Scholar]
- 48.Goldgar DE, Neuhausen SL, Steele L, Fields P, Ward JH, Tran T, Ngyuen K, Stratton MR, Easton DF. A 45-year follow-up of kindred 107 and the search for BRCA2. J Natl Cancer Inst Monogr 1995:15-9; PMID:8573446 [PubMed] [Google Scholar]
- 49.Gurova KV, Hill JE, Razorenova OV, Chumakov PM, Gudkov AV. p53 pathway in renal cell carcinoma is repressed by a dominant mechanism. Cancer Res 2004; 64:1951-8; PMID:15026329; http://dx.doi.org/ 10.1158/0008-5472.CAN-03-1541 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.